

Abstract
Objective:
The aim of this study was to evaluate the efficacy of maraviroc along with darunavir/ritonavir, all once daily, for the treatment of antiretroviral-naive HIV-1 infected individuals.
Design:
MODERN was a multicentre, double-blind, noninferiority, phase III study in HIV-1 infected, antiretroviral-naive adults with plasma HIV-1 RNA at least 1000 copies/ml and no evidence of reduced susceptibility to study drugs.
Methods:
At screening, participants were randomized 1 : 1 to undergo either genotypic or phenotypic tropism testing. Participants with CCR5-tropic HIV-1 were randomized 1 : 1 to receive maraviroc 150 mg once daily or tenofovir/emtricitabine once daily each with darunavir/ritonavir once daily for 96 weeks. The primary endpoint was the proportion of participants with HIV-1 RNA less than 50 copies/ml (Food and Drug Administration snapshot algorithm) at Week 48. A substudy evaluated bone mineral density, body fat distribution and serum bone turnover markers.
Results:
Seven hundred and ninety-seven participants were dosed (maraviroc, n = 396; tenofovir/emtricitabine, n = 401). The Data Monitoring Committee recommended early study termination due to inferior efficacy in the maraviroc group. At Week 48, the proportion of participants with HIV-1 RNA less than 50 copies/ml was 77.3% for maraviroc and 86.8% for tenofovir/emtricitabine [difference of −9.54% (95% confidence interval: −14.83 to −4.24)]. More maraviroc participants discontinued for lack of efficacy, which was not associated with non-R5 tropism or resistance. Discontinuations for adverse events, Category C events, Grade 3/4 adverse events and laboratory abnormalities were similar between groups.
Conclusion:
A once-daily nucleos(t)ide-sparing two-drug regimen of maraviroc and darunavir/ritonavir was inferior to a three-drug regimen of tenofovir/emtricitabine and darunavir/ritonavir in antiretroviral-naive adults.
Keywords: darunavir, emtricitabine, HIV-1, maraviroc, nucleos(t)ide-sparing regimen, tenofovir, treatment-naive
Introduction
Treatment guidelines for management of treatment-naive HIV-1 infected individuals recommend the use of two nucleos(t)ide reverse transcriptase inhibitors (NRTIs), along with one other drug from a different antiretroviral drug class [1–5]. In spite of the proven efficacy of NRTI-containing regimens, mitochondrial toxicity can complicate the use of this class of drugs [6]. There are several drug-specific adverse effects, including bone and renal toxicity for tenofovir (TDF), hypersensitivity reactions with abacavir and bone marrow suppression with zidovudine [7,8].
NRTI-sparing regimens, including efavirenz or raltegravir (RAL) combined with a protease inhibitor for treatment-naive HIV-1 infected individuals, have demonstrated that inclusion of NRTIs was not a prerequisite for a successful antiretroviral regimen. However, virologic failure and/or development of resistance mutations were more common in the NRTI-sparing groups in several studies [9–12].
Maraviroc (MVC) is efficacious and well tolerated with a clearly defined and favourable long-term safety profile at doses of 300 mg (or equivalent) twice daily [13–15]. MVC concentrations are increased by potent cytochrome P450 3A4 inhibitors, including darunavir/ritonavir (DRV/r) and most other boosted protease inhibitors; therefore, the dose is reduced to 150 mg when used in the presence of these drugs [16].
Three pilot studies evaluating MVC along with a ritonavir-boosted protease inhibitor in a two-drug NRTI-sparing regimen over 48 weeks have been completed [17–19]. Study A4001078 evaluated MVC 150 mg once daily (q.d.) along with atazanavir/ritonavir (ATV/r) versus TDF/emtricitabine (FTC) along with ATV/r, with 74.6 and 83.6% of individuals achieving HIV-1 RNA less than 50 copies/ml, respectively. No resistance to any component of either treatment regimen or change in tropism was observed in virus from these individuals. However, more individuals in the MVC group discontinued due to adverse events, mostly hyperbilirubinemia secondary to ATV/r, which may have adversely affected the study outcome. The imbalance between the groups may have been due to the drug–drug interaction between TDF and ATV that reduces ATV exposure with fewer cases of hyperbilirubinemia resulting in discontinuation in this group [17]. The VEMAN study evaluated MVC 150 mg q.d. along with lopinavir/ritonavir (LPV/r) or TDF/FTC along with LPV/r in 50 individuals, with 100% of MVC-treated individuals achieving HIV-1 RNA less than 50 copies/ml [19]. In the MIDAS study, 92% of treatment-naive individuals who received MVC 150 mg q.d. along with DRV/r had HIV-1 RNA less than 50 copies/ml [18].
The MODERN (Maraviroc Once-daily with Darunavir Enhanced by Ritonavir in a New regimen) study was designed to assess the safety and efficacy of MVC 150 mg q.d. versus TDF/FTC, both in combination with DRV/r, in antiretroviral-naive individuals infected with R5 HIV-1.
Materials and methods
Study design and population
MODERN was designed as a 96-week, multicentre, international, double-blind, randomized, comparative, phase III study conducted between September 2011 and January 2014 at 138 sites in Europe, the United States, Australia and Canada (ClinicalTrials.gov identifier: NCT01345630). It was conducted in compliance with the Declaration of Helsinki and International Conference on Harmonisation Good Clinical Practice Guidelines, and all local regulatory requirements were followed. The protocol was approved by institutional review boards/independent ethics committees at all sites and written informed consent was provided by all individuals. An independent Data Monitoring Committee (DMC) reviewed the data every 4 months with a formal interim analysis for futility when nearly 50% of individuals reached Week 24. Participants were antiretroviral-naive, HIV-1 infected, at least 18 years old and had HIV-1 RNA at least 1000 copies/ml, R5 HIV-1, no evidence of active hepatitis B virus infection and no resistance to DRV/r, TDF or FTC. As the study was also designed to prospectively compare genotypic versus phenotypic tropism testing results to predict response to MVC, all participants were initially randomized to undergo tropism testing with one of two assays [a genotypic tropism test (GTT) (Siemens Healthcare Diagnostics, Berkeley, California, USA), or the phenotypic Trofile test from Monogram Biosciences (South San Francisco, California, USA)]. Participants identified as having R5 virus were then randomized to treatment with MVC (150 mg q.d.) or TDF/FTC (300/200 mg q.d.) both combined with DRV/r (800/100 mg q.d.) and stratified by screening plasma HIV-1 RNA (≥ or <100 000 copies/ml).
