

Abstract
Ephrin type‐B receptor 2 (EphB2) is a member of the receptor tyrosine kinase family and plays an important role in learning and memory functions. In patients with Alzheimer's disease (AD) and in mouse models of AD, a reduction in the hippocampal EphB2 level is observed. It was recently reported that normalization of the EphB2 level in the dentate gyrus rescues memory function in a mouse model of AD, suggesting that drugs that restore EphB2 levels may be beneficial in the treatment of AD. Amyloid β (Aβ) oligomers, which are believed to be key molecules involved in the pathogenesis of AD, induce EphB2 degradation through their direct binding to EphB2. Thus, compounds that inhibit the binding of Aβ oligomers to EphB2 may be beneficial. Here, we screened for such compounds from drugs already approved for clinical use in humans. Utilizing a cell‐free screening assay, we determined that dihydroergotamine mesilate, bromocriptine mesilate, cepharanthine, and levonorgestrel inhibited the binding of Aβ oligomers to EphB2 but not to cellular prion protein, another endogenous receptor for Aβ oligomers. Additionally, these four compounds did not affect the binding between EphB2 and ephrinB2, an endogenous ligand for EphB2, suggesting that the compounds selectively inhibited the binding of Aβ oligomers to EphB2. This is the first identification of compounds that selectively inhibit the binding of Aβ oligomers to EphB2. These results suggest that these four compounds may be safe and effective drugs for treatment of AD.
Keywords: Alzheimer's disease, amyloid β oligomer, ephrin type‐B receptor 2
Abbreviations
- ABTS
2,2′‐azino‐bis(3‐ethylbenzothiazoline‐6‐sulfonic acid)
- AD
Alzheimer's disease
- Aβ
amyloid β peptide
- BRO
bromocriptine mesilate
- BSA
bovine serum albumin
- CEP
cepharanthine
- DIH
dihydroergotamine mesilate
- DMEM
Dulbecco's modified Eagle medium
- DMSO
dimethyl sulfoxide
- EphB2
ephrin type‐B receptor 2
- HFIP
1,1,1,3,3,3‐hexafluoro‐2 propanol
- HRP
horseradish peroxidase
- LCA
lithocholic acid
- LEV
levonorgestrel
- LTP
long‐term potentiation
- PBS
phosphate‐buffered saline
- PrPC
cellular prion protein
- SD
standard deviation
Alzheimer's disease (AD) is a progressive neurodegenerative disease and the most common cause of dementia. Because the world's population is aging, the number of AD patients has been increasing; however, there are currently no disease‐modifying treatments for AD. Thus, development of disease‐modifying drugs is important.
One hallmark pathological feature of AD is senile plaques, which are composed of amyloid β peptide (Aβ). Monomeric Aβ easily self‐assembles to form soluble oligomers, protofibrils, and fibrils. A correlation between the amount of soluble Aβ oligomers and cognitive impairment in AD patients has been reported 1. In addition, soluble Aβ oligomers inhibit long‐term potentiation (LTP) 2, 3 which is involved in learning and memory functions 4. Therefore, Aβ oligomers are believed to play an important role in the learning and memory dysfunction observed in patients with AD.
Some endogenous receptors for Aβ oligomers, such as ephrin type‐B receptor 2 (EphB2) and cellular prion protein (PrPC), appear to be involved in the inhibitory effects of Aβ oligomers on LTP 5, 6, 7. EphB2, a member of the receptor tyrosine kinase family 8, is a receptor for ephrinB ligands (B1, B2, and B3) and ephrinA5 9 and functions in synaptic plasticity and synapse formation 10, 11, 12. Mice lacking EphB2 exhibit LTP impairment and memory dysfunction 11, 12. Reduced EphB2 levels are observed in postmortem hippocampal tissue from patients with AD 13 and in mouse models of AD 5, 13, 14. Aβ oligomers directly bind to EphB2 and induce its internalization and proteasome‐mediated degradation 5. In addition, artificial expression of EphB2 in the dentate gyrus rescues LTP and memory function in a mouse model of AD 5. These results suggest that inhibiting Aβ oligomer binding to EphB2 may be therapeutically beneficial in the treatment of AD.
The number of drugs reaching the marketplace each year is decreasing, mainly due to unexpected adverse effects being revealed at advanced stages of clinical trials. To overcome this problem, we took advantage of a recent strategy for drug discovery and development that focuses on examining drugs already approved for use in humans (i.e., drug repositioning). In this strategy, compounds with clinically beneficial pharmacological activity are screened from a library of approved drugs already in clinical use to be developed for new indications. One major advantage of this strategy is a decreased risk for unexpected adverse effects in humans because the safety of these drugs has already been well characterized in humans 15.
