

Abstract
One major class of disease-causing RNAs is expanded repeating transcripts. These RNAs cause diseases via multiple mechanisms, including: (i) gain-of-function, in which repeating RNAs bind and sequester proteins involved in RNA biogenesis and (ii) repeat associated non-ATG (RAN) translation, in which repeating transcripts are translated into toxic proteins without use of a canonical, AUG, start codon. Herein, we develop and study chemical probes that bind and react with an expanded r(CGG) repeat (r(CGG)exp) present in a 5′ untranslated region that causes fragile X-associated tremor/ataxia syndrome (FXTAS). Reactive compounds bind to r(CGG)exp in cellulo as shown with Chem-CLIP-Map, an approach to map small molecule binding sites within RNAs in cells. Compounds also potently improve FXTAS-associated pre-mRNA splicing and RAN translational defects, while not affecting translation of the downstream open reading frame. In contrast, oligonucleotides affect both RAN and canonical translation when they bind to r(CGG)exp, which is mechanistically traced to a decrease in polysome loading. Thus, designer small molecules that react with RNA targets can be used to profile the RNAs to which they bind in cells, including identification of binding sites, and can modulate several aspects of RNA-mediated disease pathology in a manner that may be more beneficial than oligonucleotides.
Introduction
Although RNA targets in the transcriptome are numerous, there is a dearth of small molecule chemical probes that can be used to study RNA function and dysfunction. Despite great interest in this area, the development of such compounds is difficult(1) owing to a lack of fundamental information, or design principles, that could enable the development of compounds that selectively target an RNA in a cell.(2, 3) One approach to design compounds that affect function is to study RNA motif–small molecule interactions, thereby identifying small molecule “modules” that bind regions of an RNA of interest. Affinity and selectivity of the modules can be improved by linking them together to bind two or more regions in the desired RNA simultaneously. Indeed, such a bottom-up approach has been used to design compounds that target repeating transcripts and other RNAs.(4, 5)
The use of small molecules to modulate RNA function is of particular interest for studying various aspects of disease pathology. Repeating transcripts cause diseases via a gain-of-function mechanism or by translation into toxic proteins with or without the use of a start codon (Figure 1).(6) A common defect caused by RNA repeat gain-of-function is dysregulation of alternative pre-mRNA splicing.(7) For example, fragile X-associated tremor/ataxia syndrome (FXTAS) is caused by an expanded r(CGG) repeat (r(CGG)exp) that binds and sequesters various proteins including DiGeorge syndrome critical region 8 protein (DGCR8), Src-associated in mitosis 68 kDa (Sam68), and others.(8, 9) Sequestration of these proteins causes dysregulation of microRNA processing and alternative pre-mRNA splicing.(8, 9) As has been demonstrated for the RNAs that cause the myotonic dystrophies (DM), amyotrophic lateral sclerosis and frontal temporal dementia (ALS/FTD), and FXTAS, expanded repeating RNAs are also translated without the use of a start codon, or repeat associated non-ATG (RAN) translation.(10-12) RAN translation produces toxic polymeric proteins that appear to contribute to disease.
Figure 1.
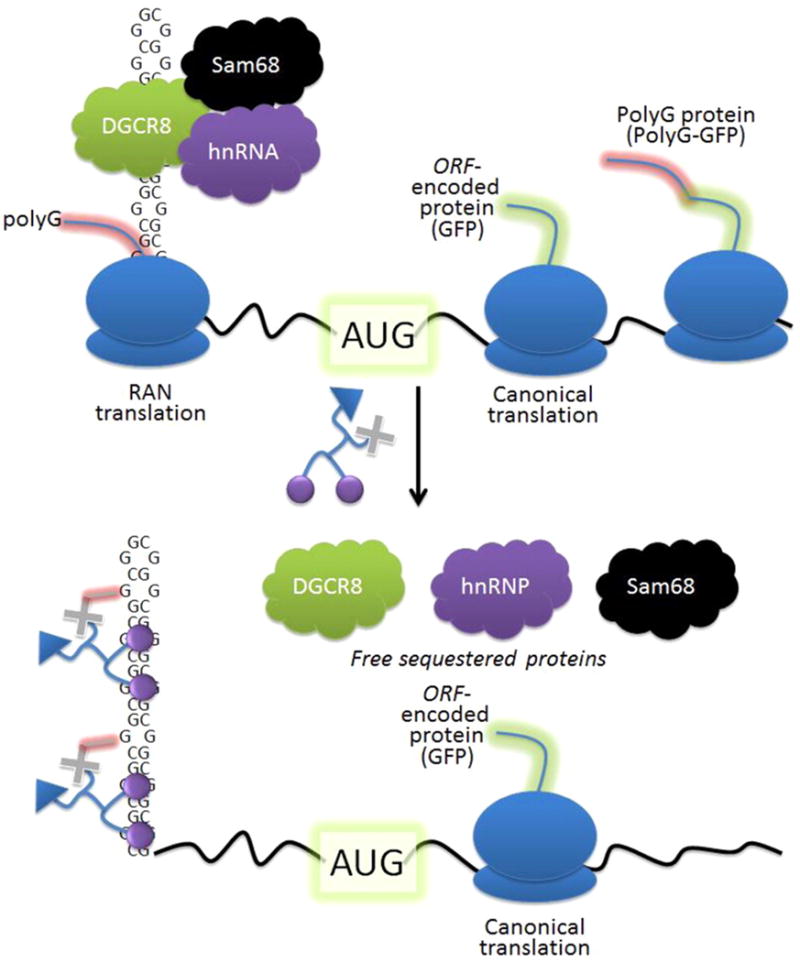
Modes of toxicity associated with r(CGG)exp, the causative agent of FXTAS. (Top) The repeating RNA folds into a hairpin structure that binds and sequesters proteins that regulate RNA processing. Additionally, repeating transcripts are translated without a start codon in a mechanism called RAN translation, producing toxic polymeric proteins that contribute to pathogenesis. (Bottom) Small molecules that bind to and react with r(CGG)exp and free bound proteins improve defects in RNA processing and inhibit production of RAN, but not normal, translation products.
We sought to determine if we could design small molecules that address both modes of toxicity using r(CGG)exp as a model system (Figure 1). Ideally, the designed compound would improve alternative pre-mRNA splicing defects and inhibit RAN translation while having no effect on translation of the downstream open reading frame (ORF). It is particularly important that translation of this downstream ORF is not affected; r(CGG)exp is located in the 5′ untranslated region (UTR) of the fragile X mental retardation 1 (FMR1) gene, which encodes fragile X mental retardation protein (FMRP). FMRP is important for regulation of protein synthesis.(13) In fragile X syndrome (FXS), which is also caused by a r(CGG) expansion, albeit larger than those that cause FXTAS, FMRP is silenced; mice that do not produce FMRP have learning defects and hyperactivity.(14) There are likely spatial aspects that govern whether RAN and/or canonical translation is inhibited, as has been previously shown in engineered systems.(15, 16) Thus, a focus of the present studies is to examine such aspects in systems in which translational events resemble those found in disease-associated mRNAs and how such events can be advantageously modified.
