

Abstract
Objectives:
To identify the genomic mechanisms that result in PARK2 large gene deletions.
Methods:
We conducted mutation screening using PCR amplification of PARK2-coding regions and exon-intron boundaries, followed by sequencing to evaluate a large series of 244 unrelated Portuguese patients with symptoms of Parkinson disease. For the detection of large gene rearrangements, we performed multiplex ligation-dependent probe amplification, followed by long-range PCR and sequencing to map deletion breakpoints.
Results:
We identified biallelic pathogenic parkin mutations in 40 of the 244 patients. There were 18 different mutations, some of them novel. This study included mapping of 17 deletion breakpoints showing that nonhomologous end joining is the most common mechanism responsible for these gene rearrangements. None of these deletion breakpoints were previously described, and only one was present in 2 unrelated families, indicating that most of the deletions result from independent events.
Conclusions:
The c.155delA mutation is highly prevalent in the Portuguese population (62.5% of the cases). Large deletions were present in 42.5% of the patients. We present the largest study on the molecular mechanisms that mediate PARK2 deletions in a homogeneous population.
Parkinson disease (PD) is the second most common neurodegenerative disorder, the etiology of which remains mostly unknown.1 Most PD cases are sporadic, although the discovery of genes linked to familial forms has provided valuable insights into disease mechanisms. Among recessive forms, PARK2 mutations are the most common cause of parkinsonism, being responsible for autosomal recessive juvenile Parkinson disease (AR-JP).2 AR-JP is genetically heterogeneous and, apart from age at onset, is clinically indistinguishable from idiopathic PD, presenting with rigidity, bradykinesia, and resting tremor, usually before the age of 40 years.3,4
PARK2, located on chromosome 6q25.2–q27, encodes parkin, an E3 ubiquitin ligase. The loss of this ubiquitin ligase activity appears to be the mechanism responsible for the pathogenesis of AR-JP.5 PARK2 is composed of 12 exons surrounded by large intronic regions and spans approximately 1.38 Mb. Mutations have been identified across the entire gene and include all mutation types.6 PARK2 is the 17th largest gene of the human genome and is located within a large common fragile site (CFS), FRA6E,7 a 3.6-Mb region of instability, susceptible to form gaps, breaks, and rearrangements when cells are exposed to certain conditions such as DNA replication inhibitors,8–10 which may explain the large frequency of PARK2 deletions.
In this study, we aimed to identify the breakpoints of 17 different deletions to understand further the mechanisms favoring the occurrence of these rearrangements and evaluated the frequency of PARK2 mutations in patients with clinical suspicion of early-onset parkinsonism.
METHODS
Patients and mutation analysis.
We evaluated 244 unrelated Portuguese patients with symptoms of PD referred to our center for molecular study of PARK2. The mean age at onset was 34.3 years (range, 6 months to 64 years).
We conducted mutation screening, performing PCR amplification of the entire PARK2-coding region and exon-intron boundaries, using the HotStarTaq Master Mix Kit (Qiagen, Venlo, the Netherlands), followed by bidirectional direct sequencing with the BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Waltham, MA) and loaded on an ABI-PRISM 3130xl Genetic Analyzer (Applied Biosystems). To detect large gene rearrangements, we performed multiplex ligation-dependent probe amplification (MLPA) using the SALSA MLPA kit P051 (MRC Holland) according to the manufacturer's instructions and analyzed fragments on an ABI-PRISM 3130xl Genetic Analyzer using 500-LIZ (Applied Biosystems), as a size standard, and GeneMarker v1.90 (SoftGenetics, State College, PA).
Standard protocol approvals, registrations, and patient consents.
All study participants gave informed consent before participating in this study. We stored DNA samples at the Center for Predictive and Preventive Genetics–authorized biobank.
Long-range PCR and breakpoint analysis.
Because of the large size of PARK2 introns, we genotyped several single-nucleotide polymorphisms (SNPs), located in the introns flanking each deletion to narrow down their extension. SNPs were obtained from the HapMap Genome Browser. We performed SNP genotyping using SNAPShot. For SNPs that seemed to be in the homozygous state using the SNAPshot technique and in patients with heterozygous deletions, we performed dosage analysis by quantitative real-time PCR to confirm or exclude homozygosity for that particular SNP. After reducing the possible extension of these deletions, we used the primer pairs closest to the deletion breakpoint for long-range PCR amplification. Because the predicted amplicons were larger than 2 kb, we performed PCR amplification using the Expand Long Template PCR System (Roche Diagnostics, Basel, Switzerland) and/or Ranger Mix (Bioline, Taunton, MA).
