

Abstract
Mutations detected in cancers are often divided into “drivers” and “passengers.” We suggest that this classification is potentially misleading for purposes of early detection and prevention. Specifically, some mutations are frequent in tumors and thus appear to be drivers, but are poor predictors of cancer; other mutations are individually rare and thus appear to be passengers, but may collectively explain a large proportion of risk. The assumptions bundled into the terms “driver” and “passenger” can lead to misunderstandings of neoplastic progression, with unintended consequences including overdiagnosis, overtreatment, and failure to identify the true sources of risk. We argue that samples from healthy, benign, or neoplastic tissues are critical for evaluating the risk of future cancer posed by mutations in a given gene.
Keywords: driver, passenger, overdiagnosis, natural selection, hitchhiking
Current cancer literature divides mutations detected in cancers into “drivers” and “passengers.” From Stratton et al. (1): A driver mutation is causally implicated in oncogenesis. It has conferred growth advantage on the cancer cell and has been positively selected in the microenvironment of the tissue in which the cancer arises. […] A passenger mutation has not been selected, has not conferred clonal growth advantage and has therefore not contributed to cancer development. Passenger mutations are found within cancer genomes because somatic mutations without functional consequences often occur during cell division.
This perspective has been useful in understanding advanced cancers. However, for cancer prevention or early detection it is necessary to look forward in time and consider outcomes other than cancer. In a prospective view, a clone of mutant cells in a healthy individual may die off, persist without expanding, form a non-invasive benign tumor or neoplasm, or progress to cancer. Early detection and preventative treatment rely on the ability to distinguish these trajectories. For these purposes the driver/passenger paradigm is an oversimplification.
We recommend the use of more specific terms for mutations suspected of involvement in cancer. A mutation or epigenetic change can be termed advantageous when it confers a somatic growth or survival advantage in a given environment, so that cells possessing it increase in number at the expense of other cells, or disadvantageous when it leads to reduced somatic growth or survival. A mutation can be termed predisposing when it tends toward a cancer phenotype (tissue invasion and metastasis), either by conferring one of the hallmarks of cancer (2) or by destabilizing the genome and thus promoting the occurrence of other mutations.
A classical driver is both advantageous and predisposing, but these two qualities need not be coupled, as shown in Table 1. If we limit our categories to drivers and passengers, mutations which are advantageous but not predisposing are likely to be misclassified as drivers and falsely treated as causal to cancer. Conversely, mutations which are predisposing but not advantageous are likely to be misclassified as passengers and falsely dismissed as irrelevant to cancer. Disadvantageous mutations may be disregarded completely, even though their presence can influence the survival of a mutant clone. It is important to disentangle these issues, but because the study of cancer is dominated by the driver/passenger paradigm, current experiments are seldom designed to reveal them.
Table 1.
Mutations classified by their putative role in cancer. Gray boxes indicate mutations whose understanding may be impeded by the driver/passenger paradigm. The two upper boxes can be distinguished using non-cancer samples because mutations which are both advantageous and predisposing will show high relative risk, whereas mutations which are only advantageous will show little or no elevation in relative risk. Similarly, the two lower boxes can be distinguished because mutations which are predisposing but not advantageous, while they tend to be uncommon, will show elevated relative risk when they do occur, whereas mutations which are neither advantageous nor predisposing will show no elevation in relative risk.