Previously, we designed the small molecule 2H-5 (Figure 2A) to target r(CGG)exp by mining interactions in a RNA motif-small molecule database.(17) The compound contains two RNA-binding modules (H) that are appropriately spaced to target two 5′CGG/3′GGC motifs in r(CGG)exp simultaneously (Figure 2A). Indeed, 2H-5 improves pre-mRNA splicing defects caused by sequestration of Sam68 at μM concentrations and does not affect translation of a model downstream ORF.(17) In this study, we sought to improve 2H-5's potency by appending it to a reactive module akin to our previously reported studies of a designed small molecule that targets r(CUG)exp,(3) the causative agent of DM type 1. To engender 2H-5 with the ability to react with cellular RNA targets, it was coupled to a nitrogen mustard (CA or MA, Figure 2A).(3) Since such compounds prefer to react with the N-7 of G,(18) we hypothesized that r(CGG)exp might be an ideal RNA to which to extend this approach. A biotin tag was also incorporated onto 2H-5 to allow for the facile isolation of the RNA targets it binds in cells (via Chemical Cross-Linking and Isolation by Pull-down, or Chem-CLIP),(3) affording 2H-5-CA-Biotin and 2H-5-MA-Biotin (Figure 2A). We also synthesized 2P-5-CA-Biotin and 2P-5-MA-Biotin, control compounds that lack the RNA-binding modules, to study the effect on selective targeting of r(CGG)exp. (Please see the Supporting Information for details of chemical syntheses, in particular Schemes S-1–3 and Figure S-1.) Importantly, the reactive compounds bind the desired target in cells and potently improve FXTAS-associated pre-mRNA splicing and RAN translational defects, while not affecting translation of the downstream ORF.
Figure 2.
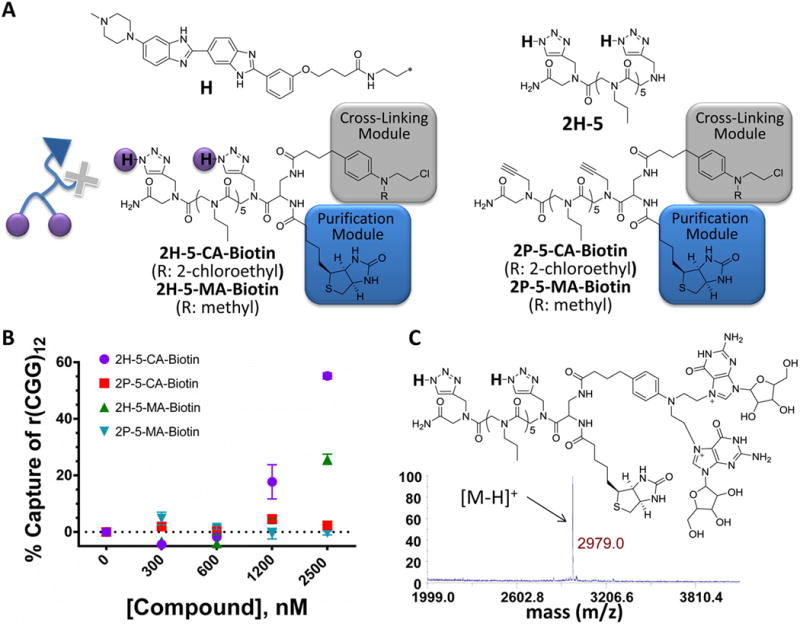
Structures of the compounds used in these studies. (A) 2H-5-CA-Biotin is comprised of two RNA-binding modules (purple circles), a reactive module (gray box), and a biotin purification module (blue box) to improve potency and enable identification of cellular targets. 2H-5 noncovalently binds r(CGG)exp and was described previously.(17) 2P-5-CA-Biotin and 2P-5-MA-Biotin lack RNA binding modules. 2H-5-MA-Biotin has a single reactive group. (B) Studying the reaction of designer small molecules with RNA in vitro. (C) MS analysis of the 2H-5-CA-Biotin-r(CGG)12 adduct generated in vitro.
Results and Discussion
Studying the Reaction of Designer Small Molecules with RNA in Vitro
Compounds 2H-5-CA-Biotin, 2H-5-MA-Biotin, 2P-5-CA-Biotin, and 2P-5-MA-Biotin were tested for selective reaction with r(CGG)12. The compounds contain two RNA-binding modules (H), a reactive module (CA or MA), and a purification tag (Biotin) (Figure 2A). Control compounds (P) without RNA-binding modules were also tested. The CA module on 2H-5-CA-Biotin can react with two sites (typically an intrastrand cross-link)(19) within a single target RNA because two N-chloroethyl groups are present. In contrast, 2H-5-MA-Biotin only has one reactive N-chloroethyl group and is thus incapable of forming intrastrand cross-links.(19) Comparative studies of these two compounds facilitate an understanding of how different reactivity modes can affect biological activity and target recognition.
Initial in vitro assays were completed by using 32P-labeled r(CGG)12. In these experiments, the biotin tag installed on the compounds allowed capture of reacted RNAs with streptavidin-coated resin. This allowed us to determine the amount of RNA that reacted with a given small molecule via scintillation counting. These studies showed that the order of reaction was 2H-5-CA-Biotin > 2H-5-MA-Biotin > 2P-5-CA-Biotin > 2P-5-MA-Biotin (Figure 2B). Thus, addition of the RNA-binding modules enhances the ability of the compounds to react with RNA in vitro, and the type of reactive module used influences compound reactivity.
The products of the in vitro reaction between r(CGG)12 and 2H-5-CA-Biotin or 2P-5-CA-Biotin were further studied. After incubation with both 2H-5-CA-Biotin and 2P-5-CA-Biotin, r(CGG)12 was treated with P1 nuclease, and the reaction products were identified by mass spectral analysis. The observed mass of the reaction products corresponds to a 2H-5-CA-Biotin adduct that reacted with two guanosines (Figure 2C), consistent with the preferred reactivity of chlorambucil (N-7 of guanine).(18) Importantly, no adduct formation between r(CGG)12 and 2P-5-CA-Biotin was detected. Collectively, these studies show that 2H-5-CA-Biotin reacts with r(CGG) repeats in vitro and that the RNA-binding modules (H) are required.
Studying the Cellular Activity of Designer Compounds: Inhibition of RAN Translation
Next, we studied if the above compounds inhibit translation events of a r(CGG)exp-containing transcript, including both RAN and canonical translation. For these studies, we employed a cellular model system in which r(CGG)88 was embedded in the 5′ UTR of green fluorescent protein (GFP), which was previously designed and validated by the Todd and Paulson groups.(11) The construct was developed such that the orientation of r(CGG)88 in the 5′ UTR relative to the GFP ORF mimics that between r(CGG)exp and the ORF in FMR1. Two translation products are observed in cells: the canonical translation product, GFP, and the RAN translation product, polyG-GFP. The two products can be resolved by SDS-PAGE and visualized by Western blot with an anti-GFP antibody as previously described.(11)
Indeed, 2H-5-CA-Biotin decreases the RAN translation product by ∼40% at 500 nM but does not significantly affect translation of GFP initiated at the canonical translational start site (Figures 3A and S-2). Further, a dose response is observed. Although 2H-5-MA-Biotin also inhibits RAN translation but not canonical translation, it is less potent (Figure 3A); at 1 μM, 2H-5-MA-Biotin and 2H-5-CA-Biotin inhibit 23% and 47% of RAN translation, respectively. Interestingly, the extent of inhibition of RAN translation correlates with in vitro reactivity toward 32P-labeled r(CGG)12 for each compound (Figure 2B). We also employed a construct in which the ATG start codon was removed from the r(CGG)88-GFP construct (Figure 4A). As expected, only the RAN translation product was observed, and its abundance was similar to the amount of RAN product observed for the plasmid containing an ATG start codon (Figure S-6). In this system, 2H-5-CA-Biotin inhibits ∼65% of RAN translation at 1 μM compound concentration (Figure 4B).
Figure 3.