We separated DNA fragments of interest on 0.8% agarose gels, excised and purified with the Illustra GFX PCR DNA and Gel Band Purification Kit (GE Healthcare, Little Chalfont, United Kingdom) according to the manufacturer's instructions. Isolated and purified fragments were sequenced with the BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems) and loaded on an ABI-PRISM 3130xl Genetic Analyzer (Applied Biosystems); deletion breakpoints were narrowed down by primer walking.
The nucleotide sequence positions described are based on the human reference sequence (GRCh37). We evaluated sequence identities of nucleotide sequences encompassing each breakpoint using the National Center for Biotechnology Information BLASTN tool and RepeatMasker with default parameters to identify interspersed repeats.
RESULTS
PARK2 mutations in patients with parkinsonism.
This mutational analysis of 244 Portuguese participants confirmed the PD clinical diagnosis in 16.4% (40/244) of the patients. We identified 18 different mutations, including missense mutations, small and large deletions, and a splicing mutation (table 1). We found homozygous parkin mutations in 67.5% of the patients, and large deletions were present in 42.5% of the cases. The most frequent mutation was a 1-base pair (bp) deletion, c.155delA, which was present in 62.5% of the patients. We observed 2 novel mutations, a 1-bp deletion (c.1030delG) and an indel (c.1072-1073delCTinsA), both predicted to result in an altered reading frame and a premature stop codon (p. E344Sfs*91 and p. L358Rfs*77).
Table 1.
Overview of molecular and clinical information from 40 patients with a molecular diagnosis of autosomal recessive juvenile Parkinson disease
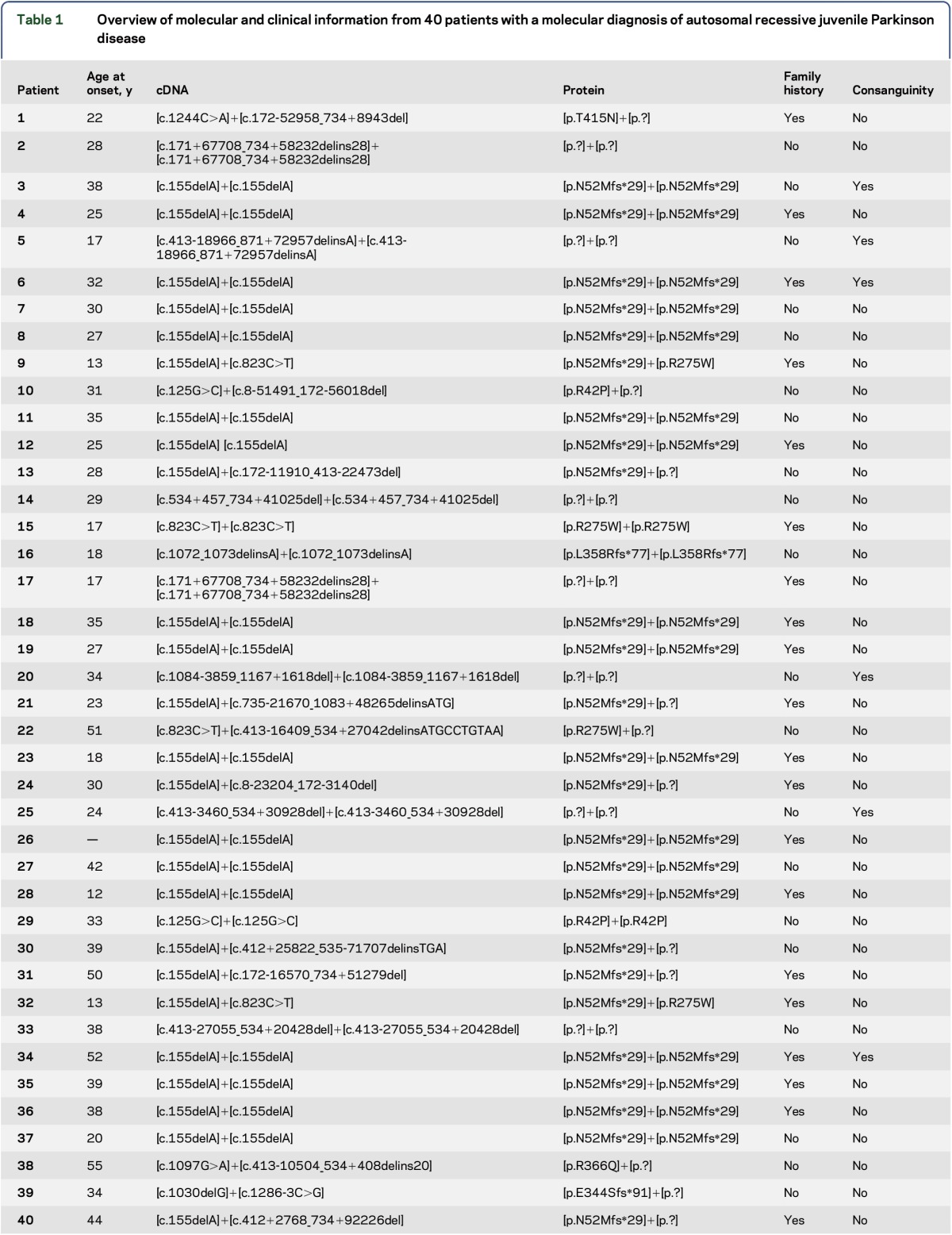