| Tends toward invasion and metastasis (“predisposing”) | Does not tend toward invasion and metastasis (“non-predisposing”) | |
| Growth advantage (“advantageous”) | Drivers Elevated relative risk |
Drivers? CDKN2A in esophageal adenocarcinoma Notch1 in skin Non-elevated relative risk |
| No growth advantage (“non-advantageous”) |
Passengers? BRCA2, MSH2 Elevated relative risk |
Passengers Non-elevated relative risk |
Mutations which are advantageous but not predisposing
When such mutations occur in a somatic cell they confer a growth or survival advantage on their cell lineage, but do not otherwise contribute to tissue invasion or metastasis. An example is provided by CDKN2A inactivation in Barrett’s esophagus (BE), a metaplastic condition which can give rise to esophageal adenocarcinoma (EA). Cells with deletions or loss of heterozygosity in CDKN2A are selectively favored early in the development of BE (3). However, individuals whose BE segment contains CDKN2A deletions or losses of heterozygosity (4) or point mutations (5) are no more likely to develop EA than those without. Furthermore, this is not an isolated instance: of 15 loci recurrently mutated in EA, 13 did not distinguish EA from low-risk, non-dysplastic BE (5), demonstrating that “driver” and “locus frequently mutated in cancer” are not synonymous. Similarly, healthy sun-exposed eyelid skin contains large numbers of clones with mutations in genes identified as skin-cancer drivers, particularly NOTCH1, and many of these clones appear to possess a growth advantage (6).
Benign tumors may result from the action of advantageous mutations in the absence of predisposing ones: the cell lineage overgrows (e.g. melanocytic nevus, colorectal adenoma), but it does not develop the hallmarks of cancer (melanoma, colorectal carcinoma). A number of mutations generally regarded as drivers are commonly found in benign tumors (7).
Mutations which are predisposing but not advantageous
Mutations in this category do not confer a somatic growth advantage, but they move the cell toward development of a cancer phenotype. They may be selectively neutral or give a growth or survival disadvantage. Tumor suppressor loci whose loss increases the somatic mutation rate provide examples of this category; conceptually, an increased mutation rate in itself should not confer a survival or growth advantage, although it opens the door to later mutations which may. Mutations in BRCA2, a gene involved in recombination and repair, lead to a high risk of breast and ovarian cancer when inherited in the germ line and thus present in all somatic cells. However, cell lines with deletion of both copies of BRCA2 show a consistent growth disadvantage compared to wild-type lines (8). This may help explain why BRCA2 mutations are not significantly enriched in sporadic breast cancers (9,10). Similarly, hereditary non-polyposis colon cancer (HNPCC) is associated with mutations in the mismatch-repair gene MSH2. However, inactivation of one copy of MSH2 had no growth advantage when tested in mouse cell lines (11) and inactivation of the second copy had no growth advantage in cell culture (12). The microsatellite instability seen in MSH2-deficient cells can eventually lead to a growth advantage by inducing mutations in TGFBR2 (13), but when the clone first arises it will behave neutrally.
Although the importance of predisposing non-advantageous mutations is well established in familial cancers, they are often disregarded in non-familial cancer. Table S4 in Vogelstein et al. (14) lists 45 loci for which mutations predispose to cancer when inherited in the germ line but are rare in somatic tumors, noting that they are listed “for completeness.” It is assumed that a lineage whose genotype confers no advantage cannot expand, and without clonal expansion the probability of developing the subsequent mutations required for progression is low. However, cells with no advantage over their neighbors can and do expand as clonal populations as part of normal processes such as tissue turnover and renewal and wound healing. They may also expand due to presence of a non-predisposing advantageous mutation with which the predisposing mutation hitchhikes (15); the lineage containing both might be said to have a driver phenotype, but neither mutation on its own behaves as a driver.
The search for drivers has led to discovery of a “long tail” of genes with functional mutations in a small minority of cancers of a given type (16). Some of these may be non-advantageous predisposing mutations that can expand only under specific circumstances (tissue growth, hitchhiking) and that are causal in those tumors that do possess them. If such mutations are classified as passengers we may have a paradoxical situation in which a substantial proportion of cancer risk is due to “passenger” mutations.