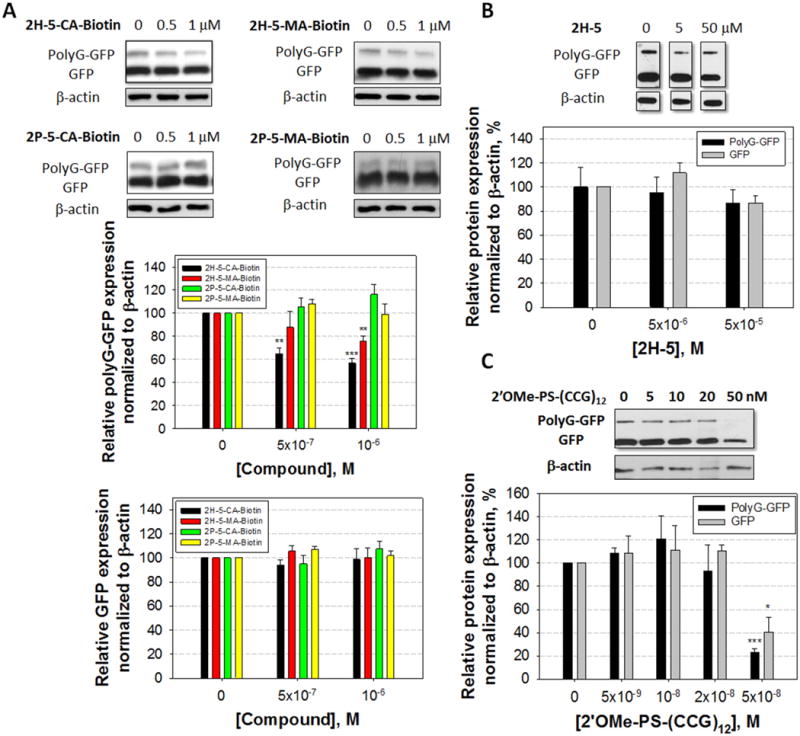
Effects of small molecules and an oligonucleotide on protein translation in cells. (A) Effect of 2H-5-CA-Biotin, 2H-5-MA-Biotin, 2P-5-CA-Biotin, and 2P-5-MA-Biotin on RAN translation of r(CGG)exp and canonical translation of the downstream ORF that encodes GFP, as determined by Western blotting. (B) Effect of 2H-5 on RAN and canonical translation. (C) Effect of a 2′OMe phosphorothioate oligonucleotide, 2′OMe-PS-(CCG)12, on RAN and canonical translation. *, **, ***, and **** denote p < 0.05, p < 0.01, p < 0.001, and p < 0.0001, respectively, as compared to the untreated sample with a two-tailed Student t test (n ≥ 3 for compounds and n = 3 for 2′OMe-PS-(CCG)12). Values are reported as mean ± standard error.
Figure 4.
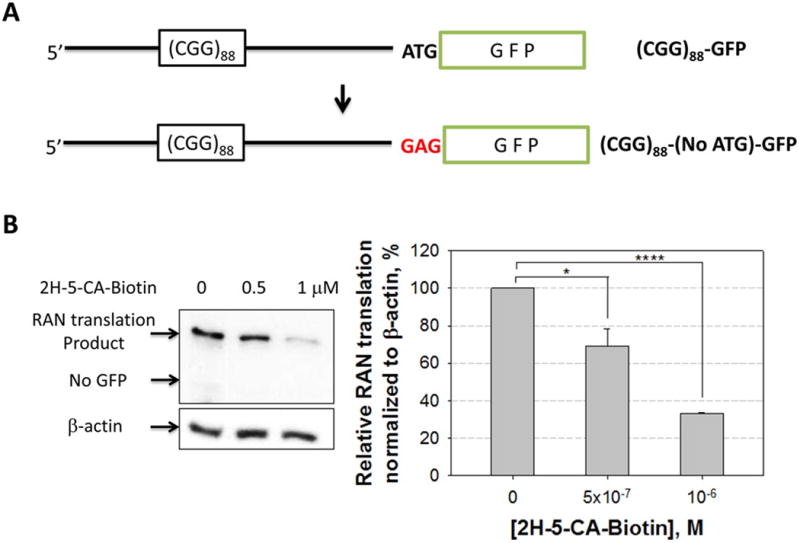
Modification of (CGG)88-GFP to afford a construct without an ATG start codon and the effect of 2H-5-CA-Biotin on RAN translation in the absence of AUG. (A) Schematic of the (CGG)88-(No ATG)-GFP construct. (B) Western blot image and quantification of relative amounts RAN translation products in the presence or absence of 2H-5-CA-Biotin when the AUG start codon is removed. * and **** denote p < 0.05 and p < 0.0001, respectively, as determined by a two-tailed Student t test (n = 3). Values are reported as mean ± standard error.
To further study the features of compounds that allows for inhibition of RAN translation, we studied the control compound that lacks the RNA-binding modules, 2P-5-CA-Biotin, and the noncovalent binder 2H-5 (Figures 2B and 3A). Neither canonical nor RAN translation were inhibited when cells were treated with 50 μM 2H-5 or 1 μM 2P-5-CA. Collectively, these studies show that both a reactive module and RNA-binding modules are required for inhibition of RAN translation in this system (Figure 3A and 3B). We also tested the effect of 2H-5-CA-Biotin on r(CGG)88-GFP mRNA levels (Figure S-3) and translation of GFP when the 5′ UTR lacks r(CGG)exp (Figure S-4). No statistically significant changes were observed for protein expression or mRNA abundance. The compound exhibited no significant cytotoxicity via a MTT assay at the concentrations used in biological assays (Figure S-5). Collectively, these studies suggest that the effects are due to reaction at specific sites within the mRNA, likely r(CGG)exp in the 5′ UTR, vide infra. It also shows that it is possible to inhibit RAN translation of an expanded repeat embedded in a 5′ UTR with a small molecule without significantly affecting translation of the downstream ORF.
We also studied if a 2′OMe phosphorothioate oligonucleotide (2′OMe-PS-(CCG)12) that is complementary to r(CGG)exp modulates RAN and canonical translational events. The oligonucleotide inhibits both RAN and canonical translation (Figure 3C), and selective inhibition of RAN translation could not be obtained despite using a 2-fold alteration in treatment concentrations. We also tested the effect of the oligonucleotide on r(CGG)88-GFP mRNA levels; these data showed that there was no effect on the mRNA's abundance (Figure S-7). The oligonucleotide had no effect on translation of GFP if the 5′ UTR lacks r(CGG)exp (Figure S-8). It has been previously observed that the binding of oligonucleotides to the 5′ UTR can affect translation.(20) It is likely that the oligonucleotide inhibits both RAN and canonical translation by either preventing ribosome loading or inhibiting ribosomal read-through, vide infra.
Studying the in Cellullo Activity of Designer Compounds: Improvement of FXTAS-Associated Pre-mRNA Splicing Defects
Since 2H-5-CA-Biotin improves one FXTAS-associated defect (RAN translation), we next determined if the compound improves alternative pre-mRNA splicing defects. The ability of 2H-5-CA-Biotin to improve FXTAS-associated pre-mRNA splicing defects was evaluated using a plasmid that encodes r(CGG)60 and a mini-gene that reports on the alternative splicing of survival motor neuron 2 (SMN2) mRNA.(21) It was previously shown that the alternative splicing of SMN2 exon 7 is dysregulated in FXTAS by sequestration of Sam68 by r(CGG)exp.(8) In the presence of r(CGG)60, exon 7 is included in ≈60% of the mature mRNA, while in the absence of r(CGG)60, exon 7 is included in ≈20% (Figure 5A).
Figure 5.