Study evaluations
After screening and baseline, visits were scheduled at Weeks 4, 8, 12, 16, 20, 24, 36, 48, 60, 72, 84, 96 and follow-up (28 days after last study dose). The primary endpoint was the proportion (%) of participants with HIV-1 RNA less than 50 copies/ml at Week 48 using the Food and Drug Administration (FDA) snapshot algorithm. Secondary endpoints included changes in CD4+ and CD8+ T-cell counts (assessed by flow cytometry) at Week 48.
Participants were discontinued if they experienced treatment failure, defined as meeting any of the five criteria as follows (and confirmed at a subsequent visit):
Decrease in HIV-1 RNA less than 1 log10 from baseline after Week 4 unless HIV-1 RNA was less than 50 copies/ml;
HIV-1 RNA more than 1 log10 above the nadir value after Week 4;
HIV-1 RNA at least 50 copies/ml at any time after Week 24;
HIV-1 RNA at least 50 copies/ml after suppression to less than 50 copies/ml on two consecutive visits;
Decrease in HIV-1 RNA 2 log10 or less from baseline on or after Week 12 unless HIV-1 RNA was less than 400 copies/ml.
Samples for genotypic and phenotypic resistance analyses (PhenoSense GT and Entry assays; Monogram Biosciences, and TRUGENE HIV-1 Genotyping Kit; Siemens Healthcare Diagnostics) and for determination of tropism (Trofile or GTT) were tested/assessed at the initial failure event (or at a later time point before discontinuation if the sample was unsuitable), provided that HIV-1 RNA was at least 400 copies/ml. Resistance was assessed using the proprietary algorithms for PhenoSense GT and TRUGENE HIV-1. For MVC, findings of maximal percentage inhibition less than 95% or half-maximal inhibitory concentration (IC50) fold change at least 3 relative to the comparator virus were considered to be evidence of resistance.
Safety was assessed at all visits and included monitoring of all adverse events and serious adverse events (SAEs), vital signs and laboratory parameters. Adverse events were assessed and graded according to Division of AIDS (DAIDS) toxicity scales.
No pharmacokinetic analysis was originally planned; therefore, no specific pharmacokinetic samples or dose times were collected. A post hoc pharmacokinetic analysis to determine MVC, DRV and ritonavir concentrations using available archived plasma samples was performed for 313 of 396 participants in the MVC group (for details, see Supplemental Digital Content 1) in order to determine whether lower than expected drug concentrations may have potentially contributed to the reduced efficacy of the MVC arm.
In a subset of participants who were willing to participate at the centres able to perform the evaluations, dual-energy X-ray absorptiometry (DEXA) scans of the lumbar spine, femoral neck and hip were completed at baseline and weeks 48 and 96 or early termination to measure percentage changes in bone mineral density (BMD) and body fat distribution. The serum bone turnover markers osteocalcin and C-terminal telopeptide of type 1 collagen (CTx) were also evaluated.
Health outcomes research assessments were also conducted (see Supplementary Digital Content 1).
Statistical analysis
A sample size of 804 participants was targeted to yield a power of at least 90% assuming that the true response rates (HIV-1 RNA <50 copies/ml) in the two groups at Week 48 were at least 75% and the one-sided Type I error rate (alpha) was 2.5%. The difference in proportions between the MVC and the TDF/FTC groups and the two-sided 95% confidence bound were determined using the stratum-adjusted Mantel–Haenszel method. If the lower bound of the two-sided 95% confidence interval (CI) was greater than or equal to −10%, MVC would be considered noninferior to TDF/FTC. Efficacy analyses were performed on the full analysis set (FAS) population, which included all participants who received at least one dose of study drug. Continuous measurements were summarized using descriptive statistics and discrete data by counts. All secondary endpoints involving efficacy and safety were analysed using the FAS population as treated.
The futility decision boundary for the week 24 DMC interim analysis was based on the Haybittle–Peto stopping criterion, which tested for inferiority of MVC versus TDF/FTC at the one-sided significance level of 0.0005. The sponsor, investigators and participants were blinded to the results of these analyses.
For the DEXA substudy, a sample size of 109 individuals per group was chosen to detect a 2% difference in outcomes between the groups with the assumptions of standard deviation being 4.5% and power being 90% at the two-sided 5% level of significance. However, by the time the targeted sample size for the primary endpoint was reached (804), the number of participants enrolled in the substudy fell short of the predetermined target.
Results
Study participants and demographic data
A total of 1423 participants were screened, 813 were randomized and 797 received at least one dose of study medication (FAS population) (Fig. 1). Demographic and baseline characteristics were similar between the groups. At baseline, the mean age was 37.1 years, 8.8% were female and 18.7% were classified as nonwhite. One hundred and forty-six participants (18.3%) had HIV-1 RNA at least 100 000 copies/ml, while 83 (10.4%) had a CD4+ cell count of less than 200 cells/μl. The mean (standard deviation) CD4+ cell count at baseline was 382 (173) cells/μl in the MVC group and 380 (171) cells/μl in the TDF/FTC group.
Fig. 1.
. Disposition of individuals at Week 48.
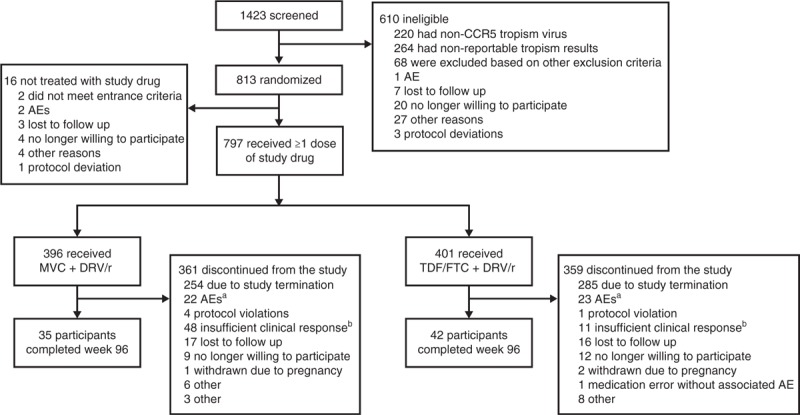
AE, adverse event; MVC, maraviroc; DRV/r, darunavir/ritonavir; TDF/FTC, tenofovir/emtricitabine. aOf the AEs leading to discontinuation, 12 in each group were considered to be related to study treatment. bInvestigator decision to discontinue for lack of efficacy; individual did not necessarily meet criteria for protocol-defined virologic failure.
