

CONSPECTUS
Characterization of monocopper intermediates in enzymes and other catalysts that attack strong C–H bonds is important for unraveling oxidation catalysis mechanisms and, ultimately, designing new, more efficient catalytic systems. Because initially formed 1:1 Cu/O2 adducts resulting from reactions of Cu(I) sites with O2 react relatively sluggishly with substrates with strong C–H bonds, it has been suggested that reductive O–O bond scission might occur instead to yield more reactive [CuO]+ or protonated [CuOH]2+ cores. Experimental and theoretical studies of [CuO]+ species in the gas phase have provided key insights into the possible reactivity of these species, but detailed information is lacking for discrete complexes with the [CuO]+ or [CuOH]2+ core in solution or the solid state. We describe herein our recent efforts to address this issue through several disparate approaches. In one strategy based on precedent from studies of enzymes and synthetic compounds with iron-α-ketocarboxylate motifs, reactions of O2 with Cu(I)-α-ketocarboxylate complexes were explored, with the aim of identifying reaction pathways that would implicate the intermediacy of a [CuO]+ species. A second approach focused on the reaction of N-oxides with Cu(I) complexes, with the goal being to elicit O–N bond heterolysis to yield [CuO]+ complexes. For both strategies, the course of the reactions depended on the nature of the supporting bidentate N-donor ligand, and indirect evidence in support of the sought-after [CuO]+ intermediates was obtained in some instances.
In the final approach discussed herein, strongly electron donating and sterically encumbered pyridine-dicarboxamide ligands (L) enabled the synthesis of [LCu(II)OH]− complexes, which upon one-electron oxidation formed complexes with the [CuOH]2+ core that were characterized in solution. Rapid hydrogen atom abstraction (HAT) from dihydroanthracene (DHA) was observed, yielding LCu(II)OH2. The O–H bond dissociation enthalpy (BDE) of ~90 kcal/mol for this complex was determined through evaluation of its pKa (~19) and the [LCu(II)OH]−/LCu(III)OH reduction potential (approximately −0.08 V vs Fc/Fc+). Thus, the poor oxidizing power of the complex is offset by the high basicity of the hydroxide moiety to yield a strong O–H bond. This high BDE provided a thermodynamic rationale for the rapid HAT rate from DHA and suggested that stronger C–H bonds could be attacked. Indeed, using an inert solvent (1,2-difluorobenzene), substrates with C–H bond strengths as high as 99 kcal/mol were shown to react with the [CuOH]2+ complex, and a linear log k vs C–H BDE plot supported similar HAT pathways across the series. Importantly, these results provided key evidence in favor of the possible intermediacy of this core in oxidation catalysis, and we suggest that because it is a more energetically accessible intermediate than the [CuO]+ moiety, it should be considered as an alternative in proposed mechanisms for oxidations by enzymes and other synthetic systems.
Graphical Abstract
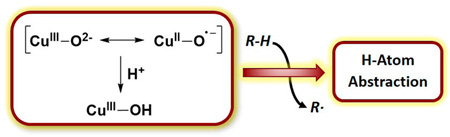
INTRODUCTION
Oxidation reactions promoted by copper centers in enzymes and by other catalysts are critical for transforming organic molecules for life processes and synthetic applications.1,2 A longstanding research objective has been to understand the mechanisms of such oxidation reactions, and in particular, to reveal how copper centers react with O2 or other oxidants and to determine the nature of the key resulting copper–oxygen intermediates responsible for attacking substrate C–H bonds.3 In general, it is postulated that Cu(I) centers in enzymes and other catalytic systems react with O2 to yield initial 1:1 Cu/O2 adducts; higher nuclearity species are important in multicopper systems,4 but we focus here only on mononuclear sites. A number of 1:1 Cu/O2 adducts have been identified in synthetic studies using suitably designed supporting ligands, and they have been described as end-on (1) or side-on (2) copper(II)–superoxide or copper(III)–peroxide species on the basis of computational and experimental evidence (Scheme 1).5 Armed with the precedent provided by these synthetic efforts, as well as more direct experimental and computational data on the catalytic systems themselves, a functional role for 1 and 2 in a variety of enzymes has been proposed.1,6 This role commonly involves a hydrogen atom transfer (HAT) from a substrate C–H bond in a key rate-determining step of the catalytic mechanism. Yet, while HAT reactions of discrete, well-characterized examples of complexes with moieties 1 and 2 have been observed, they typically only occur at reasonable rates with substrates having relatively low C–H bond dissociation free energies (weaker bonds than the catalytically relevant substrates).5,7 As a result, questions have been raised about the feasibility of intermediates 1 and 2 as oxidants in catalytic systems that transform substrates with strong C–H bonds such as, for example, particulate methane monooxygenase (pMMO), for which multicopper oxidants have been proposed, including dicopper species with monocopper–oxygen units.3c,8
Scheme 1.
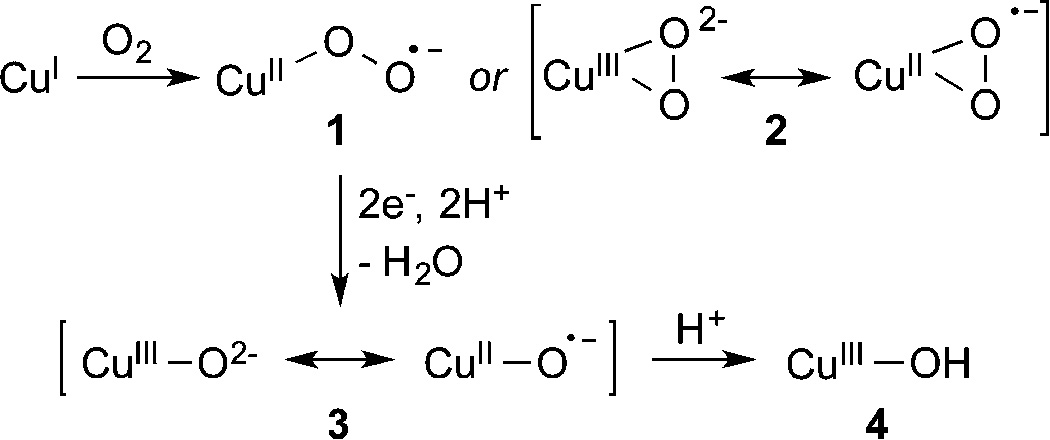
Proposed Cu/O2 Activation Pathways and Intermediates
An obvious alternative to 1 and 2 as intermediates that is inspired by the mechanistic paradigm for iron-containing catalysts9 is addition of protons and electrons prior to attack on substrate, resulting in scission of the O–O bond to yield a high valent metal–oxo species that would be a more potent oxidant (3, Scheme 1). While examples of well-characterized mononuclear iron(IV)–oxo complexes now abound, and advances in our understanding of their reactivity have been substantial,10 much less is known about copper congeners having a [CuO]+ core (3). Many proposals for the involvement of [CuO]+ species, as well as the protonated version [CuOH]2+ (4), in oxidation reactions have appeared, but supporting evidence is sparse and indirect, and such species have not been observed directly as discrete complexes in condensed phase.11 Compelling evidence for [CuO]+ has come from gas phase studies supplemented by theoretical calculations.12 These studies suggest extraordinarily high reactivity and strong thermodynamic driving forces for HAT reactions with C–H bonds by a species best described as a triplet copper(II)–oxyl radical. Computational work supports this bonding picture and similarly high reactivity for putative [CuO]+ intermediates in enzymes such as dopamine β-monooxygenase (DβM),13 peptidylglycine α-amidating monooxygenase (PHM),14 and lytic polysaccharide monooxygenase (LPMO).15 Inspired by these provocative findings and the overarching goal of defining the nature of copper–oxygen oxidants involved in metalloenzyme and other catalysts, we targeted complexes containing the [CuO]+ core for synthesis and characterization.