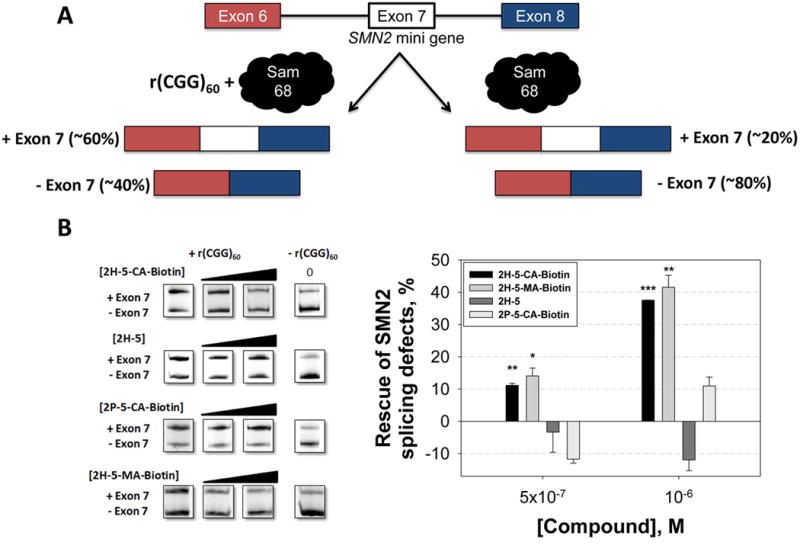
Improvement of FXTAS-associated pre-mRNA splicing defects by 2H-5-CA-Biotin. (A) Schematic of the alternative pre-mRNA splicing of SMN2 mini-gene in the presence and absence of r(CGG)exp. (B) Cellular efficacy of 2H-5-CA-Biotin, 2H-5, and 2P-5-CA-Biotin against FXTAS as assessed by improvement in SMN2 pre-mRNA splicing defects. ** and *** denote p < 0.01 and p < 0.001, respectively, as compared to the untreated sample by a two-tailed Student t test (n = 3). Values are reported as the mean ± standard error.
Treatment of cells with 1 μM of 2H-5-CA-Biotin improved the SMN2 splicing defect by ∼40%, similarly to 2H-5-MA-Biotin (Figure 5B). Neither compound affected SMN2 splicing in cells that did not express r(CGG)60 (Figure S-9). Further, no significant effect on SMN2 splicing was observed when cells were treated with 1 μM 2H-5 or 2P-5-CA-Biotin. The observed bioactivity of 2H-5-CA-Biotin was not due to changes in r(CGG)60 abundance as there was no effect on transcription as determined by qRT-PCR (Figure S-10). In agreement with our RAN translation studies (Figure 3), these studies show that both covalent binding and the RNA-binding modules are required for the observed bioactivity (Figure 5B).
Chem-CLIP Evaluation of the Cellular Targets of the Designer Small Molecules
Compounds were evaluated for the RNA targets they reacted with in cells by using a reactive profiling approach named Chem-CLIP (Chemical Cross-Linking and Isolation by Pull-down),(3) a small molecule approach similar to cross-linking and immunoprecipitation (CLIP).(22) For these studies, we employed the model cellular model system described above for RAN translation.(11) Cells were treated with 2H-5-CA-Biotin, 2H-5-MA-Biotin, or 2P-5-CA-Biotin, and cellular targets were isolated using streptavidin-coated resin. Bound targets were released from the resin and analyzed via qRT-PCR (Figure 6A).(3) Relative to the starting total RNA, a 24-fold enrichment of r(CGG)88-GFP was observed in the pulled down fractions from cells treated with 2H-5-CA-Biotin. Compound 2H-5-MA-Biotin was also able to enrich r(CGG)88-GFP by ∼6-fold. There was no enrichment in r(CGG)88-GFP upon treatment with 2P-5-CA-Biotin, consistent with our in vitro reactivity studies (Figure 2B). Only a minor enrichment of the GFP mRNA was observed when cells that express GFP lacking r(CGG)exp were treated with 2H-5-CA-Biotin (Figure 6A). Taken together, these studies show that 2H-5-CA-Biotin reacts with the intended target in cellulo.
Figure 6.
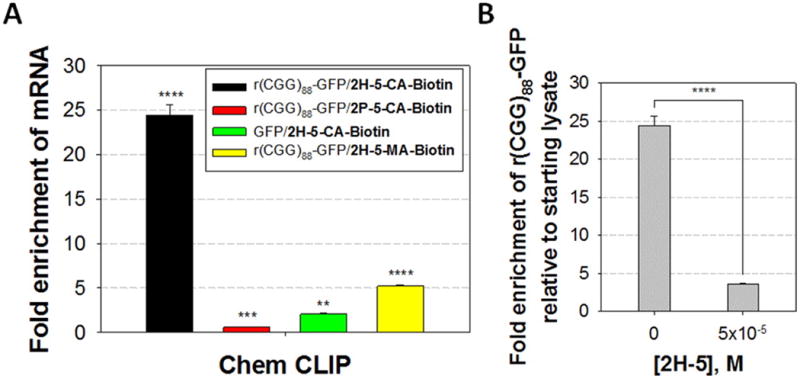
Identification of the cellular RNA targets of 2H-5-CA-Biotin, 2H-5-MA-Biotin, 2H-5, and 2P-5-CA-Biotin. (A) Results of Chem-CLIP pull-down of RNA targets in cells upon exposure to reactive compounds. An ∼24- and ∼6-fold enrichment of r(CGG)88-GFP relative to starting lysate was observed in the pulled down fraction from cells treated with 2H-5-CA-Biotin (500 nM) and 2H-5-MA-Biotin (1 μM), respectively. No enrichment was observed when cells were treated with 2P-5-CA-Biotin. Further, r(CGG)88 is required for pull down by 2H-5-CA-Biotin as an ∼13-fold decreased enrichment of GFP mRNA was observed in cells that express a construct lacking r(CGG)exp in the 5′ UTR. (B) Results of competitive Chem-CLIP experiment. Cells were cotreated with 2H-5 and 2H-5-CA-Biotin. The depletion of the r(CGG)88-GFP target confirms that 2H-5 binds to r(CGG)88-GFP. **, ***, and **** denote p < 0.01, p < 0.001, and p < 0.0001, respectively, (compared to the starting lysate for A) as determined by a two-tailed Student t test (n = 3). Values are reported as the mean ± standard error.
To assess the extent to which the noncovalent binder binds to r(CGG)88-GFP in cellulo, we completed a Competitive Chem-CLIP (C-Chem-CLIP) experiment. In C-Chem-CLIP, cells are cotreated with 2H-5 and 2H-5-CA-Biotin, and depletion of RNA targets captured by 2H-5-CA-Biotin is assessed (indicates binding of 2H-5). The results indicate that 2H-5 can compete with 2H-5-CA-Biotin for binding to r(CGG)exp in cells, confirming that both the covalent and the noncovalent compound engage the target (Figure 6B). Thus, noncovalent recognition of 5′(CGG)exp in cells by 2H-5 is not sufficient to affect a translational event, whereas covalent reaction is. This observation may be due to the degree of thermodynamic stabilization of r(CGG)exp upon compound binding; that is, a minimum amount of stabilization is required to prevent ribosomal read-through or loading. We therefore measured the thermodynamic stability imparted to r(CGG)12 by 2H-5-CA-Biotin and 2H-5 by optical melting. These experiments showed that reaction of 2H-5-CA-Biotin with r(CGG)12 as compared to the RNA alone increases the Tm by >10 °C (Figure S-11). In contrast, binding of 2H-5 has a negligible effect on Tm. Taken together, 2H-5-CA-Biotin inhibits either: (i) ribosome loading onto r(CGG)exp; and/or (ii) ribosomal read-through by forming a steric or thermal block within the repeats that does affect ribosome loading onto the downstream ORF. That is, the ribosome can shunt through to the canonical AUG start codon. The latter mechanism is supported by studies in which oligonucleotides that target an ORF only inhibit translation when they are conjugated to psoralen and hence cross-linked with the mRNA; noncovalent binders had no effect on translation.(20) These studies are also consistent with 2H-5's inability to inhibit RAN translation despite binding to r(CGG)exp in cellulo; binding does not enhance the RNA's thermodynamic stability sufficiently enough to impede ribosomal read-through.