The most common mutation, c.155delA, is a small deletion that causes the alteration of the open reading frame starting in the amino acid asparagine in position 52 and results in a stop codon 29 amino acids later (p.N52Mfs*29), leading to loss of most of the protein.
Seventeen patients showed large gene rearrangements, and we observed at least 9 different deletions either in homozygosity or heterozygosity. The most common deletions were those of exon 4 and of exons 3–6 (table 1).
Breakpoint determination and deletion mechanisms.
To explore the mechanisms underlying these large rearrangements and to confirm MLPA results, we determined the exact breakpoints of 17 deletions using an SNP approach to narrow down the deletion breakpoint. We describe localization of the breakpoints found in these patients and the responsible mechanisms in table 2.
Table 2.
Overview of 17 mapped deletions and responsible mechanisms
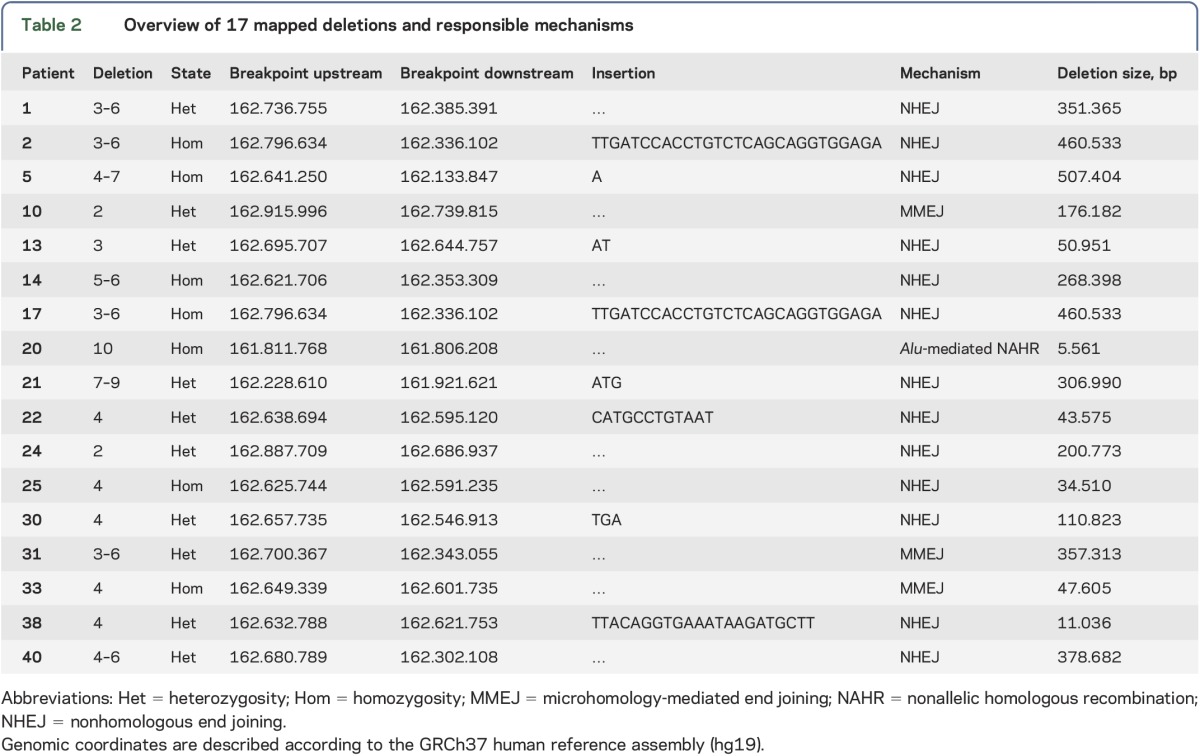
We found that nonhomologous end joining (NHEJ) was the mechanism responsible for 76.5% of the large deletions. Three cases presented with microhomology domains in the junctions, and the deletions resulted from microhomology-mediated end joining (MMEJ). Only one of the deletions could be explained by nonallelic homologous recombination (NAHR) mediated by Alu sequences. We identified other repetitive elements (table 3) that were not present at both sides of the breakpoint, and thus, unlikely to be directly involved in the rearrangement.
Table 3.
Overview of repetitive elements found in breakpoint analysis
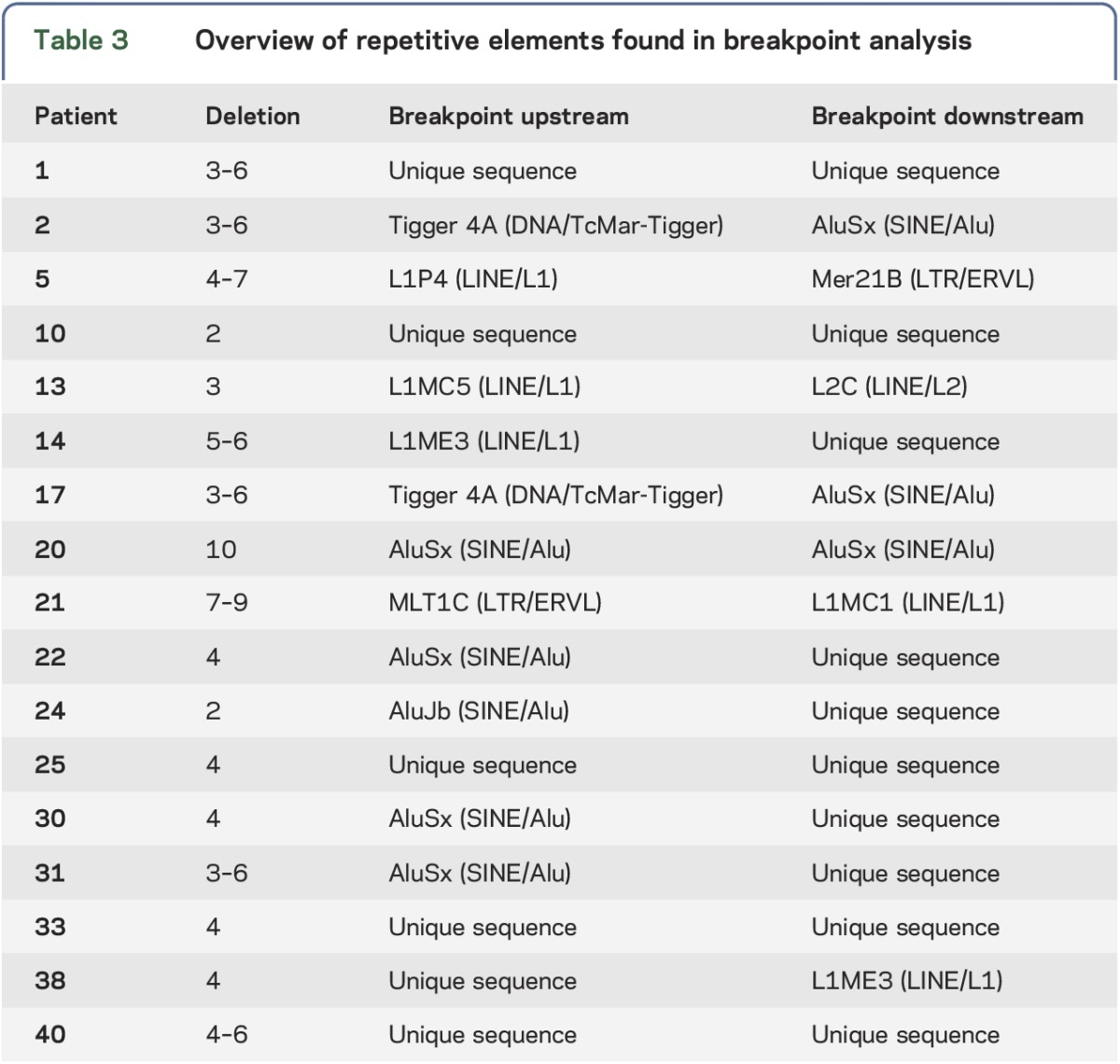