In this Account, we summarize our efforts to prepare complexes with the [CuO]+ and [CuOH]2+ cores. We first describe initial work aimed at accessing [CuO]+ complexes through oxidative decarboxylation of Cu(I)-α-ketoacid complexes and reactions of Cu(I) complexes with oxo-transfer reagents, before turning to more recent efforts to prepare and understand the properties of [CuOH]2+ compounds. The recently demonstrated ability of these [CuOH]2+ cores to attack strong C–H bonds has provided an experimental foundation for the hypothesis that such species are possible reactive intermediates in oxidation catalysis.
DECARBOXYLATION OF Cu(I)–α-KETOACID COMPLEXES
An intriguing route to Fe(IV)–oxo species followed by nonheme iron enzymes involves reaction of O2 with an Fe(II)–α-ketocarboxylate (5, M=Fe, Scheme 2).9b–dThe result is ejection of CO2 and O–O bond homolysis to form the reactive Fe(IV)=O that attacks substrate C–H bonds. We envisioned that a target [CuO]+ species could be formed via a similar pathway starting from a Cu(I)–α-ketocarboxylate complex (5, M = Cu). Choice of a proper supporting ligand was viewed as key in order to observe decarboxylation and enable trapping of what was expected to be an extraordinarily reactive and likely quite unstable [CuO]+ core.
Scheme 2.
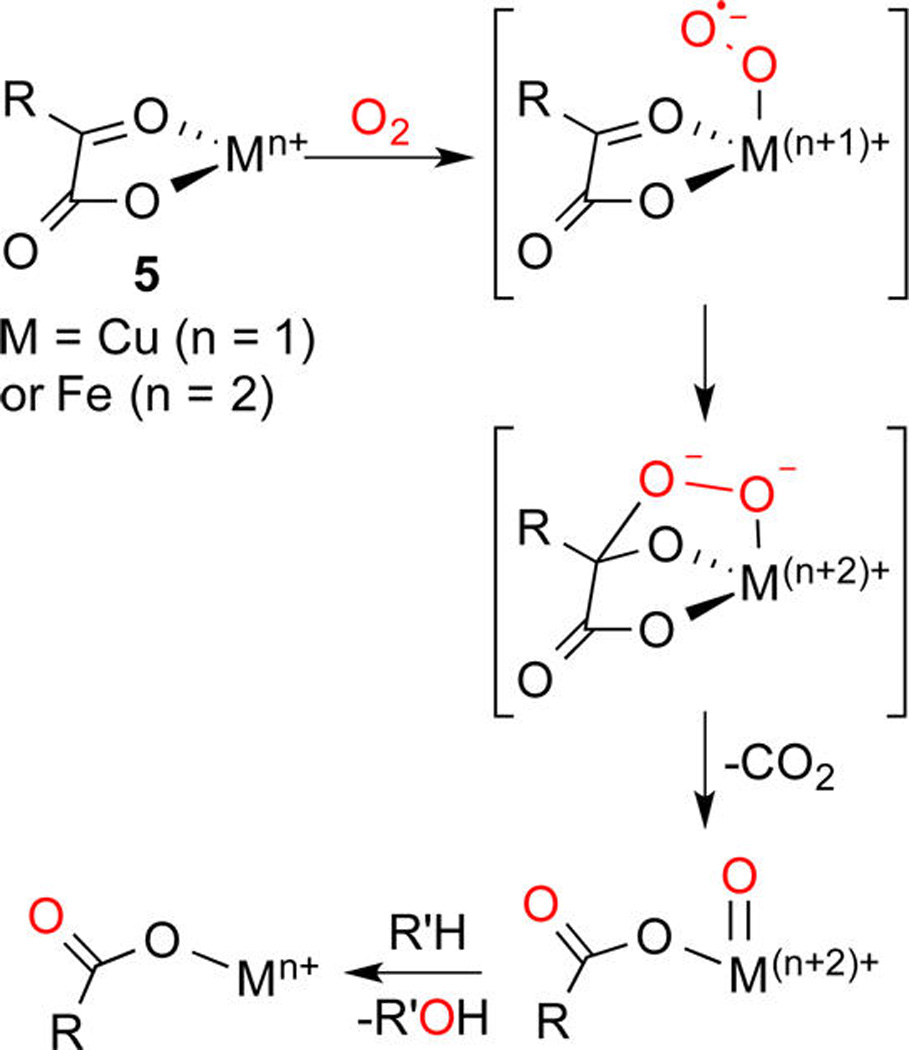
General Mechanism for Reaction of a Metal α-Ketocarboxylate Complex with O2
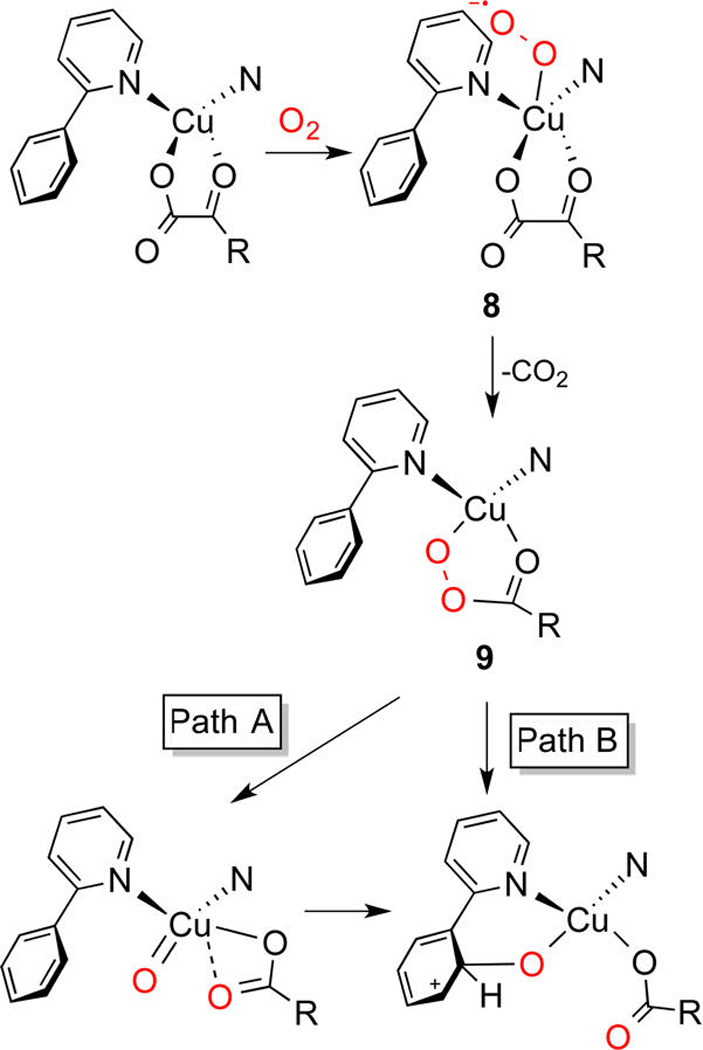
In one approach,16 we used Me4pda and tBu2Me2eda (Scheme 3), strongly electron donating bidentate diamines known to promote Cu(I)/O2 reactions.17 The model α-ketocarboxylates, benzoyl formate (BF) or p-nitrobenzoyl formate (nitro-BF), were found to bind in mono- or bidentate fashion via their carboxylate moieties to the (tBu2Me2eda)Cu(I) moiety (Scheme 3). Oxygenation of solutions of the complexes (tBu2Me2eda)Cu-(BF) or -(nitro-BF) at room temperature failed to induce decarboxylation, but modest yields (11–16%) of the appropriate benzoic acid resulting from decarboxylation were isolated in the presence of excess cyclohexene or CH3CN. To explain these results, two possible mechanisms were considered (Scheme 3). In path A, binding of the additive (S) to the Cu(I) center enhances decarboxylation in some unknown manner, while in path B, displacement of BF or nitro-BF by the additive is followed by formation of a peroxo- or bis(oxo)dicopper complex that then reacts with the α-ketocarboxylate and induces decarboxylation. To test the feasibility of path B, solutions of [LCu(CH3CN)]OTf in CH2Cl2 at −80 °C were oxygenated to yield either a mixture of the peroxodicopper(II) and bis(oxo)dicopper(III) complexes (L = tBu2Me2eda) or the bis(oxo)dicopper(III) species alone (L = Me4pda), as reported previously.3,17 Subsequent addition of BF to these complexes resulted in decarboxylation, demonstrating the feasibility of path B, an undesired route in efforts to traverse the mechanism shown in Scheme 2.