Chem-CLIP-Map Enables Mapping of Small Molecule Binding Sites in Cellulo
Given that Chem-CLIP allowed for the targeted pull-down of the r(CGG)88-GFP transcript, we sought to determine the site of reaction. This could be completed by primer extension, as has been described to identify sites of chemical modification by dimethyl sulfate (DMS) or kethoxol to map RNA secondary structure.(23) Such an approach is intractable for mapping small molecule binding sites in r(CGG)exp, or other repeating transcripts, due to their highly structured, GC-rich nature that complicates read-through by reverse transcriptase. Thus, we mapped the ligand-binding site by using an alternative approach named Chem-CLIP-Map. In this approach, cells are treated with a reactive compound, and total RNA is harvested. The harvested RNA is then cleaved by treatment with an antisense oligonucleotide and RNase H (Figure S-12).(24) The cleaved RNA fragments are then captured onto streptavidin resin and quantified by qRT-PCR to identify the region(s) within the mRNA that reacted with compound (Figure 7). In this application, the antisense oligonucleotide binds between r(CGG)exp and the AUG start codon. If the compound reacts with r(CGG)exp as expected, then the 5′ region of the RNA should be pull-down by streptavidin resin, while the remainder of the RNA (the ORF) should not.
Figure 7.
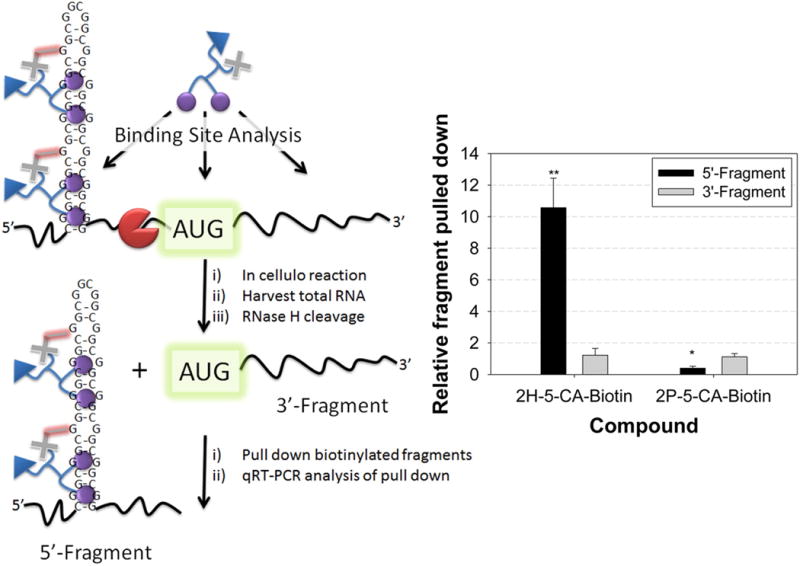
Binding site mapping for reactive compounds. (A) Schematic of Chem-CLIP-Map. (B) Results for 2H-5-CA-Biotin and 2P-5-CA-Biotin. The enrichment of the 5′ fragment by 2H-5-CA-Biotin indicates the small molecule binds to the 5′ UTR in the mRNA. * and ** denote p < 0.05 and p < 0.01, respectively, as compared to samples before pull-down by a two-tailed Student t test (n = 2). Values are reported as the mean ± standard error.
As expected, 2H-5-CA-Biotin reacted preferentially with the 5′ end of the r(CGG)88-GFP mRNA, where the repeat is present (Figure 7). No enrichment was observed for 2P-5-CA-Biotin. Thus, the RNA-binding modules engender the compound with its reaction preference for the r(CGG)exp-containing mRNA in cells. These studies support a mechanism for inhibition of FXTAS-associated defects in which the small molecule reacts with r(CGG)exp, displacing bound proteins or preventing their binding altogether (improvement of alternative pre-mRNA splicing defects) and simultaneously inhibiting ribosome recognition/loading of r(CGG)exp (RAN translation) but not the downstream ORF.
Studying the Mechanism of Compound Inhibition of RAN Translation via Polysome Profiling
Polysome profiling is a powerful tool to study mRNAs bound by the ribosome and undergoing active translation. We therefore used polysome profiling to further investigate the modes of action of 2H-5-CA-Biotin and 2′OMe-PS-(CCG)12. COS7 cells were transfected with the (CGG)88-GFP plasmid and treated with 2H-5-CA-Biotin or 2′OMe-PS-(CCG)12. A sucrose gradient was then used to separate RNAs bound to single ribosomes and polysomes (Figure 8A and 8B). The distribution of r(CGG)88-GFP in each fraction was then determined by qRT-PCR. Compared to the untreated control, no significant change in the distribution of r(CGG)88-GFP was observed after treatment with 500 nM 2H-5-CA-Biotin (Figures 8C and S-13). In contrast, treatment with 2′OMe-PS-(CCG)12 decreased the amount r(CGG)88-GFP mRNA associated with active polysomes by 27% (Figures 8C and S-13), consistent with its inhibition of both RAN and canonical translation events shown in Figure 3C. The control transcript (GFP mRNA lacking r(CGG)exp) showed no significant change in its distribution after 2H-5-CA-Biotin or 2′OMe-PS-(CCG)12 treatment. We further studied the effect of 2H-5-CA-Biotin on ribosome loading using a r(CGG)88-GFP that lacks a start codon. As GFP is not translated in this system, we expected a larger effect on polysome loading upon compound treatment as compared to the r(CGG)88-GFP construct containing a start codon. Indeed, 2H-5-CA-Biotin reduced the amount of the transcript that lacks the start codon associated with polysomes by 10% relative to untreated sample (Figure S-14), indicating the reactive compound inhibits RAN translation in this system and does so by at least in part by reducing the amount of mRNA bound by ribosomes.
Figure 8.
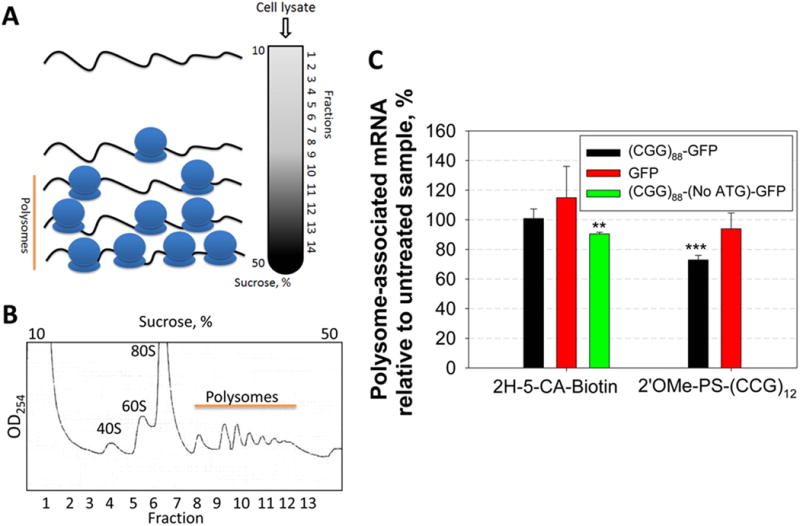
Polysome profiling of cells treats with 2H-5-CA-Biotin and 2′OMe-PS-(CCG)12. (A) Schematic of polysome profiling. (B) Representative polysome profile. (C) Plot of the number of polysomes associated with r(CGG)88-GFP upon treatment with 2H-5-CA-Biotin and 2′OMe-PS-(CCG)12. While no change in the amount of r(CGG)88-GFP and GFP (control) mRNA associated with translationally active polysomes was observed after 2H-5-CA-Biotin treatment, the oligonucleotide decreased the number of associated polysomes by ∼30%. 2H-5-CA-Biotin diminished the number of polysomes associated with r(CGG)88-(No ATG)-GFP, which lacks a start codon by ∼10%. ** denotes p < 0.01 and *** denotes p < 0.001, as compared to the untreated sample with a two-tailed Student t test (n = 2).