Data Monitoring Committee review
During a routine DMC review shortly before the Week 48 time point, the DMC requested a review of the efficacy data after the last participant's Week 48 visit. Following the DMC's preliminary review of the 48-week primary clinical efficacy data, the study was terminated on the basis of inferiority of the MVC group. As a consequence of this early termination, only a small number of participants completed Week 96; thus, virologic response and other endpoints at Week 96 were not evaluated.
Virologic and immunologic response
The proportion of participants with HIV-1 RNA less than 50 copies/ml (FDA snapshot) at Week 48 was lower in the MVC group than in the TDF/FTC group: 77.3 versus 86.8%, respectively (Fig. 2a). The point estimate of the difference in proportion between treatment with MVC + DRV/r and TDF/FTC + DRV/r was −9.54% (95% CI −14.83 to −4.24). As the lower bound of the two-sided 95% CI was lower than the preset noninferiority margin of −10%, the MVC group was inferior to the TDF/FTC group. When stratified by baseline HIV-1 RNA or CD4+ cell count, the proportion of responders at Week 48 was also lower for MVC in all subgroups (Fig. 2b). The 2 CCR5-tropism assays (GTT or Trofile) were similar in predicting a positive treatment outcome (<50 copies/ml at Week 48) for both MVC (80.7% for genotype versus 74.4% for Trofile) and TDF/FTC (86.5% for genotype versus 87.0% for Trofile). Detailed results of the comparisons between the two tropism assays will be published separately. The mean (standard deviation) change from baseline in CD4+ cell count at Week 48 in the MVC group was 195 (176) cells/μl compared with 194 (176) cells/μl in the TDF/FTC group.
Fig. 2.
. Virologic response at Week 48 by treatment group.
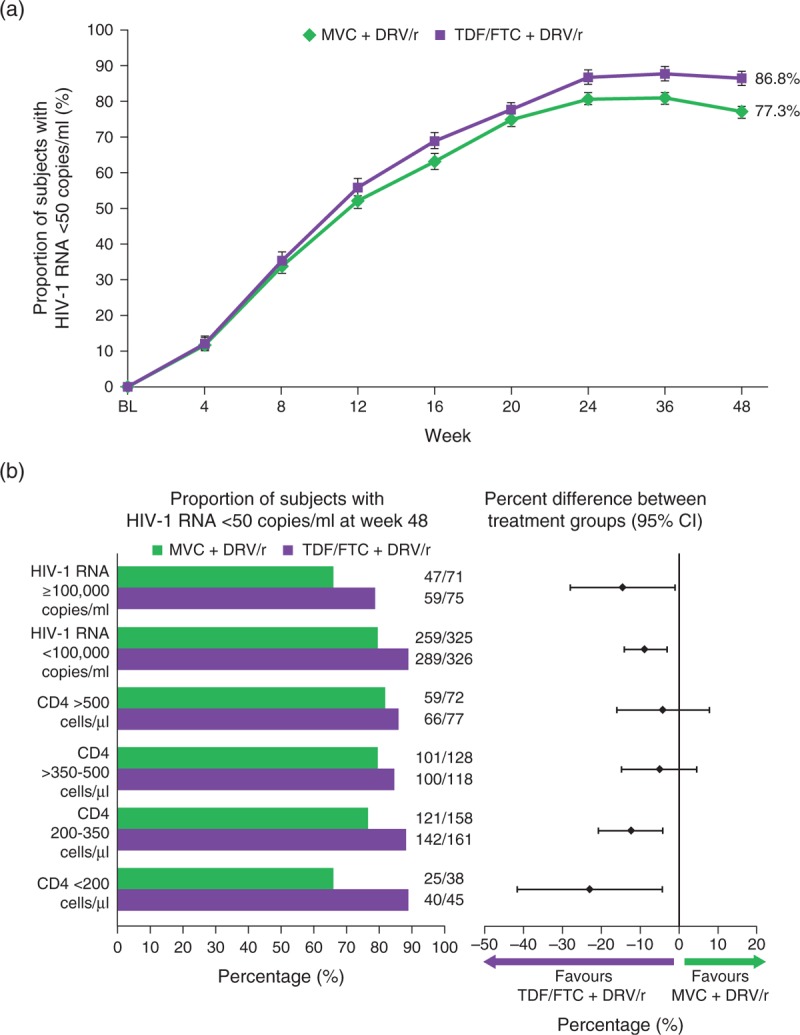
(a) Percentage of individuals with HIV-1 RNA <50 copies/ml with standard errors over time (FAS population) using the FDA snapshot algorithm of MSDF. Plasma HIV-1 RNA concentration was determined using a real-time HIV-1 RNA assay with a lower limit of quantification of 40 copies/ml (Abbott Molecular Inc, Des Plaines, Illinois, USA). (b) Proportion of individuals with HIV-1 RNA <50 copies/ml at Week 48 stratified by baseline HIV-1 RNA (< or ≥100 000 copies/ml) and CD4+ cell count (<200, 200–350, >350–500 and >500 cells/μl), and percentage difference between treatment groups (95% CI) at Week 48 stratified by screening viral load (< or ≥100 000 copies/ml) and baseline CD4+ (<200, 200–350, >350–500 and >500 cells/μl) (FAS population) using the FDA snapshot algorithm of MSDF. CI, confidence interval; DRV/r, darunavir/ritonavir; FAS, full analysis set; FDA, Food and Drug Administration; MVC, maraviroc; MSDF, missing, switch or discontinuation = failure; TDF/FTC, tenofovir/emtricitabine.
Resistance
Through Week 48, there were 47 participants with confirmed protocol-defined treatment failure (PDTF) (MVC: n = 37, 9.3%; TDF/FTC: n = 10, 2.5%). An additional three patients in each treatment group met PDTF criteria during the study but had HIV-1 RNA less than 400 copies/ml and showed a response at Week 48.