Scheme 3.
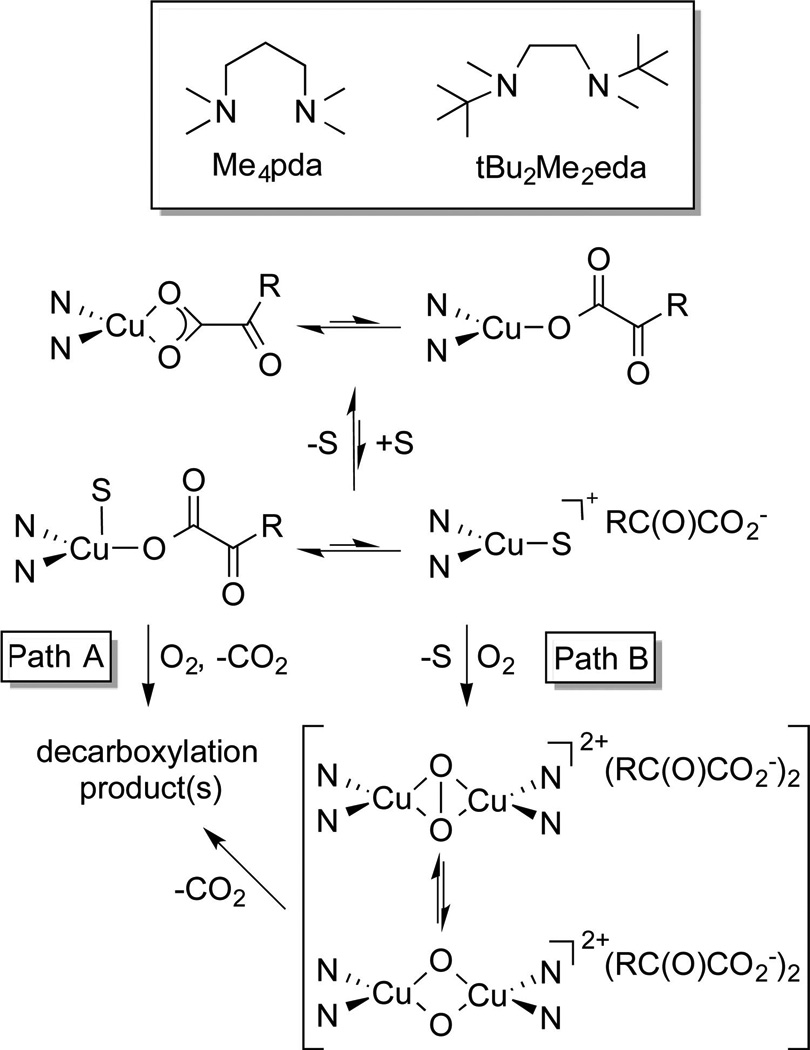
Proposed Mechanisms for Decarboxylation of Cu(I)–α-Ketocarboxylate Complexes Supported by Diamine Ligandsa
aN = diamine donor atoms; R = phenyl or para-nitrophenyl; S = cyclohexene or CH3CN.
To obviate Cu(I)/O2 reactivity in the absence of a coordinated α-ketocarboxylate, we used the less electron donating pyridylimine ligands 6 (Scheme 4), with various phenyl groups strategically placed in the pyridyl 2-position in order to trap a transient oxidizing intermediate.18 Oxygenation of the Cu(I)–BF complexes of 6 (R = H; X = H or OMe) followed by aqueous workup revealed formation of benzoic acid, signifying decarboxylation, and the product of hydroxylation of the arene of the supporting ligand (7). The extent of hydroxylation was enhanced for X = OMe, consistent with attack by an electrophilic copper–oxygen species. Notably, the complex (6)Cu(O3SCF3) (R = Me, X = H) was unreactive with O2, arguing against involvement of an initially formed peroxo- or bis(oxo)dicopper species in the hydroxylation reaction.
Scheme 4.
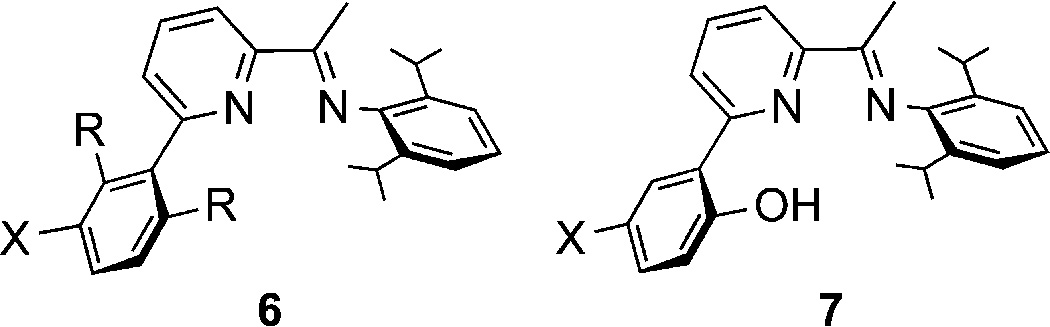
Pyridyl-imine Ligands 6 (R = X = H; R = Me, X = H; R = H, X = OMe) and the Decarboxylation Product 7 (R = X = H; R = H, X = OMe)
The hydroxylation mechanism was explored for a variety of derivatives of 6 using density functional theory (DFT) and multireference second-order perturbation theory (CASSCF/CASPT2) calculations.18,19 The results support the pathway shown in Scheme 5, with the formation of the Cu(I)–peracid intermediate 9 from the initial adduct 8 being quite exergonic (>40 kcal/mol). Importantly, two pathways for the arene hydroxylation were found, one involving direct attack at the arene π system concomitant with peracid O–O bond cleavage along a singlet surface (path B), and the other involving O–O bond scission followed by attack at the arene by the resulting triplet [CuO]+ core (path A). A preference for the latter pathway was indicated by its smaller barrier (12 vs 18 kcal/mol), a provocative finding in support of the intermediacy of the [CuO]+ core in this system.