Disadvantageous mutations
The driver/passenger definition given above does not mention disadvantageous mutations, yet a substantial proportion of functional somatic mutations are expected to be disadvantageous in a given environment. Some of these will persist in clonal populations due to hitchhiking with an advantageous mutation or being present in a rapidly expanding tissue, while others will be eliminated by natural selection. From the perspective of advanced cancer disadvantageous mutations may seem irrelevant, but McFarland et al. (17) shows evidence from computer simulation that negatively selected passengers may be involved in tumor stasis and regression. Correct evaluation of the risk posed by a mutant clone in a healthy individual may therefore require evaluation of the deleterious mutations it carries.
Why does this matter?
There is a tendency to assume that if a mutation is not a driver, it must be a passenger–and if it is not a passenger, it must be a driver. In reality the situation is more complex, and this oversimplification can lead to unintended clinical consequences.
Molecular screening for cancer risk relies on finding mutations which are both typical of cancer and rare in individuals who neither have cancer nor are likely to develop it in their lifetimes. There is no question that CDKN2A lesions are typical of EA, and a cancer-only analysis would identify CDKN2A as a driver in EA. However, use of CDKN2A as a tool for cancer risk prediction would lead to massive overdiagnosis and overtreatment. It can be argued that CDKN2A lesions promote development of BE, which is itself a risk factor; but if so, CDKN2A is a “driver” which fails to drive to cancer in the majority of cases (4) despite being able to spread across the BE segment (5) and as such is not a useful predictor of future cancer. Similarly, the fact that NOTCH1 mutations are abundant in skin cancers and show a marked growth advantage does not make them suitable targets for risk prediction given their high frequency in healthy skin; and a treatment which killed NOTCH1 mutant cells would, as (7) points out, do unacceptable collateral damage to healthy tissue.
Advantageous mutations that occur frequently in the run-up to a particular cancer are likely to pass all tumor-based tests for driver status: frequency, enrichment for mutations of functional effect over silent ones, and presence in many or most cells in the tumor (18). Sequencing of cancer genomes is simply not sufficient to identify loci which can be used for risk screening: it is essential to know the frequency of putative driver mutations in healthy, neoplastic, and benign-tumor tissues as well, as illustrated in Figure 1. The non-predisposing status of CDKN2A mutations in EA was discovered only because individuals with BE who did not progress to EA were evaluated as controls (4,5). Ubiquitous NOTCH1 mutations in healthy sun-exposed skin were also a major surprise (6).
Figure 1.
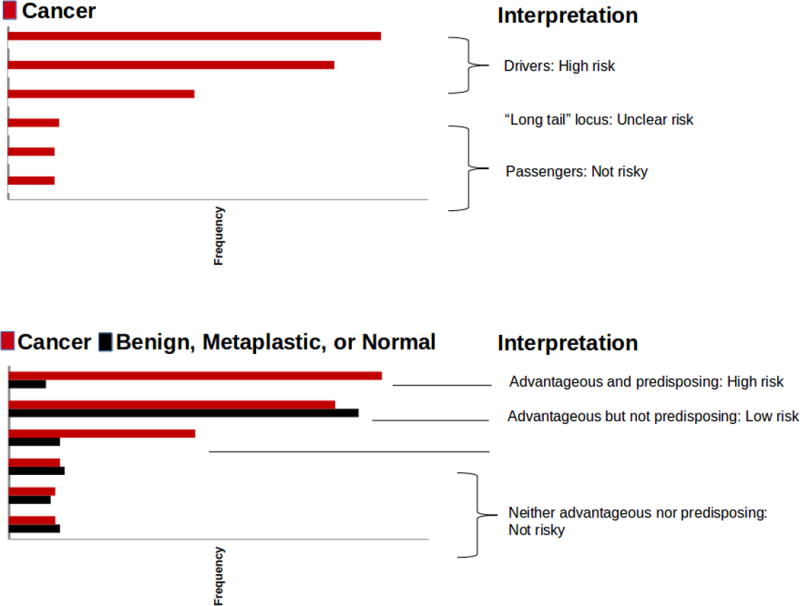
Cancer-only analysis gives incomplete information about risk of cancer. All mutations shown are assumed to be functional. Upper panel, analysis using only cancer samples. Mutations seen at high frequency are assumed to confer a high risk of progression to cancer; those seen at low frequency are assumed to confer low risk. Lower panel, analysis using cancer samples as well as samples from benign tumors, neoplastic tissues, or healthy tissue, so that relative risk can be assessed. Mutations seen at high frequency in both cancer and non-cancer samples are likely to be advantageous non-predisposing mutations of low risk. Mutations seen at intermediate frequency in cancer samples (the “long tail”) but rarely in non-cancer samples are likely to be predisposing non-advantageous mutations which can confer high risk if the cells bearing them are numerous. Thus, use of both cancer and non-cancer samples allows a more accurate assessment of risk.