Only 17 MVC-treated and three TDF/FTC-treated participants had sufficient plasma HIV-1 RNA (>400 copies/ml) for virologic analysis at or after PDTF. Non-R5 virus was not observed at or after failure in either group when the tropism assay used for randomization was also used to assess tropism at failure. Maraviroc susceptibility was retained in MVC-treated participants who experienced PDTF with R5-tropic virus. No resistance to DRV/r or TDF/FTC was recorded.
In general, participants in the study failed with low HIV-1 RNA concentrations. Nineteen of 37 MVC-treated participants (51%) and seven of 10 TDF-treated participants (70%) had plasma HIV-1 RNA 200 copies/ml or less at the time of treatment failure (Table 1). In addition, there were 18 MVC-treated participants and seven TDF-treated participants with plasma HIV-1 RNA determined after the failure confirmation visit; in six and three instances, respectively, the participants had attained full virologic suppression while still receiving study treatment.
Table 1.
HIV-1 RNA values in individuals who discontinued with confirmed protocol-defined treatment failurea.
| n (%) | MVC + DRV/r | TDF/FTC + DRV/r |
| Total N | 37 | 10 |
| 50–100 | 10 (27%) | 3 (30%) |
| 101–200 | 9 (24%) | 4 (40%) |
| 201–300 | 1 (3%) | 0 |
| 301–400 | 4 (11%) | 1 (10%) |
| >400 | 13 (35%) | 2 (20%) |
DRV/r, darunavir/ritonavir; MVC, maraviroc; PDTF, protocol-defined treatment failure; TDF/FTC, tenofovir/emtricitabine.
aHIV-1 RNA values at the time of initial protocol-defined treatment failure are shown.
Pharmacokinetics
Median Week 4 plasma concentrations of MVC, DRV or ritonavir were similar between participants with HIV-1 RNA less than 50 copies/ml at Week 48 and those with HIV-1 RNA at least 50 copies/ml at Week 48 (for detailed results, tables and figures, see Supplemental Digital Content 2).
Safety
Maraviroc along with DRV/r was well tolerated with a safety profile comparable to that of TDF/FTC and DRV/r. Over 48 weeks, rates of SAEs and nonserious adverse events; discontinuations due to adverse events; and rates of DAIDS Grade 3 or 4 AEs and laboratory abnormalities were similar between the groups (Table 2). There were no deaths and no new safety findings.
Table 2.
Summary of treatment-emergent adverse events (all causalities), adverse events occurring in >5% of individuals and Grade 3/4 laboratory abnormalities.
| MVC + DRV/r (N = 396) N (%) | TDF/FTC + DRV/r (N = 401) N (%) | |
| Individuals with AEs | 360 (90.9) | 365 (91.0) |
| Individuals with SAEs | 41 (10.4) | 40 (10.0) |
| Individuals with Grade 3 or 4 AEs | 65 (16.4) | 71 (17.7) |
| Individuals discontinued due to AEs | 24 (6.1) | 24 (6.0) |
| Most common AEs (>5% of individuals in any group) | ||
| Diarrhoea | 89 (22.5) | 135 (33.7) |
| Nasopharyngitis | 48 (12.1) | 55 (13.7) |
| Upper respiratory tract infection | 40 (10.1) | 45 (11.2) |
| Rash | 38 (9.6) | 30 (7.5) |
| Nausea | 34 (8.6) | 45 (11.2) |
| Fatigue | 27 (6.8) | 46 (11.5) |
| Cough | 27 (6.8) | 30 (7.5) |
| Bronchitis | 25 (6.3) | 24 (6.0) |
| Gastroenteritis | 23 (5.8) | 17 (4.2) |
| Depression | 28 (7.1) | 30 (7.5) |
| Insomnia | 15 (3.8) | 25 (6.2) |
| Grade 3/4 laboratory abnormalities | ||
| Alanine aminotransferase | 9 (2.3) | 6 (1.5) |
| Total bilirubin | 3 (0.8) | 1 (0.3) |
| Creatine kinase | 18 (4.5) | 22 (5.5) |
| LDL cholesterol | 50 (12.6) | 24 (6.0) |
AE, adverse event; DRV/r, darunavir/ritonavir; LDL, low-density lipoprotein; MVC, maraviroc; SAE, serious adverse event; TDF/FTC, tenofovir/emtricitabine.
Forty-one (10.4%) and 40 (10.0%) participants in the MVC and TDF/FTC groups, respectively, experienced at least one treatment-emergent SAE up to 30 days after last dose of study drug. Five SAEs were considered to be related to study drugs. Four occurred in the MVC group and two were considered possibly related to MVC by the investigator: one of abnormal weight loss and one of Hodgkin's lymphoma. The other two treatment-related SAEs in the MVC group were rash considered to be related to DRV/r and concomitant treatment with ritonavir and carbamazepine that led to drug toxicity. The event in the TDF/FTC group was abnormal weight loss, considered to be related to TDF/FTC by the investigator.
Seven Category C events were reported, four of Kaposi sarcoma in the MVC group and one case each of Kaposi sarcoma, cytomegalovirus infection and cerebral toxoplasmosis in the TDF/FTC group. Nine participants in the MVC group experienced malignancies, compared with three in the TDF/FTC group (Table 3).
Table 3.
Individuals with treatment-emergent malignancies.
| Study drug | Individual | Malignancy | Baseline CD4+ cell count | Study day | SAE | Related |
| MVC | 1 | Kaposi sarcoma | 138 | 196 | N | N |
| 2 | Basal cell carcinoma | 513 | 139 | N | N | |
| 3 | Hodgkin's lymphomaa | 343 | 226 | Y | Yb | |
| 4 | Kaposi sarcoma | 339 | 137 | N | N | |
| 5 | Castleman's diseasea,c | 185 | 33 | Y | N | |
| 6 | Hodgkin's lymphoma | 402 | 65 | Y | N | |
| 7 | Lung adenocarcinoma | 259 | 365 | Y | N | |
| 8 | Lymphomaa | 250 | 315 | Y | N | |
| 9 | Kaposi sarcoma | 485 | 237 | Y | N | |
| TDF/FTC | 10 | Kaposi sarcomaa | 179 | 334 | Y | N |
| 11 | Testicular cancer | 345 | 59 | Y | N | |
| 12 | Basal cell carcinoma | 338 | 465 | N | N |
MVC, maraviroc; SAE, serious adverse event; TDF/FTC, tenofovir/emtricitabine.
aDiscontinued from study drug due to event.
bInvestigator causality initially attributed to study drug but later considered to be likely due to immune reconstitution inflammatory syndrome and Epstein–Barr virus infection.
cInitially reported as Kaposi sarcoma and plasmablastic lymphoma but later grouped under the term Castleman's disease.