Mechanistic Considerations for 2H-5-CA-Biotin and 2′OMe-PS-(CCG)12
Taken together, our data beg the questions: (i) how does 2H-5-CA-Biotin selectively inhibit RAN translation?; and (ii) why does the oligonucleotide inhibit both RAN and canonical translation? (Figure 9). The lack of selectivity by the oligonucleotide is likely due to its formation of an extraordinarily stable secondary structure 5′ to the canonical start codon. In order for a stable secondary structure to inhibit initiation, it must be close to the 5′ cap.(25, 26) Stable secondary structures distal to the cap can inhibit translation/elongation by preventing unwinding by helicase eIF4A, provided that their ΔG° is at least −50 kcal/mol.(25, 26) Since binding of 2′OMe-PS-(CCG)12 occurs distal to the 5′ cap, >100 nucleotides downstream, it is therefore likely that 2′OMe-PS-(CCG)12 inhibits ribosome read-through/processivity during both RAN translation and ribosome shunting to the canonical AUG start codon. Indeed, the predicted free energy of the duplex formed between a single 2′OMe-PS-(CCG)12 and r(CGG)12 (up to seven oligonucleotides could bind to each transcript) is −101.7 kcal/mol. In contrast, the binding of our small molecule thermodynamically stabilizes the repeat but does not significantly affect ribosome loading. This suggests: (i) initiation of RAN and canonical translation is not inhibited; (ii) the cross-link and thermodynamic stabilization created by the small molecule inhibit elongation of ribosomes bound to r(CGG)exp, likely preventing unwinding by helicase eIF4a; and (iii) the small molecule does not prevent scanning of the ribosome to identify the canonical AUG start site since canonical translation is not affected. Ribosome shunting has been previously observed to bypass stable secondary structures to identify the canonical AUG start codon.(27, 28)
Figure 9.
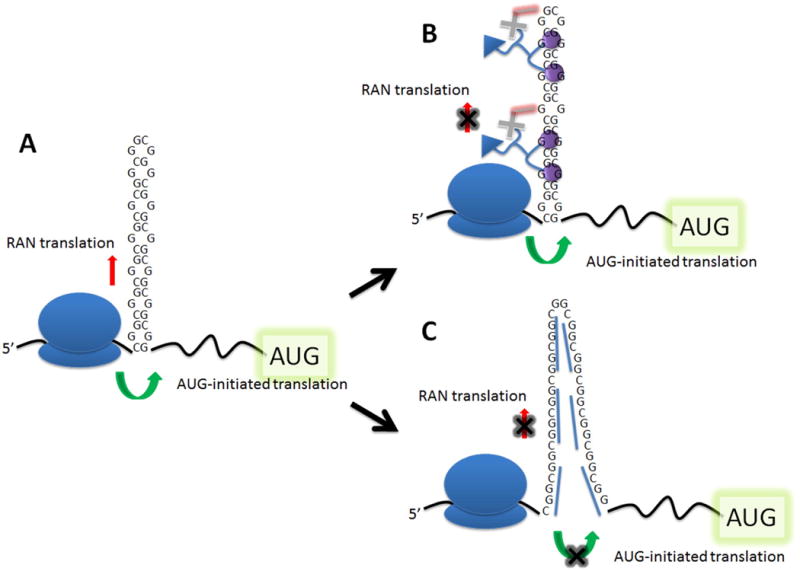
Possible mechanisms of inhibition of RAN translation in FXTAS by 2H-5-CA-Biotin and 2′OMe-PS-(CCG)12. (A) Schematic of RAN and canonical translational events in FXTAS. (B) Schematic of a potential mode of action for the selective inhibition of RAN translation by 2H-5-CA-Biotin. (C) Schematic of a potential mode of active for the inhibition of RAN and canonical translation events by 2′OMe-PS-(CCG)12.
Conclusion
In summary, we have demonstrated that a designer small molecule that binds and reacts with an RNA target covalently, 2H-5-CA-Biotin, is a more potent inhibitor than a related compound that binds noncovalently (2H-5). In particular, 2H-5-CA-Biotin improves pre-mRNA splicing defects and selectively inhibits RAN translation in a FXTAS cellular model. Compounds that modulate noncanonical translational events could have broad applicability. For example, proteome-wide profiling in viruses and human cell lines has shown that many peptides are translated without the use of start codons.(11) There are many speculations about the functional consequences of these proteins; one way to study them is to develop inhibitors of their production, as shown herein.
Materials and Methods
General
The RNA used in in vitro reactions (r(CGG)12) was purchased from Dharmacon and deprotected per the manufacturer's recommended protocol. DNA primers used for RT-PCR were purchased from Integrated DNA Technologies, Inc (IDT). Western blot images were quantified with QuantityOne version 4.6.5 software. Mass spectra were collected on an ABI 4800 MALDI-TOF spectrometer. Statistical significance was computed by using a student t test and *, **, ***, and **** indicate p < 0.05; p < 0.01; p < 0.001; p < 0.0001, respectively.
Reaction of Small Molecules with r(CGG)12
The r(CGG)12, 5′-GCGGCGGCGGCGGCGGCGGCGGCGGCGGCGGCGGCGGC, was 5′-end-labeled with 32P using [γ-32P] ATP. To determine if 2H-5-CA-Biotin, 2P-5-CA-Biotin, 2H-5-MA-Biotin, and 2P-5-MA-Biotin react with r(CGG)12 in vitro, 5 μL of 32P-labeled r(CGG)12 (∼1,000,000 cpm) was diluted in a total volume of 300 μL of 1× PBS (10 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, and 2.7 mM KCl, pH 7.4). The RNA was folded by heating at 60 °C for 5 min and slowly cooling to room temperature. Compound was then added at various concentrations, and the solutions were incubated for at least 4 h at room temperature. Next, 10 μL of streptavidin resin (high capacity streptavidin agarose beads; Thermo Scientific) was added to the samples which were then incubated for 30 min at room temperature. After centrifugation, the supernatant was removed, and the resin was washed with 1× PBST (1× PBS + 0.1% Tween 20). The amount of radioactivity in the supernatant, wash, and associated with the beads was measured using a Beckman Coulter LS6500 liquid scintillation counter.
MS Analysis of the 2H-5-CA-Biotin-r(CGG)12 Adduct
The r(CGG)12 RNA (1 μM) was folded in 1× PBS by heating at 95 °C followed by slowly cooling to room temperature. To this solution was added 2H-5-CA-Biotin (1 μM) and 2P-5-CA-Biotin (1 μM) in a total volume of 10 μL. The sample was incubated at 37 °C for 24 h. The RNA was digested with nuclease P1 (1 unit) at 37 °C for 19 h, and the solution was then desalted by ZipTip Pipette Tip (EMD Millipore). The products of the reaction were analyzed by MALDI-TOF mass spectrometry.
Quantifying RAN and Canonical Translation Products by Western Blot
COS7 cells were grown as monolayers in 96-well plates in 1× DMEM containing 10% FBS and 1× Glutagro (growth medium). After the cells reached 90–95% confluency, they were transfected with 150 ng of a plasmid encoding (CGG)88-GFP using Lipofectamine 2000 (Invitrogen) per the manufacturer's standard protocol. The compound of interest was added to the transfection cocktail, which was then applied to the cells. Approximately 5 h post-transfection, the transfection cocktail was removed and replaced with growth medium containing the compound, and the cells were incubated at 37 °C for 18–20 h. Cells were lysed in the plate using 100 μL/well of mammalian protein extraction reagent (MPER, Pierce Biotechnology) containing 1 μL of halt protease inhibitor cocktail (Thermo Scientific). Cellular proteins were separated by SDS-PAGE and then transferred to a PVDF membrane. Western blotting was completed using anti-GFP (Santa Cruz) or anti-β-actin (Sigma-Aldrich) as primary antibodies and anti-IgG-horseradish peroxidase conjugate as the secondary antibody. Chemiluminescent signal was generated by SuperSignal West Pico Chemiluminescent substrate (Thermo Scientific), and the blot was imaged using X-ray film.