Scheme 5.
Computed Pathways for Arene Hydroxylation of Cu(I)–α-Ketocarboxylate Complexes Supported by 6a
aFull ligand structure not shown.
REACTIONS OF Cu(I) COMPLEXES WITH OXO TRANSFER REAGENTS
Another method for preparing metal–oxo complexes is to react low valent precursors with oxo transfer reagents, such as R3NO, pyridine-N-oxides, or PhIO. Of particular relevance are reports of reactions of PhIO with Cu complexes that have been proposed to involve [CuO]+ intermediates11d,e and catalytic arene hydroxylations by Cu/Me3NO systems, for which Cu(II)–ONMe3 adducts were proposed on the basis of theory to generate [CuO]+ or, upon protonation, [CuOH]2+ intermediates via O–N bond homolysis.11a,b,20 Hypothesizing that similar intermediates could be generated from heterolytic scission of the O–N bond in Cu(I)–OX adducts (X = NMe3, pyridine), we examined the reactions of various oxo transfer reagents with Cu(I) complexes of the ligands 6 (Scheme 4, R = Me, X = H), 10, and 11 (Scheme 6).21 For complexes of 6, 10 (Y = CF3), and 11, novel examples of stable Cu(I)–OX adducts formed, but O–N heterolysis could not be induced (no decomposition at 80 °C in THF for 1 d). In contrast, reaction of Me3NO with the Cu(I) complex of the more electron donating 10 (Y = Me) at low temperature yielded a bis(oxo)dicopper species,22 a motif previously only accessible from O2.3 While it is tempting to postulate that this product resulted from dimerization of [CuO]+ species, it is also possible that it resulted from O–N cleavage after dimerization of a Cu(I)–ONMe3 intermediate.
Scheme 6.
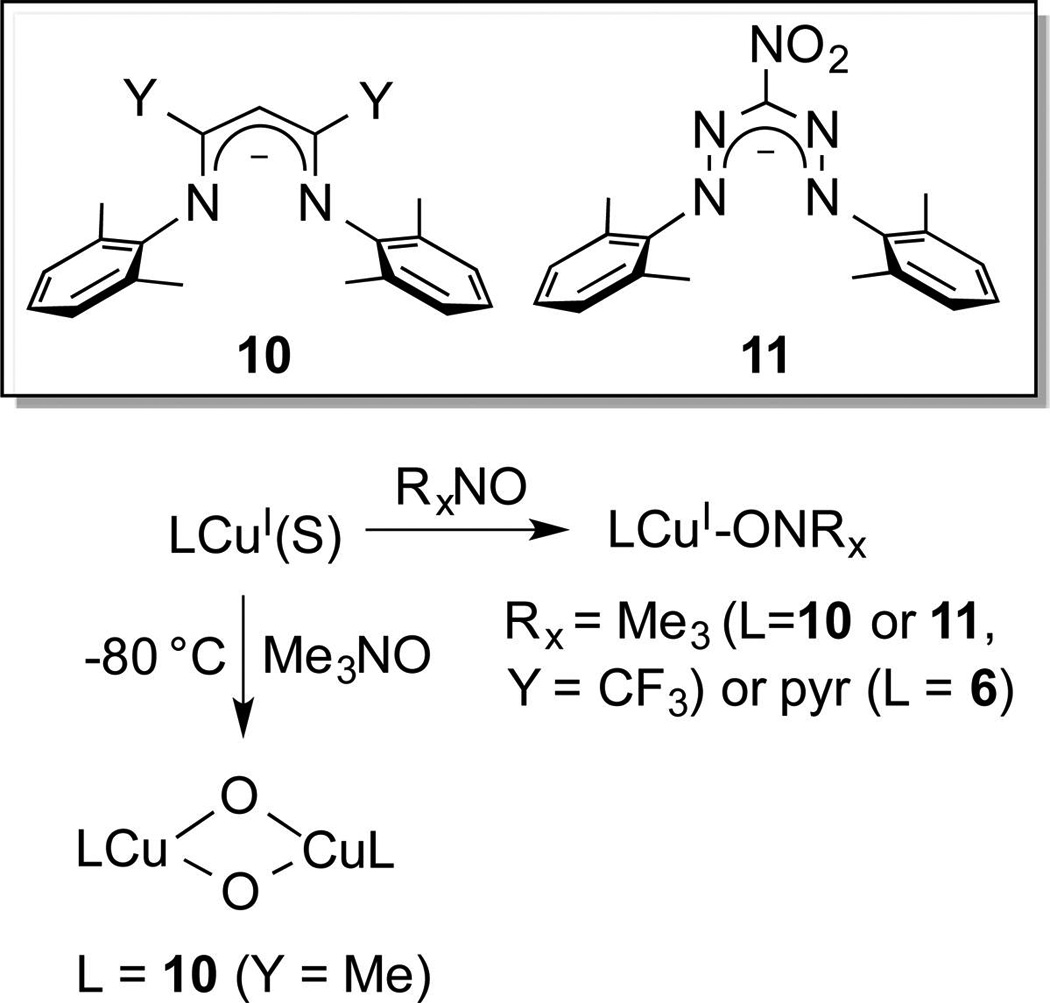
Reactions of Cu(I) Complexes with N-Oxides (S = CH3CN or O3SCF3)
COMPLEXES WITH A [CuOH]2+ CORE
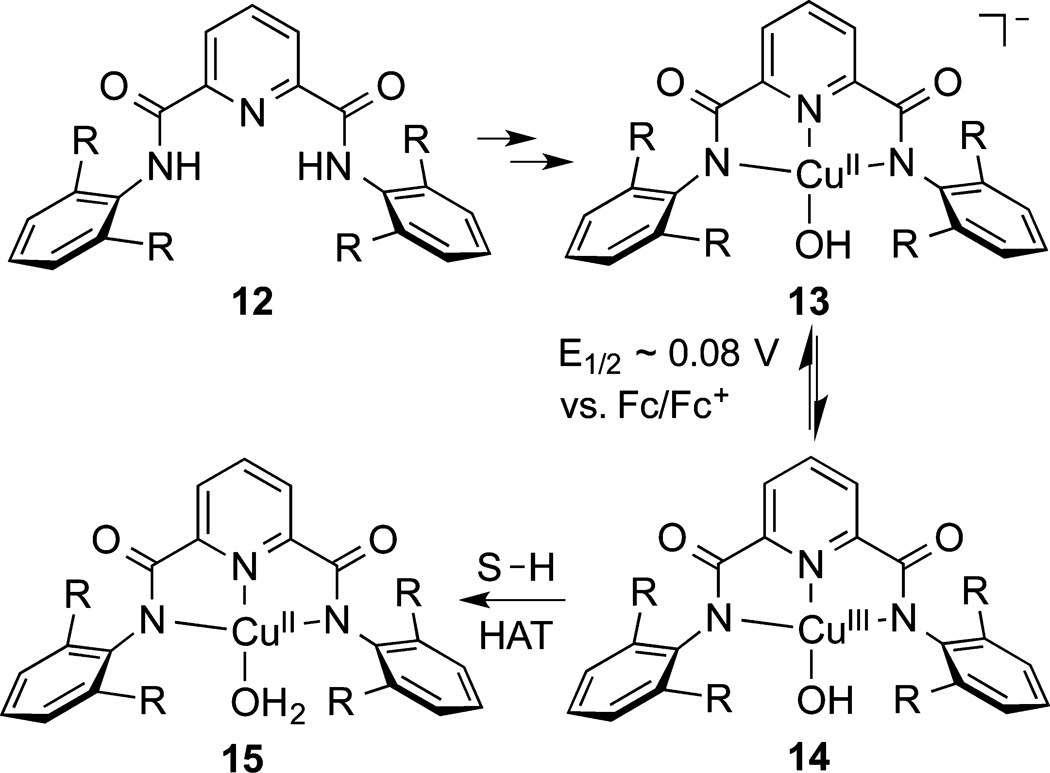
In another approach, we postulated that a protonated version of the elusive [CuO]+ core, [CuOH]2+, might be accessed through simple one-electron oxidation of a Cu(II)–OH precursor and, if supported by a suitably electron donating ligand, could be stabilized sufficiently to be identified. Inspired by precedent for stabilization of Cu(III) complexes by amide ligands23 and a prior report of the synthesis of a Ni(II)–OH complex using 12 (R = Me),24 we prepared complexes 13 (Scheme 7, R = Me or iPr).25,26 Oxidation of solutions of 13 by FcPF6 at low temperature resulted in the development of deep purple color and intense absorption features at ~540 nm (ε ≈ 12 000 M−1 cm−1) that bleached upon warming. The species giving rise to these characteristics were identified as Cu(III)–OH complexes 14 on the basis of (a) spectrophotometric titration and cyclic voltammetry results that indicated they were formed via a reversible one-electron oxidation (E1/2 = −0.076 V vs Fc/Fc+ in acetone), (b) EPR silence, (c) DFT calculations indicative of a singlet ground state with shortened metal–ligand bond distances and no evidence for ligand-based oxidation, (d) TD-DFT calculations that identified the intense absorption feature as a predominantly arene π-to-Cu(dx2−y2) ligand-to-metal charge transfer transition, and (e) Cu K-edge X-ray absorption edge energies for 14 (R = iPr) that are shifted by >1 eV relative to precursor 13.