There was no evidence of an increased risk of hepatotoxicity in the MVC group. Nine participants in the MVC group and six in the TDF/FTC group met the liver chemistry stopping criteria. Most of these patients had hepatitis virus coinfection (three with hepatitis A coinfection, two with hepatitis B coinfection and six with hepatitis C coinfection). Three participants in the MVC group and seven in the TDF/FTC group were discontinued due to treatment-emergent renal impairment (decreased glomerular filtration rate, increased serum creatinine or renal failure). Creatinine clearance rates (Cockcroft–Gault equation) decreased by −3.4 ml/min for MVC-treated participants compared with −9.3 ml/min for TDF/FTC-treated participants (P = 0.0001). Assessment of glomerular filtration rate (estimated using the chronic kidney disease epidemiological collaboration equation) showed minimal changes in both groups. Metabolic and cardiovascular events were reported infrequently, with only one myocardial infarction in the TDF/FTC group, and ratios of high-density to low-density lipoprotein cholesterol and total cholesterol to high-density lipoprotein cholesterol remained unchanged in both groups. The results of the substudy on BMD, bone turnover and body fat distribution, as well as the health outcomes assessments are presented in Supplementary Digital Content 2.
Discussion
There is an increasing interest in identifying simplified antiretroviral treatment regimens to improve adherence, preserve future treatment options and reduce toxicities. Nucleos(t)ide-sparing, two-drug regimens containing ritonavir-boosted protease inhibitors have not been extensively studied in antiretroviral-naive individuals. Such regimens may offer opportunities to avoid NRTI toxicities [6–8] and maintain future options.
MODERN demonstrated that a two-drug NRTI-sparing regimen of MVC 150 mg q.d. along with DRV/r was inferior to TDF/FTC along with DRV/r, with treatment failure the main reason for the difference in response rate. Importantly, there was no development of resistance to any of the drugs. This outcome is consistent with results from the A4001078 pilot study of MVC along with ATV/r [17].
Similarly, in the RADAR study, RAL along with DRV/r resulted in 62.5% of participants achieving HIV-1 RNA less than 50 copies/ml, compared with 83.7% for TDF/FTC and DRV/r, while the ACTG A5262 study reported high rates of virologic failure and emergence of resistance mutations in participants receiving RAL and DRV/r [12,20]. In contrast, the SPARTAN study found that 74.6% of participants receiving ATV/RAL achieved HIV-1 RNA less than 50 copies/ml, compared with 63.3% of those receiving ATV/r and TDF/FTC. However, more participants receiving ATV/RAL developed resistance mutations [11]. In the PROGRESS study, virologic response rates between the RAL along with LPV/r and TDF/FTC along with LPV/r groups were similar, but three participants with treatment failure in the RAL group developed integrase and/or LPV mutations, while only one individual in the TDF/FTC group developed M184V [10]. More recently, data from the NEAT study demonstrated noninferiority at 96 weeks (using a composite primary endpoint) in participants receiving RAL along with DRV/r compared with those receiving TDF/FTC along with DRV/r, but more treatment failures and resistance mutations were seen in the RAL group [21].
The reasons for the consistent trend towards lower virologic response rates and/or higher risk for resistance mutations in these two-drug NRTI-sparing studies are not clear. In some, but not all studies, poorer response rates were observed in participants with HIV-1 RNA more than 100 000 copies/ml [22]. Another possible explanation is that three drugs are needed to ensure a good virologic response in some patients and, therefore, two-drug antiretroviral regimens are inferior to regimens containing three drugs. Altogether, data from these studies indicate that, in general, two-drug nucleos(t)ide-sparing, ritonavir-boosted protease inhibitor containing dual-therapy regimens are inferior in treatment-naive participants.
Interestingly, the majority of individuals in both treatment arms in MODERN failed treatment with low plasma HIV-1 RNA and several went on to achieve HIV-1 RNA less than 50 copies/ml at a later time point. In addition, there was no emergence of resistance-associated mutations observed. This may be a reflection of the stringent PDTF criteria specified in the protocol, leading to patients with minor blips in HIV-1 RNA being classified as virologic failures. The greater number of individuals with PDTF in the MVC arm compared with TDF may in part be explained by a slightly slower response rate with MVC, which in turn might reflect the activation of viral production by latent reservoirs of infected cells by MVC as well as redistribution of virus into plasma, both of which have been reported [23–27]. Furthermore, a higher incidence of low level viremia or blips has been described in patients receiving protease inhibitor-based HAART or boosted protease inhibitor monotherapy (including DRV) as part of simplification strategies [28,29]. In most studies, viral load blips have not been found to be predictive of an increased risk of persistent viral load rebound [29]. Therefore, the detection of low-level plasma HIV-1 RNA may not be an appropriate marker of virologic failure with MVC, especially in combination with a protease inhibitor.
Modified Medication Adherence Self-Report Inventory questionnaire data collected in this study indicated that response rates were lower in participants with less than 95% self-reported adherence; however, this was seen in both groups. The absence of prospective pharmacokinetic analysis is a limitation of the study; however, data from the post hoc pharmacokinetic analysis revealed no evidence that lower concentrations accounted for a lack of efficacy. Maraviroc is a sensitive substrate for both cytochrome P450 3A and P-glycoprotein and coadministration with DRV/r, or most other boosted protease inhibitors, increases MVC plasma concentrations [16,30]. The average MVC plasma concentration (Cavg) associated with near maximal efficacy in exposure–response analyses from the MOTIVATE and MERIT studies was at least 100 and ≥75 ng/ml, respectively, while the in-vivo IC50 has been estimated at 7.65 ng/ml [31–34]. In a pharmacokinetic substudy of Study A4001078, the median (range) MVC Cavg was 180 (80.3–305) ng/ml (n = 15), thus exceeding the in-vivo IC50 by more than 10-fold [17]. A pharmacokinetic study conducted by Mora-Peris et al.[34] showed that the geometric mean (95% CI) for MVC Cavg was 125 (99–158) ng/ml (n = 11). Irrespective of efficacy outcome, the pharmacokinetics in MODERN was consistent with the 24-h dose interval in the pharmacokinetic study previously mentioned.