Determination of r(CGG)88-GFP mRNA Expression Levels by qRT-PCR
COS7 cells were transfected with the (CGG)88-GFP plasmid and treated with compound as described above. Total RNA was harvested by using a Quick-RNA MiniPrep Kit (Zymo Research) per the manufacturer's recommended protocol including an on-column DNA digestion. cDNA was generated from 50 ng of RNA using a qScript cDNA Synthesis Kit (Quanta Biosciences) per the manufacturer's protocol. qPCR was performed on an ABI 7900 HT Real-Time PCR System (Applied Biosystems). Primer pairs for (CGG)88-GFP (GFP-F/R) and a β-actin (hACTBF and hACTBR) are provided in Table S-1. (CGG)88-GFP mRNA levels were normalized to β-actin.
Cell Viability, MTT Assay
COS7 cells in 96-well plates were transfected with the (CGG)88-GFP plasmid and treated with compound as described above. MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide, Life Technologies) assays were completed per the manufacturer's recommended protocol.
Construction of a Plasmid Expressing r(CGGexp)-GFP Lacking a Start Codon
The plasmid without ATG start codon was derived from (CGG)88 +1 GFP plasmid.(21) A small fragment of the (CGG)88+1 GFP plasmid (75 nucleotides) containing the ATG start codon was removed by digestion with NheI and BseRI, and the vector was purified by agarose gel electrophoresis. This small fragment was replaced with an insert in which the ATG start codon was mutated, generated by PCR-amplification of the (CGG)88 + 1 GFP plasmid with the following primers: forward: 5′-CTCCCGGCGCTAGCAAGGGCTGAAGAGACCGAGGTG-3′ (the bold text denotes the modified sequence) and reverse: 5′-GATGGGCACCACCCCGGTGAACAG-3′. The new fragment was doubly digested with NheI and BseRI and ligated back into vector. The sequence of the construct was verified by Eton Bioscience.
Improvement of pre-mRNA Splicing Defects in a FXTAS Cell Culture Model Using RT-PCR
Improvement of pre-mRNA splicing defects by various compounds was tested in cellulo as previously described with minor modifications.(21) Briefly, COS7 cells were grown as monolayers in 96-well plates in growth medium. After the cells reached 90–95% confluency, they were transfected with 200 ng of total plasmid (150 ng of a plasmid encoding r(CGG)60 and 50 ng of the SMN2 mini-gene) using lipofectamine 2000 (Invitrogen) per the manufacturer's standard protocol. The compound of interest was added to the transfection cocktail, which was then applied to the cells. Approximately 5 h post-transfection, the transfection cocktail was removed and replaced with growth medium containing the compound, and the cells were incubated at 37 °C for 18–20 h. Total RNA was harvested as described above. A sample of RNA was subjected to RT-PCR as previously described,(17) and the pre-mRNA splicing products were separated by denaturing polyacrylamide gel electrophoresis. The primer sequences for the SMN2 mini-gene (SMN2-F/R) are provided in Table S-1.
Chem-CLIP and C-Chem-CLIP to Pull-Down the Cellular Targets of Compounds
COS7 cells were grown in growth medium (1× DMEM, 10% FBS, and 1× GlutaMax (Invitrogen)) as monolayers in a 75 cm2 flask to ∼95% confluency. Cells were transfected with the plasmid of interest ((CGG)88-GFP or GFP) using lipofectamine 2000 (Invitrogen) per the manufacturer's recommended protocol. The compound of interest was added to the transfection cocktail (final concentration of 500 nM for 2H-5-CA-Biotin and 2P-5-CA-Biotin and 1 μM for 2H-5-MA-Biotin), which was then applied to the cells. For C-Chem-CLIP, cells were treated with 500 nM 2H-5-CA-Biotin and 50 μM 2H-5. After 5 h incubation, the transfection cocktail was removed and replaced with growth medium containing compound. The cells were then incubated at 37 °C for 18–20 h. Total RNA was extracted by using Trizol reagent (Ambion) according to the manufacturer's protocol. After RQ1 DNase (Promega) treatment, total RNAs were phenol:chloroform extracted and ethanol precipitated. Approximately 100 μg of total RNA was incubated with streptavidin beads (100 μL, Sigma-Aldrich) in 1× PBS for 2 h at room temperature with gentle shaking. The solution was removed, and the beads were washed with 5 × 200 μL H2O for 5 min each, until the presence of RNA was no longer detected in the wash solution as determined by absorbance at 260 and 280 nm using a Nanodrop 2000C spectrophotometer (Thermo Scientific). Bound RNA was released from the beads by heating the beads in 1× Elution Buffer (100 μL, 95% formamide, 10 mM EDTA, pH 8.2) at 65 °C for 5 min. The released RNA was purified by a Quick-RNA MiniPrep Kit (Zymo Research), and cDNA was generated from 50 ng of RNA using a qScript cDNA Synthesis Kit per the manufacturer's protocol. qPCR was performed on an ABI 7900 HT Real-Time PCR System. Primer sequences for GFP mRNA are provided in Table S-1.
Mapping the Binding Sites of Small Molecules in Cellulo (Chem-CLIP-Map)
COS7 cells were transfected with the (CGG)88-GFP plasmid and treated with compound followed by isolation of total RNA as described above. After RQ1 DNase treatment, total RNAs were phenol:chloroform extracted and ethanol precipitated. Next, 10 μg of total RNA (100 μL) in RNase H Buffer (20 mM Hepes, pH 7.5, 100 mM KCl, 20 mM MgCl2, and 0.1 mM DTT) was annealed with 500 pmoles of antisense oligonucleotide at 37 °C with slow cooling to room temperature over 20 min. RNase H (Life Sciences) (5 U) was added, and the mixture was incubated at 37 °C for 30 min. After, the mixture was heated at 65 °C for 20 min to deactivate the RNase H, and then the samples were incubated with streptavidin beads (100 μL, Sigma-Aldrich) for 1 h at room temperature with gentle shaking. The solution was removed, and the beads were washed with 5 × 200 μL H2O for 5 min each, until the presence of RNA was no longer detected in the wash solution as determined by absorbance at 260 and 280 nm. Bound RNA was released from the beads by heating the beads in 1× Elution Buffer at 65 °C for 5 min. The released RNA was purified by ethanol precipitation, and cDNA was generated from 20 ng of RNA using a qScript cDNA Synthesis Kit per the manufacturer's protocol. qPCR was performed on an ABI 7900 HT Real-Time PCR System. The sequences of antisense oligonucleotide and primers (a-d) are provided in Table S-1.
Polysome Profiling
COS7 cells in 100 mm dishes were transfected with the construct of interest ((CGG)88-GFP or (CGG)88-(No ATG)-GFP) and treated with compound as described above. About 18 h post-transfection, 100 μg/mL (final concentration) cycloheximide (CHX) was added into the dish, and cells were incubated at 37 °C for 10 min. Cells were washed twice for 5 min with 10 mL of 1× PBS supplemented with 100 μg/mL CHX and then twice for 5 min with 5 mL of 1× PBS supplemented with 100 μg/mL CHX and 1 mM PMSF. Cells were scraped into 1 mL 1× PBS with 100 μg/mL CHX and 1 mM PMSF, pelleted by centrifugation, and resuspended in 400 μL lysis buffer (20 mM Hepes, pH 7.5, 100 mM KCl, 5 mM MgCl2, 0.3% NP40, RNasin, protease inhibitor, 100 μg/mL CHX, and 1 mM DTT). Lysed cells were pelleted by centrifugation at 13,200 rpm in a cold microfuge. The resulting supernatant was layered onto 10 mL linear 10–50% sucrose gradients containing 20 mM Hepes pH 7.4, 5 mM MgCl2, 100 mM KCl, 300 mM DTT, and 100 μg/mL CHX. The samples were centrifuged at 40,000 rpm for 2 h at 4 °C. The gradients were fractionated using a fraction collector (Brandel Inc.,) and the absorbance of cytosolic RNA was recorded at 254 nm by an inline UV monitor. Total RNAs were extracted from 100 μL of each fraction by using a Quick-RNA MiniPrep Kit (Zymo Research). A sample of RNA was subjected to qRT-PCR with primers (GFP-F/R) as described above.