In the present study, from a library of approved drugs already in clinical use, we screened for compounds that inhibited the binding of Aβ oligomers to EphB2. We identified dihydroergotamine mesilate (DIH), bromocriptine mesilate (BRO), cepharanthine (CEP), and levonorgestrel (LEV). Although these compounds inhibited the binding of Aβ oligomers to EphB2, they did not inhibit the binding of an endogenous ligand, ephrinB2, to EphB2. This is the first time that compounds that selectively inhibited the binding of Aβ oligomers to EphB2 have been identified.
Materials and methods
Materials
Biotin‐conjugated Aβ42 (biotin‐Aβ) was obtained from AnaSpec Inc. (Fremont, CA, USA). EphB2 receptor ectodomains fused to the Fc portions of human IgG (EphB2‐Fc) and biotinylated ephrinB2‐Fc were purchased from R&D systems Inc. (Minneapolis, MN, USA). His‐tagged human PrPC was obtained from Merck Co. (Darmstadt, Germany). Bovine serum albumin (BSA) and dimethyl sulfoxide (DMSO) were purchased from Sigma‐Aldrich Co. (St. Louis, MI). Horseradish peroxidase‐conjugated streptavidin (HRP‐streptavidin) was obtained from GE Healthcare UK Ltd. (Little Chalfont, UK). The 2,2′‐azino‐bis(3‐ethylbenzothiazoline‐6‐sulfonic acid) (ABTS) was obtained from Kirkegaard & Perry Laboratories, Inc. (Gaithersburg, MD, USA). The antibodies against Aβ (6E10) and PrPC (ICSM35) were purchased from Covance Inc. (Emeryville, CA, USA) and D‐Gen Co. (London, UK), respectively. Phenol red‐free Dulbecco's modified Eagle medium (DMEM) and 1,1,1,3,3,3‐hexafluoro‐2 propanol (HFIP) were obtained from Wako Pure Chemical Industries Ltd. (Osaka, Japan). The library of drugs approved for clinical use was obtained from our laboratory stock 16.
Aβ monomer and oligomer preparation
Aβ monomers and oligomers were prepared as described previously 16. Biotin‐Aβ was dissolved in HFIP and incubated at room temperature for 1 h and then on ice for 10 min. The HFIP was evaporated and the peptides were dissolved in DMSO for a final concentration of 10 mm. The peptides were diluted in phenol red‐free DMEM to give a final concentration of 100 μm and subjected to sonication (AIWA Co., Tokyo, Japan) for 10 min.
For oligomer preparation, the Aβ solution described above was incubated at 37 °C for 16 h and then centrifuged at 20 400 g for 15 min. The supernatant was frozen in liquid nitrogen and stored at −80 °C until use.
For monomer preparation, the sample was centrifuged and stored in a manner similar to that for oligomer preparation, without the incubation at 37 °C for 16 h.
Characterization of Aβ oligomers by immunoblotting
To characterize Aβ oligomers, the supernatant was mixed with equal amounts of sample buffer and boiled at 98 °C for 5 min. Samples were applied to polyacrylamide sodium dodecyl sulfate gels and subjected to electrophoresis, after which proteins were immunoblotted with 6E10.
Cell‐free binding assay
The cell‐free binding assay was performed as described previously 16. To evaluate binding between Aβ and EphB2, EphB2‐Fc was dissolved in phosphate‐buffered saline (PBS) to a final concentration of 0.5 μg·mL−1 and applied to a 96‐well ELISA plate and left at 4 °C overnight. Each well was washed with PBS containing 0.05% Tween 80 (T‐PBS) and blocked with 2% BSA at room temperature for 1 h. Then, each well was washed with T‐PBS and incubated with both biotin‐Aβ (200 nm) and each tested drug in DMEM at room temperature for 1 h. Each well was then washed with T‐PBS and incubated with HRP‐streptavidin in PBS (1 : 2000 dilution) at room temperature for 30 min. Each well was washed with T‐PBS and incubated with ABTS. Absorbance at 412 nm was measured using a plate reader (Infinite M1000; TECAN Group Ltd., Mannedorf, Switzerland). Biotin‐Aβ binding to EphB2 was calculated as follows:
ODBlank: absorbance of well without both EphB2 and each tested drug
ODControl: absorbance of well with EphB2 but without any tested drug
ODDrug: absorbance of well with both EphB2 and each tested drug
As a quality parameter for this assay, Z′‐factor 17 was calculated as follows:
μControl: mean of ODControl
σControl: standard deviation of ODControl
μBlank: mean of ODBlank
σBlank: standard deviation of ODBlank
To evaluate binding between Aβ and PrPC, EphB2‐Fc was replaced with His‐tagged human PrPC.