Conversely, the assumption that non-advantageous mutations are mere passengers and that most cancer risk is due to classical drivers may miss key sources of risk. While mutations in BRCA2 are uncommon in sporadic breast cancers, it is likely that their heightened mutation rate contributes to the development of those cancers that do possess them. Some forms of cancer may arise in large part due to non-advantageous mutations in pathways containing many genes; in this model mutations in any given gene are low-risk as they rely on chance events to spread, but the pathway as a whole may explain a substantial proportion of cancer risk. If so, early detection may need to focus on detecting disrupted pathways rather than disrupted genes per se.
More generally, the way we think about progression to cancer needs to reflect the evolutionary process by which all cancers are believed to develop (19,20). Any predisposing mutation creates a risk of further progression; conceptually, the more cells that contain the mutation, the higher the risk. A large clone of risky cells can arise because their genotype offers a selective advantage. When the advantage and the predisposition come from the same locus this meets the classical definition of a driver locus. But clones can also become large because of their position in an expanding tissue (germ line mutations such as BRCA2 are the ultimate example of this) or because they possess other advantageous mutations. For example, the expansion of CDKN2A-mutant clones in BE may be innocuous in itself; the clones out-compete other BE lineages but do not invade or metastasize, and their growth is limited by the size of the BE segment. However, if such a clone possesses or acquires a TP53 mutation while it is expanding, the expansion process will carry TP53 to high frequency with consequent high risk. Thus, finding that a mutation possesses no growth advantage does not rule out the possibility that it is directly contributing to the development of cancer.
What does all this mean in practice? The concept of drivers and passengers is appealing because it suggests we have found the “cause” of a given cancer and can use this to develop strategies for detection and prevention. Unfortunately, the development of cancer is an evolutionary process, stochastic in nature (19), and simple categorization of genes into drivers and passengers at best fails to acknowledge this complexity and at worst misleads our thinking about the best way to approach detection and prevention without falling into the trap of overdiagnosis. We recommend that researchers attempt to evaluate both cancers and their precursors in order to appropriately evaluate the risks posed by mutations in specific genes. We also urge researchers to search for means of determining separately whether a mutation is advantageous in a given context and whether it is predisposing. This difficult task will likely require more attention paid to pre-cancerous and non-cancerous tissues: examination of the cancer genome alone cannot necessarily establish whether a mutation is predisposing. In the meantime, the driver/passenger metaphor should be used circumspectly, particularly in the context of cancer prevention and early detection, where overdiagnosis and overtreatment are increasingly being recognized as significant challenges (21).
Acknowledgments
We thank Carissa Sanchez, Patricia Galipeau, Xiaohong Li, Thomas Paulson, Carlo Maley, and Jon Yamato for helpful discussions. We thank P. Brown for supporting R. Kostadinov during this research.
Financial Support: M. Kuhner and B. Reid were supported by NIH grant P01 CA91955. B. Reid was supported by NIH grant R01 CA179949. R. Kostadinov was supported by American Cancer Society Research Scholar Grant 120237 .
Footnotes
The authors declare no conflicts of interest.