The two groups had a similar safety profile, and there were no new reported safety findings for MVC. There was a numerical imbalance in malignancies with nine participants reporting malignancies in the MVC group compared with three in the TDF/FTC group. The only malignancy that was considered possibly related to study drug by the investigator was an event of Hodgkin's disease reported in the MVC group, but on further follow-up this was considered likely related to other reasons (Epstein–Barr virus infection and immune reconstitution inflammatory syndrome). This imbalance has not been observed in other MVC studies, including during 5-year follow-up in the MOTIVATE and MERIT studies [13–15]. Altogether, the number of events was small and the types of malignancies reported were typical of an HIV-infected population [35,36].
Osteoporosis and decrease in BMD of the hip and spine are more common in HIV-infected adults than uninfected controls [37]. Declines in BMD of 2–6% have been seen in the 2 years following antiretroviral therapy initiation [38]. As was also observed in this study, low baseline CD4+ count has consistently been associated with a higher degree of bone loss in HIV-1 infected participants [38,39]. Tenofovir therapy is associated with an additional 1–2% bone loss in HIV-1 infected adults compared with other antiretrovirals [40,41]. In this study, TDF/FTC was associated with a greater hip BMD decrease and more bone turnover than MVC over 48 weeks, supporting the concept that TDF-sparing antiretroviral therapy may help to decrease bone loss. This result is also consistent with data from the ACTG 5303 study that also compared BMD changes in HIV-1 infected participants initiating MVC 150 mg q.d. versus TDF, each combined with DRV/r and FTC, which demonstrated that MVC-treated participants had a smaller decrease in BMD at both the hip and spine [42]. The mechanism(s) of TDF's negative effect on BMD is likely due to increased bone turnover, as evidenced by the increased CTx values observed in this study, and may also be affected by changes in calcium-phosphate homeostasis with net loss of phosphate, resulting in bone demineralization [43,44].
In conclusion, at Week 48, MVC dosed q.d. with DRV/r in a two-drug regimen showed inferior efficacy to TDF/FTC and DRV/r in antiretroviral-naive participants, resulting in premature discontinuation of the study following an independent DMC recommendation. No evidence of resistance to any of the study drugs was observed. The data confirmed the safety and tolerability of MVC over 48 weeks with no new safety concerns identified.
Acknowledgements
The sponsor (ViiV Healthcare) participated in the study design. The sponsor's designee (Pfizer, Inc) participated in the study design, data collection, data analysis and data interpretation. All authors had full access to all the data in the study and are responsible for the veracity and completeness of the data reported. The corresponding author had final responsibility for the decision to submit for publication.
H.-J.S. recruited participants for the study, provided input into the protocol design and data review, and served as an advisory board member. E.LeF. and A.C. contributed to the analysis and interpretation of data and drafting of the manuscript. M.S.S., C.F. and S.N. recruited participants for the study and provided input into the protocol design and data review. G.M., S.R.V., M.V., C.C., A.F.F. and J.H. contributed to the study design, acquisition of data, analysis and interpretation of data, and drafting of the manuscript. A.R.R. contributed to the analysis and interpretation of data. L.M. contributed to the study design, analysis and interpretation of data, and drafting of the manuscript. A.C. recruited participants for the study, served on the study steering committee and provided input into the protocol design and data review.
The authors would like to thank Hernan Valdez, James Goodrich, Ahmed Shelbaya, Elna van Der Ryst, Jim Demarest, Simon Portsmouth, Rebecca Zhang-Roper, Melissa Crawford, Marilyn Lewis and Barry Weatherley for their assistance with study design, conduct and analysis, and preparation of the manuscript. The authors wish to acknowledge Meredith MacPherson for editorial assistance during the development of this manuscript.
Conflicts of interest
H.-J.S. has received support for travel to meetings for the study or other purposes, for provision of writing assistance and study drug supply, and for documentation fees from ViiV Healthcare/Pfizer; has received payment for serving on advisory boards and providing lectures/developing educational presentations for AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen Cilag, Merck, Sharp & Dohme and ViiV Healthcare; and is a member of the guideline development committee of the European AIDS Society and the German AIDS Society. E.LeF. is an employee of and owns stock/stock options in ViiV Healthcare and has received consulting fees or honoraria from ViiV Healthcare. A.C. has received a grant from Pfizer; has received support for travel to meetings for the study or other purposes from ViiV Healthcare; and has received payment for board membership, consultancy fees, and payment for lectures, including service on speaker bureaus, from Gilead Sciences, MSD and ViiV Healthcare; and has received payment for development of educational presentations from ViiV Healthcare. M.S.S. has received grants/has grants pending from AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck and ViiV Healthcare. G.M., S.R.V., M.V., L.M., A.F.F. and J.H. are employees of and own stock/stock options in Pfizer. S.N. has received a grant, support for travel to an investigator's meeting and payment for board membership and for development of educational presentations from ViiV Healthcare. A.R.R. is an employee of ViiV Healthcare and owns stock/stock options in GlaxoSmithKline. C.F. has received grants/has grants pending from Cubist, EnteraHealth, Gilead Sciences, Janssen and Pfizer and has received payment for lectures, including service on speaker bureaus, from Kowa Pharmaceuticals. A.C. is an employee of ViiV Healthcare and owns stock/stock options in GlaxoSmithKline. C.C. has received consulting fees or honoraria and support for travel to meetings for the study or other purposes from Pfizer; owns stock/stock options in GlaxoSmithKline; and is a former employee of Pfizer and GSK and has performed contractual work for ViiV Healthcare. Funding for this work was provided by ViiV Healthcare.