Examples of the mapped rearrangement for the exon 4–7 deletion, exon 4 deletion, and exon 10 deletion (figure, panels A, B, and C, respectively) represent 2 different responsible mechanisms (NHEJ and NAHR) acting in 3 different ways. For the exon 4 deletion in patient 22 (figure, B) and patient 38, we can infer that the inserted region was because of a duplication of the immediately preceding region. The origin of the inserted regions for the other 6 deletions could not be identified because of their small size or, in the case of the sequence inserted in patients 2 and 17, because of their frequency in the genome.
Figure. Schematic representation of 3 breakpoints found in our patients and the suspected mechanism for these deletions.

(A) Nonhomologous end joining (NHEJ), mediated by the formation of a hairpin loop (deletion of exons 4–7; patient 5) and with the insertion of an adenine nucleotide. (B) NHEJ with the presence of a duplicated region and 2 motifs, complementary to immunoglobulin class switch repeats (deletion of exon 4; patient 22). (C) Alu-mediated nonallelic homologous recombination (NAHR) (deletion of exon 10; patient 20).
DISCUSSION
PD, which affects almost 2% of the population above the age of 65 years, is the second most commonly occurring progressive neurodegenerative disorder after Alzheimer disease. Today, the lengthening lifespan and aging population has transformed neurodegenerative diseases into an economic and societal concern.6 Mutations in the parkin coding gene are the predominant cause of monogenic forms of recessive PD with a frequency that varies across studies depending on the population, number of familial cases, age at onset, and consanguinity, among other factors.11,12 The study of these mutations is therefore of upmost importance for understanding the pathogenesis of this disease.