Scheme 7.
Synthesis and Reactivity of [CuOH]2+ Complexes 14a
aR = Me or iPr; S–H is a hydrocarbon C–H bond.
In an initial study of the reactivity of 14 (R = iPr) aimed at drawing comparisons to other metal–oxo or –hydroxo complexes, hydrogen atom abstraction (HAT) from dihydroanthracene was examined.25 Clean conversion to anthracene and the Cu(II)–OH2 complex 15 was observed, and kinetic studies revealed a second order rate law and a large kinetic isotope effect consistent with C–H bond scission in the rate-determining step via a HAT mechanism. An Eyring analysis of temperature dependent data provided the activation parameters ; from these parameters a KIE of 29 at 20 °C was calculated, which is far above the semiclassical limit. Comparison of the rate constants for reaction with DHA at −80 °C to others reported in the literature (or extrapolated via reported activation parameters) for various metal–oxo/hydroxo complexes25 revealed that the value of log k for 14 (0.93) was superseded only by a few high valent iron species (for example, log k = 2.51 for a Fe(III)OFe(IV)=Ospecies;27 log k = 1.352 for a Fe(V)=O complex28). The high rate of HAT by 14 is notable insofar as it is a rather weak oxidant accessible by oxidation of 13 with FcPF6.
In order to understand the thermodynamic basis for the high reactivity of 14 (R = iPr), we determined the bond dissociation energy (BDE) of the O–H bond in the product 15 by application of eq 1.29 The 13/14 redox potential (−0.074 V vs Fc/Fc+) and the pKa of 15 (18.8 ± 1.8) were]
| (1) |
measured in THF (CH = 66 kcal/mol), yielding BDE = 90 ± 3 kcal/mol.30 This O–H BDE is quite high relative to others reported for metal–oxo or –hydroxo complexes (see Table 1 in ref 30), with the strong basicity of 13 (accentuated in THF, as shown for neutral organic acids like phenols31) counteracting the weak oxidizing power of 14 to yield a strong thermodynamic driving force for attack at C–H bonds. In a further expression of the high basicity of 13, the less sterically hindered variant (R = Me) reacted smoothly with CH3CN to yield the product of deprotonation, a novel Cu(II)–CH2CN complex.26 The high BDE for the O–H bond of 15 is consistent with the high rate of HAT from DHA by 14 and suggests that other substrates with stronger C–H bonds could be attacked under suitable conditions. Indeed, by using relatively inert 1,2-difluorobenzene as solvent, the kinetics of the decay of 14 (R = iPr) in the presence of an excess of a range of substrates with C–H BDEs as high as 99 kcal/mol (cyclohexane) were examined. From these data, a plot of log k vs C–HBDE was constructed, the linear fit to which is consistent with a similar HAT mechanism across the series (Figure 1).30
Figure 1.
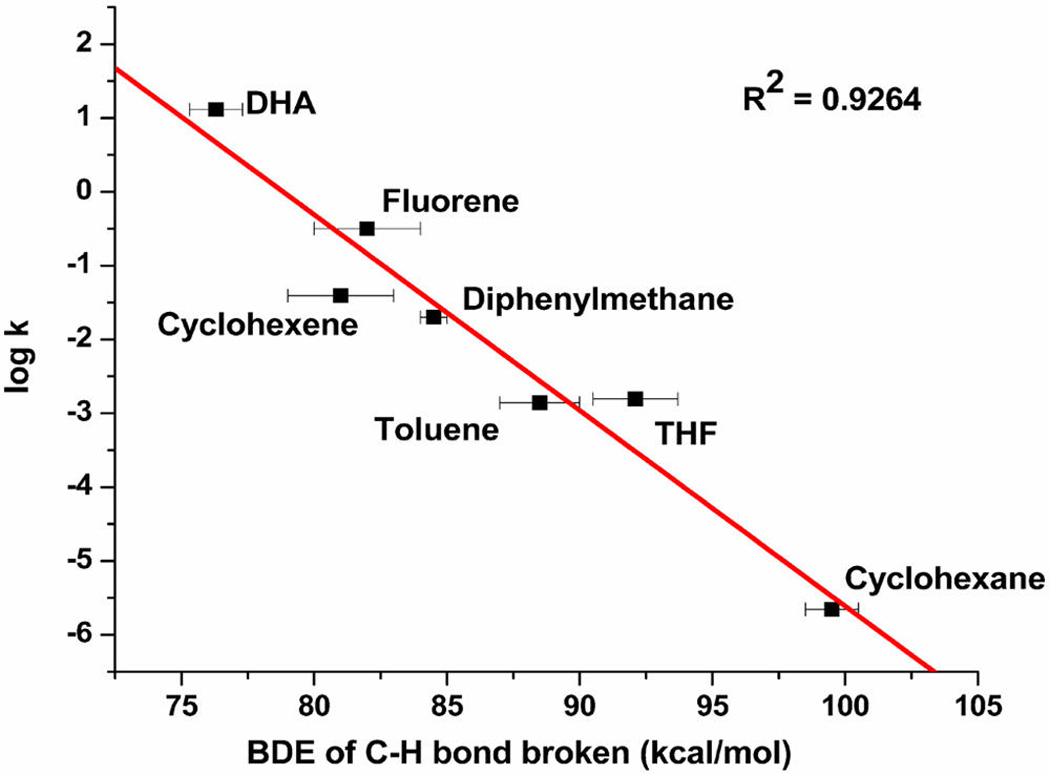
Plot of log k vs BDE for the reaction of 14 (R = iPr) with the indicated substrates. Reproduced with permission from ref 30. Copyright 2015 American Chemical Society.