Supplementary Material
References
- 1.World Health Organization. Rapid advice: antiretroviral therapy for HIV infection in adults and adolescents. November 2009. http://www.who.int/hiv/pub/arv/rapid_advice_art.pdf [Accessed 29 June 2015]. [Google Scholar]
- 2.Thompson MA, Aberg JA, Hoy JF, Telenti A, Benson C, Cahn P, et al. Antiretroviral treatment of adult HIV infection: 2012 recommendations of the International AIDS Society-USA panel. JAMA 2012; 308:387–402. [DOI] [PubMed] [Google Scholar]
- 3.Department of Health and Human Services. Panel on antiretroviral guidelines for adults and adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. 2015. http://www.aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf [Accessed 29 June 2015]. [Google Scholar]
- 4.European AIDS Clinical Society. Guidelines version 7.1. November 2014. http://www.eacsociety.org/files/guidelines-7.1-english.pdf [Accessed 29 June 2015]. [Google Scholar]
- 5.British HIV Association. Guidelines for the treatment of HIV-1 positive adults with antiretroviral therapy. November 2013. http://www.bhiva.org/documents/Guidelines/Treatment/2012/hiv1029_2.pdf [Accessed 29 June 2015]. [Google Scholar]
- 6.Brinkman K, ter Hofstede HJ, Burger DM, Smeitink JA, Koopmans PP. Adverse effects of reverse transcriptase inhibitors: mitochondrial toxicity as common pathway. AIDS 1998; 12:1735–1744. [DOI] [PubMed] [Google Scholar]
- 7.Lesho EP, Gey DC. Managing issues related to antiretroviral therapy. Am Fam Phys 2003; 68:675–686. [PubMed] [Google Scholar]
- 8.Nolan D, Reiss P, Mallal S. Adverse effects of antiretroviral therapy for HIV infection: a review of selected topics. Expert Opin Drug Saf 2005; 4:201–218. [DOI] [PubMed] [Google Scholar]
- 9.Riddler SA, Haubrich R, DiRienzo AG, Peeples L, Powderly WG, Klingman KL, et al. Class-sparing regimens for initial treatment of HIV-1 infection. N Engl J Med 2008; 358:2095–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reynes J, Trinh R, Pulido F, Soto-Malave R, Gathe J, Qaqish R, et al. Lopinavir/ritonavir combined with raltegravir or tenofovir/emtricitabine in antiretroviral-naive subjects: 96-week results of the PROGRESS study. AIDS Res Hum Retroviruses 2013; 29:256–265. [DOI] [PubMed] [Google Scholar]
- 11.Kozal MJ, Lupo S, De Jesus E, Molina JM, McDonald C, Raffi F, et al. A nucleoside- and ritonavir-sparing regimen containing atazanavir plus raltegravir in antiretroviral treatment-naive HIV-infected patients: SPARTAN study results. HIV Clin Trials 2012; 13:119–130. [DOI] [PubMed] [Google Scholar]
- 12.Taiwo B, Zheng L, Gallien S, Matining RM, Kuritzkes DR, Wilson CC, et al. Efficacy of a nucleoside-sparing regimen of darunavir/ritonavir plus raltegravir in treatment-naive HIV-1-infected patients (ACTG A5262). AIDS 2011; 25:2113–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cooper DA, Heera J, Ive P, Botes M, Dejesus E, Burnside R, et al. Efficacy and safety of maraviroc vs. efavirenz in treatment-naive patients with HIV-1: 5-year findings. AIDS 2014; 28:717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gulick RM, Fatkenheuer G, Burnside R, Hardy WD, Nelson MR, Goodrich J, et al. Five-year safety evaluation of maraviroc in HIV-1-infected treatment-experienced patients. J Acquir Immune Defic Syndr 2014; 65:78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gulick RM, Lalezari J, Goodrich J, Clumeck N, DeJesus E, Horban A, et al. Maraviroc for previously treated patients with R5 HIV-1 infection. N Engl J Med 2008; 359:1429–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kakuda TN, Abel S, Davis J, Hamlin J, Schöller-Gyüre M, Mack R, et al. Pharmacokinetic interactions of maraviroc with darunavir-ritonavir, etravirine, and etravirine-darunavir-ritonavir in healthy volunteers: results of two drug interaction trials. Antimicrob Agents Chemother 2011; 55:2290–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mills A, Mildvan D, Podzamczer D, Fätkenheuer G, Leal M, Than S, et al. Maraviroc once-daily nucleoside analog-sparing regimen in treatment-naive patients: randomized, open-label pilot study. J Acquir Immune Defic Syndr 2013; 62:164–170. [DOI] [PubMed] [Google Scholar]
- 18.Taiwo B, Acosta EP, Ryscavage P, Berzins B, Lu D, Lalezari J, et al. Virologic response, early HIV-1 decay and maraviroc pharmacokinetics with the nucleos(t)ide-free regimen of maraviroc plus darunavir/ritonavir in a pilot study. J Acquir Immune Defic Syndr 2013; 64:167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nozza S, Galli L, Antinori A, Chiappetta S, Mazzotta F, Zaccarelli M, et al. Maraviroc 150 mg daily plus lopinavir/ritonavir, a nucleoside/nucleotide reverse transcriptase inhibitor-sparing regimen for HIV-infected naive patients: 48-week final results of VEMAN study. Clin Microbiol Infect 2015; 21:510.e1–510.e9. [DOI] [PubMed] [Google Scholar]
- 20.Bedimo RJ, Drechsler H, Jain M, Cutrell J, Zhang S, Li X, et al. The RADAR study: week 48 safety and efficacy of RAltegravir combined with boosted DARunavir compared to tenofovir/emtricitabine combined with boosted darunavir in antiretroviral-naive patients. Impact on bone health. PLoS One 2014; 9:e106221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raffi F, Babiker AG, Richert L, Molina JM, George EC, Antinori A, et al. Ritonavir-boosted darunavir combined with raltegravir or tenofovir-emtricitabine in antiretroviral-naive adults infected with HIV-1: 96 week results from the NEAT001/ANRS143 randomised noninferiority trial. Lancet 2014; 384:1942–1951. [DOI] [PubMed] [Google Scholar]