Our PARK2-positive cohort showed a mean age at onset of 30 years (range, 12–55 years). This mutational study of the parkin coding gene revealed that 16.4% of these patients present with 2 mutations, thus confirming the PD clinical diagnosis. This is particularly interesting because we found several cases with parkin mutations with onset at more than 40 years of age. Also, the mean age at onset for patients in this study is lower than that reported previously (mean age 36 years) in a study considering only familial cases and with at least one of the patients with onset before the age of 54 years.13 These findings show the importance of screening PARK2 for mutations in patients with a family history of PD with later disease onset.
The mutations we identified are spread throughout the entire gene in almost every parkin domain. Of the 18 different mutations found, 3 were small insertions or deletions, 5 were point mutations, 9 were large deletions, and 1 was a splice-site mutation. Two were novel mutations predicted to be damaging to the protein: a small insertion (c.1030delG) and a splice-site mutation (c.1286-3C>G). It is interesting to note that we did not find duplications in our cohort.
Our results show that the c.155delA mutation is highly recurrent in the Portuguese population. The high frequency of this mutation in Portugal and Spain14 probably indicates a founder effect. This mutation is also commonly found in European patients along with the c.823C>T mutation.14–19 These 2 are the most common point mutations in our cohort and are located in exons 2 (ubiquitin-like domain) and 7 (RING1 domain). All missense mutations identified in our patients have been previously described.6 Last, the splice-site mutation, c.1286-3C>G, is predicted to disrupt the acceptor splice site, resulting in the loss of the last exon.