SUMMARY AND CONCLUSIONS
Studies of the reactions of Cu(I)–α-ketocarboxylate complexes with O2 and Cu(I) complexes with N-oxides led to intriguing findings, including ligand arene hydroxylation, the characterization of stable Cu(I)–N-oxide complexes, and the formation of the bis(oxo)-dicopper core via oxo-transfer. Support for the intermediacy of [CuO]+ species in some of these reactions was found, although alternative pathways could not be ruled out. Thus, direct identification in solution or solid state of complexes with the [CuO]+ core has not yet been achieved. On the other hand, by using a strongly electron donating ligand (12), complexes with the protonated variant [CuOH]2+ (14) were characterized. Importantly, we found that the [CuOH]2+ core of 14 is capable of attacking strong C–H bonds, thus providing precedent for the possible involvement of this core in oxidation catalysis. Indeed, we speculate that such a core is a more energetically accessible intermediate than the [CuO]+ moiety and should be considered as an alternative in proposed mechanisms for oxidations by enzymes and other catalytic systems. For example, in LPMO, one can envision that protonation of the putative [CuO]+ unit could yield a [CuOH]2+ core, perhaps stabilized by the anionic amide resulting from tautomerization of the “histidine brace” supporting ligand (Scheme 8). Similar processes to form a reactive [CuOH]2+ core can be envisioned for other copper oxygenases, including those that activate O2 at a single site (e.g., PHM or DβM) or at one copper ion in multicopper sites (such as pMMO8).
Scheme 8.
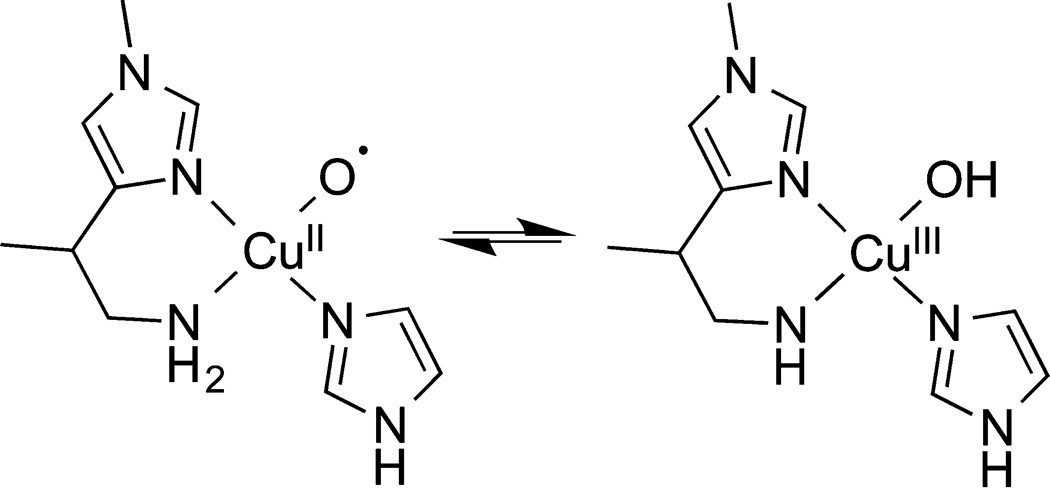
Hypothesized Tautomerization of [CuO]+ and [CuOH]2+ Cores in the Active Site of LPMO
Within an even broader context, 14 is a member of a larger class of complexes and active sites that are weak oxidants yet through enhanced basicity attain high BDE values and fast C–H bond attack rates. Invariably, these properties arise from the presence of strongly electron donating, anionic supporting ligands that stabilize a high oxidation state for the metal ion while enhancing the basicity of the oxo or hydroxo ligand. Examples include the axial thiolate in cytochrome P45032 and aromatic peroxygenase enzymes,33 the tetranionic corrolazine in Mn(V)–oxo complexes,34 and the trianionic tripodal amides in Mn– or Fe–oxo/hydroxo complexes.35 The degree to which electron and proton transfer are coupled in the reactions of these complexes can differ, and understanding the bases for mechanisms between the extremes of “pure” HAT and sequential proton and electron transfer36 is a topic of considerable current interest.37
Acknowledgments
We thank the National Institutes of Health (Grant GM47365) for financial support, and the coauthors of the cited publications from our laboratory for their important contributions to the work described herein.
Biographies
Nicole Gagnon received a B.A. degree from the College of St. Benedict, MN, in 2010. After studying at the University of Arizona, she joined the laboratory of W. B. Tolman at the University of Minnesota in 2012. She is a member of the Women in Science and Engineering group and of the Center for Metals in Biocatalysis. Her current research focus is studying macrocyclic dicopper-hydroxide complexes for C–H bond activation.