Melting Analysis of r(CGG)12
To determine the effect of the reaction of 2H-5-CA-Biotin with r(CGG)12 on the thermodynamic stability of the RNA, optical melting experiments were completed. Briefly, 10 μM of r(CGG)12 (with a trace amount of 32P-labeled r(CGG)12) in 1× PBS was folded by heating at 60 °C for 5 min and slowly cooling to room temperature. Then, 5 μM of 2H-5-CA-Biotin was added, and the solution was incubated for 4 h at room temperature. The reaction mixture was passed through a Sephadex G-25 column to remove unreacted 2H-5-CA-Biotin. The products of the reaction were then captured onto 50 μL streptavidin resin by incubating for 30 min at room temperature. The resin was then centrifuged, the supernatant was removed, and the resin was washed with 1× PBST. Then, 100 μL of 1× Elution Buffer was added to the resin and was incubated at 95 °C for 5 min to recover 2H-5-CA-Biotin-r(CGG)12 that was bound to the resin. The eluted RNA was ethanol precipitated and resuspended in 100 μL of water. A sample of r(CGG)12 was treated similarly without addition of 2H-5-CA-Biotin for comparison. To determine the stabilization afforded by binding to 2H-5, samples of r(CGG)12 and r(CGG)12 treated with 5 equivalents of 2H-5 were also prepared. Optical melting of the RNA was performed at 260 nm in 1× Melting Buffer (4 mM Na2HPO4, pH 7.0, 93 mM NaCl, and 0.5 mM EDTA) on a Beckman DU-800 UV–vis spectrophotometer using a gradient of 1 °C/min.
Supplementary Material
Acknowledgments
We thank Profs. Peter Todd and Nicolas Charlet-Berguerand for generous gifts of the plasmids used in these studies. This work was funded by the National Institutes of Health (R01 GM097455 to M.D.D.), a FRAXA postdoctoral fellowship to W–Y.Y., and The Scripps Research Institute. M.D.D. is a Camille and Henry Dreyfus Teacher-Scholar.
Footnotes
The authors declare no competing financial interest.
Supporting Information: Details for synthesis and characterization of compounds, sequences of oligonucleotides, and supplementary figures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Guan L, Disney MD. ACS Chem Biol. 2012;7:73. doi: 10.1021/cb200447r. [DOI] [PubMed] [Google Scholar]
- 2.Thomas JR, Hergenrother PJ. Chem Rev. 2008;108:1171. doi: 10.1021/cr0681546. [DOI] [PubMed] [Google Scholar]
- 3.Guan L, Disney MD. Angew Chem, Int Ed Engl. 2013;52:10010. doi: 10.1002/anie.201301639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Disney MD. Drug Discov Today. 2013;9:00263. doi: 10.1016/j.drudis.2013.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Velagapudi SP, Gallo SM, Disney MD. Nat Chem Biol. 2014;10:291. doi: 10.1038/nchembio.1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wojciechowska M, Krzyzosiak WJ. Hum Mol Genet. 2011;20:3811. doi: 10.1093/hmg/ddr299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Day JW, Ranum LP. Neuromuscul Disord. 2005;15:5. doi: 10.1016/j.nmd.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 8.Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, Schneider A, Richard S, Willemsen R, Elliott DJ, Hagerman PJ, Charlet-Berguerand N. EMBO J. 2010;29:1248. doi: 10.1038/emboj.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sellier C, Freyermuth F, Tabet R, Tran T, He F, Ruffenach F, Alunni V, Moine H, Thibault C, Page A, Tassone F, Willemsen R, Disney MD, Hagerman PJ, Todd PK, Charlet-Berguerand N. Cell Rep. 2013;3:869. doi: 10.1016/j.celrep.2013.02.004. [CAS] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MAC, Nan Z, Forster C, Low WC, Schoser B, Somia NV, Clark HB, Schmechel S, Bitterman PB, Gourdon G, Swanson MS, Moseley M, Ranum LPW. Proc Natl Acad Sci U S A. 2011;108:260. doi: 10.1073/pnas.1013343108. [Free @ PNAS] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, Renoux AJ, Chen KC, Scaglione KM, Basrur V, Elenitoba-Johnson K, Vonsattel JP, Louis ED, Sutton MA, Taylor JP, Mills RE, Charlet-Berguerand N, Paulson HL. Neuron. 2013;78:440. doi: 10.1016/j.neuron.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ash PEA, Bieniek KF, Gendron TF, Caulfield T, Lin WL, DeJesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW, Rademakers R, Boylan KB, Dickson DW, Petrucelli L. Neuron. 2013;77:639. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD, Richter JD, Darnell RB. Cell. 2011;146:247. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bakker CE, Verheij C, Willemsen R, van der Helm R, Oerlemans F, Vermey M, Bygrave A, Hoogeveen A, Oostra BA, Reyniers E, De Boule K, D'Hooge R, Cras P, van Velzen D, Nagels G, Martin JJ, De Deyn PP, Darby JK, Willems PJ. Cell. 1994;78:23. [PubMed] [Google Scholar]
- 15.Sudrik C, Arha M, Cao J, Schaffer DV, Kane RS. Chem Commun. 2013;49:7457. doi: 10.1039/c3cc43086c. [DOI] [PubMed] [Google Scholar]
- 16.Werstuck G, Green MR. Science. 1998;282:296. doi: 10.1126/science.282.5387.296. [DOI] [PubMed] [Google Scholar]
- 17.Tran T, Childs-Disney JL, Liu B, Guan L, Rzuczek S, Disney MD. ACS Chem Biol. 2014;9:904. doi: 10.1021/cb400875u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Florea-Wang D, Pawlowicz AJ, Sinkkonen J, Kronberg L, Vilpo J, Hovinen J. Chem Biodivers. 2009;6:1002. doi: 10.1002/cbdv.200800327. [DOI] [PubMed] [Google Scholar]
- 19.Wang YD, Dziegielewski J, Wurtz NR, Dziegielewska B, Dervan PB, Beerman TA. Nucleic Acids Res. 2003;31:1208. doi: 10.1093/nar/gkg215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johansson HE, Helsham GJ, Sproat BS, Hentze MW. Nucleic Acids Res. 1994;22:4591. doi: 10.1093/nar/22.22.4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Disney MD, Liu B, Yang W, Sellier C, Tran T, Charlet-Berguerand N, Childs-Disney JL. ACS Chem Biol. 2012;7:1711. doi: 10.1021/cb300135h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, Clark TA, Schweitzer AC, Blume JE, Wang X, Darnell JC, Darnell RB. Nature. 2008;456:464. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. Proc Natl Acad Sci U S A. 2004;101:7287. doi: 10.1073/pnas.0401799101. [Free @ PNAS] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arava Y, Boas FE, Brown PO, Herschlag D. Nucleic Acids Res. 2005;33:2421. doi: 10.1093/nar/gki331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gray N, Hentze M. Mol Biol Rep. 1994;19:195. doi: 10.1007/BF00986961. [DOI] [PubMed] [Google Scholar]
- 26.Pickering BM, Willis AE. Sem Cell Dev Biol. 2005;16:39. doi: 10.1016/j.semcdb.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 27.Stern-Ginossar N, Weisburd B, Michalski A, Le VT, Hein MY, Huang SX, Ma M, Shen B, Qian SB, Hengel H, Mann M, Ingolia NT, Weissman JS. Science. 2012;338:1088. doi: 10.1126/science.1227919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Slavoff SA, Mitchell AJ, Schwaid AG, Cabili MN, Ma J, Levin JZ, Karger AD, Budnik BA, Rinn JL, Saghatelian A. Nat Chem Biol. 2013;9:59. doi: 10.1038/nchembio.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.