Of the 9 identified deletions in our population, the most common were the exon 4 deletion (5 cases) and the exon 3–6 deletion (4 cases). The high frequency of these deletions is similar to the results of another study17 that showed rearrangements occurring frequently in introns 2 through 4. Large deletions are responsible for juvenile PD in almost half of the studied patients, making MLPA an essential approach in molecular diagnosis, complementary to direct sequencing.
Large deletions account for approximately 50%–60% of the PARK2 disease–causing mutations8 and have been observed worldwide. This high frequency can be partially explained by the very large introns of PARK2 that span 1.4 Mb,8 and with introns 1, 2, 6, and 7 as large as 180 kb.17 Another reason is the genomic context of PARK2. This gene is located within one of the 3 most frequently expressed CFS, namely FRA6E.20 It is interesting to note that most of the rearrangements found in PARK2 are located between exons 3 and 8, which comprise the FRA6E center, with exons 3 and 4 being among the most unstable regions and hotspots for deletion.8,21 The cause of this instability is not completely understood, although several studies show that many gross chromosomal rearrangements that accumulate in solid tumors may have originated in these fragile sites because of errors during DNA replication that could be caused by intrinsic characteristics of CFS, such as their AT sequence content and high flexibility, scarcity of replication origins, and difficulty in the transcription of long genes present in these sites.22
To date, only a small number of PARK2 deletion breakpoints have been mapped, probably because of the large gene size; despite the fact that the exonic deletions described here have been previously identified,6 all the breakpoints present in these Portuguese patients are different from those previously described.8,17,21,23–25
In the 4 patients who had an exon 3–6 deletion, only 2 share the same breakpoints. In addition, all breakpoints were different in the 5 patients who had an exon 4 deletion. Thus, it seems that the same exon deletion is not necessarily a consequence of a similar rearrangement but may stem from independent events. This finding is in agreement with the ongoing hypothesis that rearrangements are independent and recurrent events.24,26 It is also in accordance with the notion that rearrangements recurring at the same breakpoint are less frequent than nonrecurring rearrangements.8 The high frequency of repetitive elements and instability in the PARK2 locus favor the occurrence of recurrent deletion events, especially in the large PARK2 introns.17 It is curious that all of our patients with recurring deletions in homozygosity present the same breakpoints. In 3 patients, this could be explained by the presence of consanguinity. Two shared the same exon 3–6 deletion, which could indicate a recurrent event or a common founder mutation.
There are 3 major mechanisms responsible for genomic rearrangements: NAHR, NHEJ, and fork stalling and template switching. These 3 mechanisms account for the majority of genomic rearrangements in the human genome, and their distribution partially reflects the genomic architecture in the proximity of the breakpoint locus.27
Detailed analysis of the sequences flanking the breakpoints allowed us to explore causative mechanisms.
One mechanism implicated in PARK2 deletions is NAHR, mediated by Alu elements. Our analysis showed that, although there are only 526 Alu elements in PARK2, which is below the mean density of the human genome (1 per 3 kb),28 Alu-mediated NAHR seems to be the responsible mechanism for the exon 10 deletion described (figure, C) where the 2 junctions map to Alu elements of the same family (AluSx) in direct orientation. This mechanism has been previously described in PARK2 deletions.8
Regions of microhomology often contribute to NHEJ, so it is common to find them at deletion breakpoints. NHEJ is the major pathway for restoring double-strand breaks in chromosomal DNA and is a highly flexible mechanism that creates distinct breakpoint junctions, resulting in either short microhomologies (usually 1–4 bp) or inserted sequences without homology as a result of the NHEJ editing process.8 The presence of inserted sequences in the mapped deletions described here, namely a short duplication of the surrounding fragments and short sequences of unknown origin, is in agreement with features of the NHEJ mechanism. Also, this repair mechanism has already been related to parkin deletions in previous studies.24,25 Nevertheless, a second mechanism is associated with the presence of microhomology. MMEJ is also a mechanism for double-strand break repair, which uses a sequence of 5–25 bp to align the broken ends; MMEJ is frequently associated with complex rearrangements. Although MMEJ is considered a secondary mechanism, used only when NHEJ and the other mechanisms fail, it has recently been reported that MMEJ can act even when NHEJ and homologous recombination are intact.29 As 3 of the deletions reported here present a microhomology region of 5, 6, and 7 bp, we propose that this was the responsible mechanism in these cases (table 2). The other 5 cases present shorter microhomologies (table 2). Recently, 5 PARK2 deletions were mapped in Polish patients showing NHEJ and fork stalling and template switching as the responsible mechanisms.25 Here we present a higher number of deletions mapped, all different from previously described reports, and expanded the mechanisms responsible for these large gene rearrangements to include MMEJ and Alu-mediated NAHR. These 3 mechanisms, NHEJ, MMEJ, and Alu-mediated NAHR, seem to explain the identified deletions; NHEJ seems to be responsible for the majority of the deletions.