William B. Tolman is a Distinguished McKnight University Professor at the University of Minnesota. He received a B.S. degree from Wesleyan University, CT, and a Ph.D. from the University of California, Berkeley, in 1987. After a postdoctoral period, 1987–1990, at the Massachusetts Institute of Technology, he joined the faculty at the University of Minnesota. He serves as Chair of the Department of Chemistry and Editor-in-Chief of the ACS journal Inorganic Chemistry. His research interests span synthetic bioinorganic and organometallic/polymer chemistry.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- 1.Solomon EI, Heppner DE, Johnston EM, Ginsbach JW, Cirera J, Qayyum M, Kieber-Emmons MT, Kjaergaard CH, Hadt RG, Tian L. Copper Active Sites in Biology. Chem. Rev. 2014;114:3659–3853. doi: 10.1021/cr400327t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Ryland BL, Stahl SS. Practical Aerobic Oxidations of Alcohols and Amines with Homogeneous Copper/TEMPO and Related Catalyst Systems. Angew. Chem., Int. Ed. 2014;53:8824–8838. doi: 10.1002/anie.201403110. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wendlandt AE, Suess AM, Stahl SS. Copper-Catalyzed Aerobic Oxidative C-H Functionalizations: Trends and Mechanistic Insights. Angew. Chem., Int. Ed. 2011;50:11062–11087. doi: 10.1002/anie.201103945. [DOI] [PubMed] [Google Scholar]; (c) Díaz-Requejo MM, Pérez PJ. Coinage Metal Catalyzed C–H Bond Functionalization of Hydrocarbons. Chem. Rev. 2008;108:3379–3394. doi: 10.1021/cr078364y. [DOI] [PubMed] [Google Scholar]
- 3.(a) Mirica LM, Ottenwaelder X, Stack TDP. Sructure and Spectroscopy of Copper-Dioxygen Complexes. Chem. Rev. 2004;104:1013–1045. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]; (b) Lewis EA, Tolman WB. Reactivity of Copper-Dioxygen Systems. Chem. Rev. 2004;104:1047–1076. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]; (c) Himes RA, Karlin KD. Copper-Dioxygen Complex Mediated C-H Bond Oxygenation: Relevance for Particulate Methane Monooxygenase (pMMO) Curr. Opin. Chem. Biol. 2009;13:119–131. doi: 10.1016/j.cbpa.2009.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Solomon EI, Augustine AJ, Yoon J. O2 Reduction to H2O by the Multicopper Oxidases. Dalton Trans. 2008:3921–3932. doi: 10.1039/b800799c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Cramer CJ, Tolman WB. Mononuclear Cu-O2 Complexes: Geometries, Spectroscopic Properties, Electronic Structures, and Reactivity. Acc. Chem. Res. 2007;40:601–608. doi: 10.1021/ar700008c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ginsbach JW, Peterson RL, Cowley RE, Karlin KD, Solomon EI. Correlation of the Electronic and Geometric Structures in Mononuclear CoppeRII) Superoxide Complexes. Inorg. Chem. 2013;52:12872–12874. doi: 10.1021/ic402357u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kim S, Lee JY, Cowley RE, Ginsbach JW, Siegler MA, Solomon EI, Karlin KD. A N3S(thioether)-Ligated CuII-Superoxo with Enhanced Reactivity. J. Am. Chem. Soc. 2015;137:2796–2799. doi: 10.1021/ja511504n. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klinman JP. Mechanisms Whereby Mononuclear Copper Proteins Functionalize Organic Substrates. Chem. Rev. 1996;96:2541–2561. doi: 10.1021/cr950047g. [DOI] [PubMed] [Google Scholar]
- 7.Peterson RL, Himes RA, Kotani H, Suenobu T, Tian L, Siegler MA, Solomon EI, Fukuzumi S, Karlin KD. Cupric Superoxo-Mediated Intermolecular C–H Activation Chemistry. J. Am. Chem. Soc. 2011;133:1702–1705. doi: 10.1021/ja110466q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balasubramanian R, Rosenzweig A. Structural and Mechanistic Insights into Methane Oxidation by Particulate Methane Monooxygenase. Acc. Chem. Res. 2007;40:573–580. doi: 10.1021/ar700004s. [DOI] [PubMed] [Google Scholar]
- 9.(a) Sono M, Roach MP, Dawson JH. Heme-Containing Oxygenases. Chem. Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]; (b) Abu-Omar MM, Loaiza A, Hontzeas N. Reaction Mechanisms of Mononuclear Non-Heme Iron Oxygenases. Chem. Rev. 2005;105:2227–2252. doi: 10.1021/cr040653o. [DOI] [PubMed] [Google Scholar]; (c) Costas M, Mehn MP, Jensen MP, Que L., Jr Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem. Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]; (d) Krebs C, Galoníc DF, Fujimori D, Walsh C, Bollinger J. Non-Heme Fe(IV)-Oxo Intermediates. Acc. Chem. Res. 2007;40:484–492. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McDonald AR, Que L., Jr High-valent nonheme iron-oxo complexes: Synthesis, structure, and spectroscopy. Coord. Chem. Rev. 2013;257:414–428. [Google Scholar]
- 11.Illustrative examples: Reinaud O, Capdevielle P, Maumy M. CoppeRII) mediated aromatic hydroxylation by trimethylamine N-oxide. J. Chem. Soc., Chem. Commun. 1990:566–568. Blain I, Giorgi M, Riggi ID, Réglier M. Substrate-Binding Ligand Approach in Chemical Modeling of Copper-Containing Monooxygenases, 1. Intramolecular Stereoselective Oxygen Atom Insertion into a Non-Activated C–H Bond. Eur. J. Inorg. Chem. 2000:393–398. Kunishita A, Ishimaru H, Nakashima S, Ogura T, Itoh S. Reactivity of Mononuclear Alkylperoxo CoppeRII) Complex. O–O Bond Cleavage and C–H Bond Activation. J. Am. Chem. Soc. 2008;130:4244–4245. doi: 10.1021/ja800443s. Maiti D, Lee D-H, Gaoutchenova K, Würtele C, Holthausen MC, Sarjeant AAN, Sundermeyer J, Schindler S, Karlin KD. Reactions of a CoppeRII) Superoxo Complex Lead to C-H and O-H Substrate Oxygenation: Modeling Copper-Monooxygenase C-H Hydroxylation. Angew. Chem., Int. Ed. 2008;47:82–85. doi: 10.1002/anie.200704389. Maiti D, Narducci Sarjeant AA, Karlin KD. Copper–Hydroperoxo-Mediated N-Debenzylation Chemistry Mimicking Aspects of Copper Monooxygenases. Inorg. Chem. 2008;47:8736–8747. doi: 10.1021/ic800617m.
- 12.(a) Dietl N, van der Linde C, Schlangen M, Beyer MK, Schwarz H. Diatomic [CuO]+ and Its Role in the Spin-Selective Hydrogen- and Oxygen-Atom Transfers in the Thermal Activation of Methane. Angew. Chem., Int. Ed. 2011;50:4966–4969. doi: 10.1002/anie.201100606. [DOI] [PubMed] [Google Scholar]; (b) Schröder D, Holthausen MC, Schwarz H. Radical-like Activation of Alkanes by the Ligated Copper Oxide Cation (Phenanthroline)CuO+ J. Phys. Chem. B. 2004;108:14407–14416. [Google Scholar]
- 13.Yoshizawa K, Kihara N, Kamachi T, Shiota Y. Catalytic Mechanism of Dopamine β-Monooxygenase Mediated by Cu(III)-Oxo. Inorg. Chem. 2006;45:3034–3041. doi: 10.1021/ic0521168. [DOI] [PubMed] [Google Scholar]
- 14.(a) Decker A, Solomon EI. Dioxygen Activation by Copper, Heme and Non-Heme Iron Enzymes: Comparison of Electronic Structures and Reactivities. Curr. Opin. Chem. Biol. 2005;9:152–163. doi: 10.1016/j.cbpa.2005.02.012. [DOI] [PubMed] [Google Scholar]; (b) Crespo A, Marti MA, Roitberg AE, Amzel LM, Estrin DA. The Catalytic Mechanism of Peptidylglycine α-Hydroxylating Monooxygenase Investigated by Computer Simulation. J. Am. Chem. Soc. 2006;128:12817–12828. doi: 10.1021/ja062876x. [DOI] [PubMed] [Google Scholar]