Literature Cited
- 1.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Maley CC, Galipeau PC, Li X, Sanchez CA, Paulson TG, Reid BJ. Selectively advantageous mutations and hitchhikers in neoplasms: p16 lesions are selected in Barrett’s esophagus. Cancer Res. 2004;64:3414–3427. doi: 10.1158/0008-5472.CAN-03-3249. [DOI] [PubMed] [Google Scholar]
- 4.Li X, Galipeau PC, Paulson TG, Sanchez CA, Arnaudo J, Liu K, et al. Temporal and spatial evolution of somatic chromosomal alterations: a case-cohort study of Barrett’s esophagus. Cancer Prev Res. 2014;7:114–127. doi: 10.1158/1940-6207.CAPR-13-0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weaver JMJ, Ross-Innes CS, Shannon N, Lynch AG, Forshew T, Barbera M, et al. Ordering of mutations in preinvasive disease stages of esophageal carcinogenesis. Nat Genet. 2014;46:837–843. doi: 10.1038/ng.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348:880–886. doi: 10.1126/science.aaa6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marino-Enriquez A, Fletcher CDM. Shouldn’t we care about the biology of benign tumors? Nat Rev Cancer. 2014;14:701–702. doi: 10.1038/nrc3845. [DOI] [PubMed] [Google Scholar]
- 8.Al Abo M, Dejsuphong D, Hirota K, Yonetani Y, Yamazoe M, Kurumizaka H, et al. Compensatory functions and interdependency of the DNA-binding domain of BRCA2 with the BRCA1-PALB2-BRCA2 complex. Cancer Res. 2014;74:797–807. doi: 10.1158/0008-5472.CAN-13-1443. [DOI] [PubMed] [Google Scholar]
- 9.Lancaster JM, Wooster R, Mangion J, Phelan CM, Cochran C, Gumbs C, et al. BRCA2 mutations in primary breast and ovarian cancers. Nat Genet. 1996;13:238–40. doi: 10.1038/ng0696-238. [DOI] [PubMed] [Google Scholar]
- 10.The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumors. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reitmair AH, Risley R, Bristow RG, Wilson T, Ganesh A, Jang A, et al. Mutator phenotype in Msh2-deficient murine embryonic fibroblasts. Cancer Res. 1997;57:765–3771. [PubMed] [Google Scholar]
- 12.Abuin A, Zhang H, Bradley A. Genetic analysis of mouse embryonic stem cells bearing Msh3 and Msh2 single and compound mutations. Mol Cell Biol. 2000;20:149–157. doi: 10.1128/mcb.20.1.149-157.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, et al. Inactivation of the Type II TGF-β receptor in colon cancer cells with microsatellite instability. Science. 1995;268:1336–1338. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- 14.Vogelstein B, Padopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith JM, Haigh J. The hitch-hiking effect of a favourable gene. Genet Res. 1974;23:23–35. [PubMed] [Google Scholar]
- 16.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 17.McFarland CD, Korolev KS, Kryukov GV, Sunyaev SR, Mirny LA. Impact of deleterious passenger mutations on cancer progression. Proc Natl Acad Sci USA. 2013;110:2910–2915. doi: 10.1073/pnas.1213968110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raphael BJ, Dobson JR, Oesper L, Vandin F. Identifying driver mutations in sequenced cancer genomes: computational approaches to enable precision medicine. Genome Med. 2014;6:5. doi: 10.1186/gm524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res. 2012;72:4875–4882. doi: 10.1158/0008-5472.CAN-12-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Esserman LJ, Thompson IM, Reid B, Nelson P, Ransohoff DF, Welch HG, et al. Addressing overdiagnosis and overtreatment in cancer: a prescription for change. Lancet Oncol. 2014;15:e234–42. doi: 10.1016/S1470-2045(13)70598-9. [DOI] [PMC free article] [PubMed] [Google Scholar]