We describe 40 patients with autosomal recessive PD who have novel and previously reported PARK2 mutations. We present the molecular characterization of 17 large PARK2 deletions and identify NHEJ as the most frequently occurring mechanism responsible for deletions in this gene.
ACKNOWLEDGMENT
The authors thank all patients who participated in this study. They also thank all the physicians who referred patients to our center.
GLOSSARY
- AR-JP
autosomal recessive juvenile Parkinson disease
- CFS
common fragile site
- MMEJ
microhomology-mediated end joining
- MLPA
multiplex ligation-dependent probe amplification
- NHEJ
nonhomologous end joining
- NAHR
nonallelic homologous recombination
- PD
Parkinson disease
AUTHOR CONTRIBUTIONS
S. Morais: acquisition of data, analysis and interpretation, drafting and revision of manuscript. R. Bastos-Ferreira: acquisition of data, analysis and interpretation, and revision of manuscript. J. Sequeiros: revision of manuscript. I. Alonso: primary investigator, study conceptualization and design, analysis and interpretation, and revision of manuscript.
STUDY FUNDING
Supported by Fundação para a Ciência e Tecnologia cofunded by Fundo Europeu de Desenvolvimento Regional and COMPETE (Programa Operacional Factores de Competitividade), grant number PIC/IC/83232/2007.
DISCLOSURE
Ms. Morais and Ms. Bastos-Ferreira report no disclosures. Dr. Sequeiros has received travel/speaker honoraria from Fundación Jimenez Diaz and Pfizer; has served on the editorial boards of Clinical Genetics and the Journal of Community Genetics; is an employee of the University of Porto; and has received research support from Biomarin, Pfizer, Alexion, FCT (Fundação para a Ciência e Tecnologia), and the European Commission (FP6 Network of Excellence). Dr. Alonso reports no disclosures. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.de Lau LM, Breteler MM. Epidemiology of Parkinson's disease. Lancet Neurol 2006;5:525–535. [DOI] [PubMed] [Google Scholar]
- 2.Gasser T. Mendelian forms of Parkinson's disease. Biochim Biophys Acta 2009;1792:587–596. [DOI] [PubMed] [Google Scholar]
- 3.von Coelln R, Dawson VL, Dawson TM. Parkin-associated Parkinson's disease. Cell Tissue Res 2004;318:175–184. [DOI] [PubMed] [Google Scholar]
- 4.Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson's disease. Annu Rev Neurosci 2005;28:57–87. [DOI] [PubMed] [Google Scholar]
- 5.Dawson TM, Dawson VL. The role of parkin in familial and sporadic Parkinson's disease. Mov Disord 2010;25(suppl 1):S32–S39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nuytemans K, Theuns J, Cruts M, Van Broeckhoven C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat 2010;31:763–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith DI, Zhu Y, McAvoy S, Kuhn R. Common fragile sites, extremely large genes, neural development and cancer. Cancer Lett 2006;232:48–57. [DOI] [PubMed] [Google Scholar]
- 8.Mitsui J, Takahashi Y, Goto J, et al. Mechanisms of genomic instabilities underlying two common fragile-site-associated loci, PARK2 and DMD, in germ cell and cancer cell lines. Am J Hum Genet 2010;87:75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith DI, McAvoy S, Zhu Y, Perez DS. Large common fragile site genes and cancer. Semin Cancer Biol 2007;17:31–41. [DOI] [PubMed] [Google Scholar]
- 10.Denison SR, Callahan G, Becker NA, Phillips LA, Smith DI. Characterization of FRA6E and its potential role in autosomal recessive juvenile parkinsonism and ovarian cancer. Genes Chromosomes Cancer 2003;38:40–52. [DOI] [PubMed] [Google Scholar]
- 11.Lucking CB, Durr A, Bonifati V, et al. Association between early-onset Parkinson's disease and mutations in the parkin gene. N Engl J Med 2000;342:1560–1567. [DOI] [PubMed] [Google Scholar]