- 22.Achhra A, Boyd MA. Antiretroviral regimens sparing agents from the nucleoside(tide) reverse transcriptase inhibitor class: a review of the recent literature. AIDS Res Ther 2013; 10:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gutierrez C, Diaz L, Vallejo A, Hernandez-Novoa B, Abad M, Madrid N, et al. Intensification of antiretroviral therapy with a CCR5 antagonist in patients with chronic HIV-1 infection: effect on T cells latently infected. PLoS One 2011; 6:e27864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kramer VG, Schader SM, Oliveira M, Colby-Germinario SP, Donahue DA, Singhroy DN, et al. Maraviroc and other HIV-1 entry inhibitors exhibit a class-specific redistribution effect that results in increased extracellular viral load. Antimicrob Agents Chemother 2012; 56:4154–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Madrid-Elena N, Hernandez-Novoa B, Garcia-Bermejo L, Moreno S. Maraviroc (MVC) intensification can activate NFkB through CCR5 and the expression of its target genes in resting CD4+ T cells in suppressed HIV-1-infected patients [abstract MOLBPE02]. International AIDS Society Conference; 30 June–3 July 2013; Kuala Lumpur, Malaysia. [Google Scholar]
- 26.Madrid-Elena N, Hernandez-Novoa B, Sastre Turrion B, Serrano-Villar S, Solomon A, Gutierrez C, et al. Maraviroc reverts latent HIV-1 in ART suppressed patients through NFkB without viral load increase [abstract 427LB]. Conference on Retroviruses and Opportunistic Infection; 3–6 March 2014; Boston, MA. [Google Scholar]
- 27.Symons J, de Spiegelaere W, Wensing A, Drylewicz J, Middel A, Hoepelman AI, et al. Maraviroc induces HIV production in RCT and in vitro, potentially via the NFkB pathway [abstract 549]. Conference on Retroviruses and Opportunistic Infection; 23–26 February 2015; Seattle, WA. [Google Scholar]
- 28.Geretti AM, Smith C, Haberl A, Garcia-Diaz A, Nebbia G, Johnson M, et al. Determinants of virological failure after successful viral load suppression in first-line highly active antiretroviral therapy. Antivir Ther 2008; 13:927–936. [PubMed] [Google Scholar]
- 29.Torres-Cornejo A, Benmarzouk-Hidalgo OJ, Gutierrez-Valencia A, Perez-Romero P, Martin-Pena R, Ruiz-Valderas R, et al. Cellular HIV reservoir replenishment is not affected by blip or intermittent viremia episodes during darunavir/ritonavir monotherapy. AIDS 2014; 28:201–208. [DOI] [PubMed] [Google Scholar]
- 30.Abel S, Russell D, Taylor-Worth RJ, Ridgway CE, Muirhead GJ. Effects of CYP3A4 inhibitors on the pharmacokinetics of maraviroc in healthy volunteers. Br J Clin Pharmacol 2008; 65 Suppl 1:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abel S, Back DJ, Vourvahis M. Maraviroc: pharmacokinetics and drug interactions. Antivir Ther 2009; 14:607–618. [PubMed] [Google Scholar]
- 32.McFadyen L, Jacqmin P, Wade JR, Weatherley B, Chan PLS. Maraviroc (MVC) exposure efficacy relationship in treatment experienced HIV-1-infected patients [abstract P4.1/06]. 11th European AIDS Conference; 24–27 October 2007; Madrid, Spain. [Google Scholar]
- 33.McFadyen L, Jacqmin P, Wade JR, Weatherley B. Maraviroc (MVC) exposure-efficacy analysis in HIV-1-infected treatment-naïve subjects [abstract TUPE0053]. XVII International AIDS Conference; 3–8 August 2008; Mexico City, Mexico. [Google Scholar]
- 34.Mora-Peris B, Croucher A, Else LJ, Vera JH, Khoo S, Scullard G, et al. Pharmacokinetic profile and safety of 150 mg of maraviroc dosed with 800/100 mg of darunavir/ritonavir all once daily, with and without nucleoside analogues, in HIV-infected subjects. J Antimicrob Chemother 2013; 68:1348–1353. [DOI] [PubMed] [Google Scholar]
- 35.Kowalkowski MA, Mims MA, Day RS, Du XL, Chan W, Chiao EY. Longer duration of combination antiretroviral therapy reduces the risk of Hodgkin lymphoma: a cohort study of HIV-infected male veterans. Cancer Epidemiol 2014; 38:386–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spano JP, Costagliola D, Katlama C, Mounier N, Oksenhendler E, Khayat D. AIDS-related malignancies: state of the art and therapeutic challenges. J Clin Oncol 2008; 26:4834–4842. [DOI] [PubMed] [Google Scholar]
- 37.Brown TT, Qaqish RB. Antiretroviral therapy and the prevalence of osteopenia and osteoporosis: a meta-analytic review. AIDS 2006; 20:2165–2174. [DOI] [PubMed] [Google Scholar]
- 38.Brown TT, McComsey GA, King MS, Qaqish RB, Bernstein BM, da Silva BA. Loss of bone mineral density after antiretroviral therapy initiation, independent of antiretroviral regimen. J Acquir Immune Defic Syndr 2009; 51:554–561. [DOI] [PubMed] [Google Scholar]
- 39.Grant PM, Kitch D, McComsey GA, Dube MP, Haubrich R, Huang J, et al. Low baseline CD4+ count is associated with greater bone mineral density loss after antiretroviral therapy initiation. Clin Infect Dis 2013; 57:1483–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McComsey GA, Kitch D, Daar E, Tierney C, Jahed NC, Tebas P, et al. Bone mineral density and fractures in antiretroviral-naive persons randomized to receive abacavir-lamivudine or tenofovir disoproxil fumarate-emtricitabine along with efavirenz or atazanavir-ritonavir: AIDS Clinical Trials Group A5224 s, a substudy of ACTG A5202. J Infect Dis 2011; 203:1791–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moyle GJ, Stellbrink HJ, Compston J, Orkin C, Arribas JR, Domingo P, et al. 96-Week results of abacavir/lamivudine versus tenofovir/emtricitabine, plus efavirenz, in antiretroviral-naïve, HIV-1-infected adults: ASSERT study. Antivir Ther 2013; 18:905–913. [DOI] [PubMed] [Google Scholar]
- 42.Taiwo BO, Chan E, Fichtenbaum CJ, Ribaudo H, Tsibris A, Klingman KL, et al. Less bone loss with maraviroc- versus tenofovir-containing antiretroviral therapy in the AIDS Clinical Trials Group A5303 Study. Clin Infect Dis 2015; 61:1179–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grigsby IF, Pham L, Mansky LM, Gopalakrishnan R, Mansky KC. Tenofovir-associated bone density loss. Ther Clin Risk Manag 2010; 6:41–47. [PMC free article] [PubMed] [Google Scholar]
- 44.Havens PL, Kiser JJ, Stephensen CB, Hazra R, Flynn PM, Wilson CM, et al. Association of higher plasma vitamin D binding protein and lower free calcitriol levels with tenofovir disoproxil fumarate use and plasma and intracellular tenofovir pharmacokinetics: cause of a functional vitamin D deficiency?. Antimicrob Agents Chemother 2013; 57:5619–5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.