- 15.Kim S, Ståhlberg J, Sandgren M. Quantum Mechanical Calculations Suggest That Lytic Polysaccharide Monooxygenases Use a Copper-Oxyl, Oxygen-Rebound Mechanism. Proc. Natl. Acad. Sci. U.S.A. 2014;111:149–154. doi: 10.1073/pnas.1316609111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gupta A, Tolman WB. Copper/α-Ketocarboxylate Chemistry With Supporting Peralkylated Diamines: Reactivity of CoppeRI) Complexes and Dicopper-Oxygen Intermediates. Inorg. Chem. 2010;49:3531–3539. doi: 10.1021/ic100032n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Mahadevan V, DuBois JL, Hedman B, Hodgson KO, Stack TDP. Exogenous Substrate Reactivity with a [Cu(III)2O2]2+ Core: Structural Implications. J. Am. Chem. Soc. 1999;121:5583–5584. [Google Scholar]; (b) Mahadevan V, Henson MJ, Solomon EI, Stack TDP. Differential Reactivity between Interconvertible Side-On Peroxo and Bis-μ-oxodicopper Isomers Using Peralkylated Diamine Ligands. J. Am. Chem. Soc. 2000;122:10249–10250. [Google Scholar]
- 18.Hong S, Huber SM, Gagliardi L, Cramer CC, Tolman WB. CoppeRI)-α-Ketocarboxylate Complexes: Characterization and O2 Reactions That Yield Copper-Oxygen Intermediates Capable of Hydroxylating Arenes. J. Am. Chem. Soc. 2007;129:14190–14192. doi: 10.1021/ja0760426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huber SM, Ertem MZ, Aquilante F, Gagliardi L, Tolman WB, Cramer CJ. Generating CuII-Oxyl/CuIII-Oxo Species from CuI-α-Ketocarboxylate Complexes and O2: In Silico Studies on Ligand Effects and C-H-Activation Reactivity. Chem.—Eur. J. 2009;15:4886–4895. doi: 10.1002/chem.200802338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Comba P, Knoppe S, Martin B, Rajaraman G, Rolli C, Shapiro B, Stork T. CoppeRII)-Mediated Aromatic ortho-Hydroxylation: A Hybrid DFT and Ab Initio Exploration. Chem.—Eur. J. 2008;14:344–357. doi: 10.1002/chem.200700865. [DOI] [PubMed] [Google Scholar]
- 21.Hong S, Gupta AK, Tolman WB. Intermediates in Reactions of CoppeRI) Complexes with N-Oxides: From the Formation of Stable Adducts to Oxo Transfer. Inorg. Chem. 2009;48:6323–6325. doi: 10.1021/ic900435p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spencer DJE, Aboelella NW, Reynolds AM, Holland PL, Tolman WB. β-Diketiminate Ligand Backbone Structural Effects on Cu(I)/O2 Reactivity: Unique Copper-Superoxo and Bis(μ-oxo) Complexes. J. Am. Chem. Soc. 2002;124:2108–2809. doi: 10.1021/ja017820b. [DOI] [PubMed] [Google Scholar]
- 23.(a) Levason W, M.D. Spicer MD. The Chemistry of Copper and Silver in Their Higher Oxidation States. Coord. Chem. Rev. 1987;76:45–120. [Google Scholar]; (b) Anson FC, Collins TJ, Richmond TG, Santarsiero BD, Toth JE, Treco BGRT. Highly Stabilized CoppeRIII) Complexes. J. Am. Chem. Soc. 1987;109:2974–2979. [Google Scholar]
- 24.Huang D, Holm RH. Reactions of the Terminal NiII-OH Group in Substitution and Electrophilic Reactions with Carbon Dioxide and Other Substrates: Structural Definition of Binding Modes in an Intramolecular NiII⋯FeII Bridged Site. J. Am. Chem. Soc. 2010;132:4693–4701. doi: 10.1021/ja1003125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Donoghue PJ, Tehranchi J, Cramer CJ, Sarangi R, Solomon EI, Tolman WB. Rapid C–H Bond Activation by a Monocopper(III)–Hydroxide Complex. J. Am. Chem. Soc. 2011;133:17602–17605. doi: 10.1021/ja207882h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tehranchi J, Donoghue PJ, Cramer CJ, Tolman WB. Reactivity of (Dicarboxamide)MII-OH (M = Cu, Ni) Complexes - Reaction with Acetonitrile to Yield MII-Cyanomethides. Eur. J. Inorg. Chem. 2013;2013:4077–4084. doi: 10.1002/ejic.201300328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xue G, Geng C, Ye S, Fiedler A, Neese F, Que L., Jr Hydrogen Bonding Effects on the Reactivity of X-FeIII-O-FeIV=O complexes towards C–H bond Cleavage. Inorg. Chem. 2013;52:3976–3984. doi: 10.1021/ic3027896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kundu S, Van Kirk Thompson J, Shen LQ, Mills MR, Brominaar EL, Ryabov AD, Collins TJ. Activation Parameters as Mechanistic Probes in the TAML Iron(V)-Oxo Oxidations of Hydrocarbons. Chem.—Eur. J. 2015;21:1803–1810. doi: 10.1002/chem.201405024. [DOI] [PubMed] [Google Scholar]
- 29.Warren JJ, Tronic TA, Mayer JM. Thermochemistry of Proton-Coupled Electron Transfer Reagents and its Implications. Chem. Rev. 2010;110:6961–7001. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dhar D, Tolman WB. Hydrogen Atom Abstraction from Hydrocarbons by a CoppeRIII)-Hydroxide Complex. J. Am. Chem. Soc. 2015;137:1322–1329. doi: 10.1021/ja512014z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barrón D, Barbosa J. Acid-Base Behaviour of Substituted Phenolic Substances and Resolution of Acid Strength in Tetrahydrofuran. Analyt. Chim. Acta. 2000;403:339–347. [Google Scholar]
- 32.Yosca TH, Rittle J, Krest CM, Onderko EL, Silakov A, Calixto JC, Behan RK, Green MT. Iron(IV)hydroxide pKa and the Role of Thiolate Ligation in C-H Bond Activation by Cytochrome P450. Science. 2013;342:825–829. doi: 10.1126/science.1244373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang X, Ullrich R, Hofrichter M, Groves JT. Heme-Thiolate Ferryl of Aromatic Peroxygenase Is Basic and Reactive. Proc. Natl. Acad. Sci. U.S.A. 2015;112:3686–3691. doi: 10.1073/pnas.1503340112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prokop KA, de Visser SP, Goldberg DP. Unprecedented Rate Enhancements of Hydrogen-Atom Transfer to a Manganese(V)-Oxo Corrolazine Complex. Angew. Chem., Int. Ed. 2010;49:5091–5095. doi: 10.1002/anie.201001172. [DOI] [PubMed] [Google Scholar]
- 35.(a) Borovik AS. Role of Metal–Oxo Complexes in the Cleavage of C–H Bonds. Chem. Soc. Rev. 2011;40:1870–1874. doi: 10.1039/c0cs00165a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Borovik A. Bioinspired Hydrogen Bond Motifs in Ligand Design: The Role of Noncovalent Interactions in Metal Ion Mediated Activation of Dioxygen. Acc. Chem. Res. 2005;38:54–61. doi: 10.1021/ar030160q. [DOI] [PubMed] [Google Scholar]
- 36.Litwinienko G, Ingold KU. Solvent Effects on the Rates and Mechanisms of Reaction of Phenols with Free Radicals. Acc. Chem. Res. 2007;40:222–230. doi: 10.1021/ar0682029. [DOI] [PubMed] [Google Scholar]
- 37.Usharani D, Lacy DC, Borovik AS, Shaik S. Dichotomous Hydrogen Atom Transfer vs Proton-Coupled Electron Transfer during Activation of X–H Bonds (X = C, N, O) by Nonheme Iron–Oxo Complexes of Variable Basicity. J. Am. Chem. Soc. 2013;135:17090–17104. doi: 10.1021/ja408073m. [DOI] [PMC free article] [PubMed] [Google Scholar]