- 12.Kilarski LL, Pearson JP, Newsway V, et al. Systematic review and UK-based study of PARK2 (parkin), PINK1, PARK7 (DJ-1) and LRRK2 in early-onset Parkinson's disease. Mov Disord 2012;27:1522–1529. [DOI] [PubMed] [Google Scholar]
- 13.Sun M, Latourelle JC, Wooten GF, et al. Influence of heterozygosity for parkin mutation on onset age in familial Parkinson disease: the GenePD study. Arch Neurol 2006;63:826–832. [DOI] [PubMed] [Google Scholar]
- 14.Munoz E, Tolosa E, Pastor P, et al. Relative high frequency of the c.255delA parkin gene mutation in Spanish patients with autosomal recessive parkinsonism. J Neurol Neurosurg Psychiatry 2002;73:582–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abbas N, Lucking CB, Ricard S, et al. A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson's Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson's Disease. Hum Mol Genet 1999;8:567–574. [DOI] [PubMed] [Google Scholar]
- 16.Sironi F, Primignani P, Zini M, et al. Parkin analysis in early onset Parkinson's disease. Parkinsonism Relat Disord 2008;14:326–333. [DOI] [PubMed] [Google Scholar]
- 17.Hedrich K, Eskelson C, Wilmot B, et al. Distribution, type, and origin of Parkin mutations: review and case studies. Mov Disord 2004;19:1146–1157. [DOI] [PubMed] [Google Scholar]
- 18.Lohmann E, Periquet M, Bonifati V, et al. How much phenotypic variation can be attributed to parkin genotype? Ann Neurol 2003;54:176–185. [DOI] [PubMed] [Google Scholar]
- 19.Bras J, Guerreiro R, Ribeiro M, et al. Analysis of Parkinson disease patients from Portugal for mutations in SNCA, PRKN, PINK1 and LRRK2. BMC Neurol 2008;8:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao G, Smith DI. Very large common fragile site genes and their potential role in cancer development. Cell Mol Life Sci 2014;71:4601–4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clarimon J, Johnson J, Dogu O, et al. Defining the ends of Parkin exon 4 deletions in two different families with Parkinson's disease. Am J Med Genet B Neuropsychiatr Genet 2005;133B:120–123. [DOI] [PubMed] [Google Scholar]
- 22.Franchitto A. Genome instability at common fragile sites: searching for the cause of their instability. Biomed Res Int 2013;2013:730714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elfferich P, Verleun-Mooijman MC, Maat-Kievit JA, et al. Breakpoint mapping of 13 large parkin deletions/duplications reveals an exon 4 deletion and an exon 7 duplication as founder mutations. Neurogenetics 2011;12:263–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Asakawa S, Hattori N, Shimizu A, et al. Analysis of eighteen deletion breakpoints in the parkin gene. Biochem Biophys Res Commun 2009;389:181–186. [DOI] [PubMed] [Google Scholar]
- 25.Ambroziak W, Koziorowski D, Duszyc K, et al. Genomic instability in the PARK2 locus is associated with Parkinson's disease. J Appl Genet 2015;56:451–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Periquet M, Lucking C, Vaughan J, et al. Origin of the mutations in the parkin gene in Europe: exon rearrangements are independent recurrent events, whereas point mutations may result from founder effects. Am J Hum Genet 2001;68:617–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gu W, Zhang F, Lupski JR. Mechanisms for human genomic rearrangements. Pathogenetics 2008;1:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konkel MK, Batzer MA. A mobile threat to genome stability: the impact of non-LTR retrotransposons upon the human genome. Semin Cancer Biol 2010;20:211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McVey M, Lee SE. MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet 2008;24:529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]