To evaluate binding between ephrinB2 and EphB2, biotin‐Aβ in DMEM was replaced with biotinylated ephrinB2‐Fc in PBS (500 ng·mL−1).
Chemical library and screening
Each drug in the library was dissolved in water or DMSO, depending on the drug. The concentration of each drug in the library was 1 mm, 5 mm, 10 mm, 25 mm, 50 mm, 100 mm, 2 mg·mL−1, 10 mg·mL−1, or 100 mg·mL−1, depending on the drug. Each drug was diluted 1000 times for the screening (final concentration, 1 μm, 5 μm, 10 μm, 25 μm, 50 μm, 100 μm, 2 μg·mL−1, 10 μg·mL−1, or 100 μg·mL−1). We confirmed that 0.1% of water or DMSO did not affect this assay. Screening was performed in duplicate wells per each drug in the same reader plate.
Statistical analysis
All values are expressed as the mean ± standard deviation (SD). One‐way analysis of variance (anova) followed by Tukey's test was used to evaluate differences among three or more groups. Differences were considered statistically significant at P values less than 0.05.
Results and Discussion
Screening of approved drugs for compounds that inhibit the binding of Aβ oligomers to EphB2
To screen for inhibitors of Aβ oligomer binding to EphB2, we used a cell‐free binding assay. In this assay, biotin‐Aβ was applied to EphB2‐coated wells, and after washing, the amount of bound biotin‐Aβ was determined using HRP‐streptavidin. Aβ oligomers were prepared by incubation at 37 °C for 16 h, and we confirmed the formation of Aβ oligomers by immunoblotting (Fig. 1A).
Figure 1.
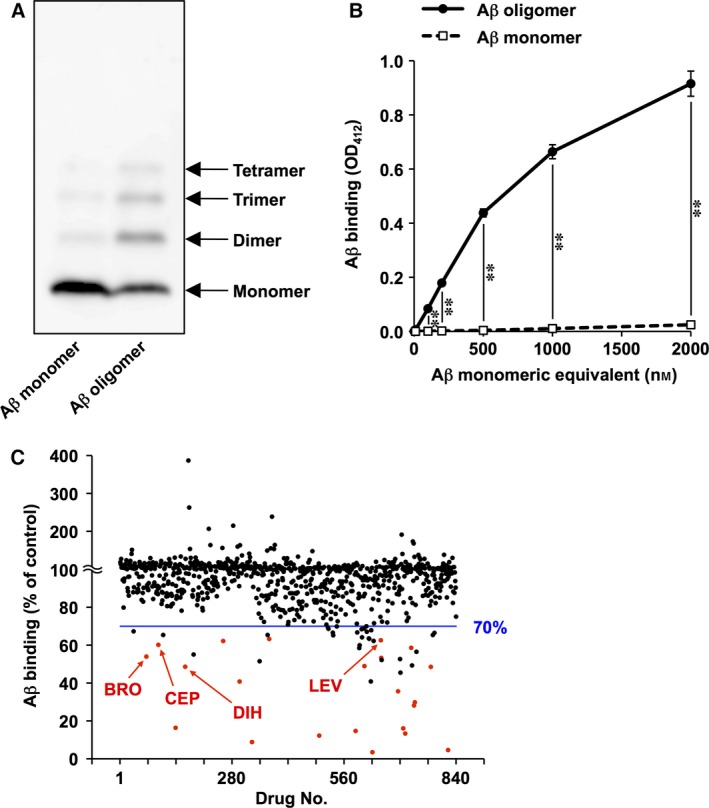
Screening of approved drugs for inhibitors of Aβ oligomer binding to EphB2. Aβ monomers and oligomers were prepared. (A) Both monomeric and oligomeric forms of biotin‐Aβ were analyzed by immunoblotting. (B) The abilities of the monomeric and oligomeric forms of biotin‐Aβ to bind to EphB2 were compared using a cell‐free binding assay. Values are mean ± SD (n = 3). **P < 0.01. (C) Each of 840 approved drugs was added simultaneously with Aβ oligomers to EphB2‐coated wells, and the amount of bound biotin‐Aβ was determined. Values are means (n = 2, duplicate wells per each drug in the same plate). Red dots indicate the 22 compounds that are listed in Table 1. Arrows indicate dihydroergotamine mesilate (DIH), bromocriptine mesilate (BRO), cepharanthine (CEP), and levonorgestrel (LEV).
We first compared the ability of Aβ monomers and oligomers to bind EphB2. We found that Aβ oligomers bound to EphB2 more efficiently than did the monomers (Fig. 1B). This result was consistent with a previous report 5 and suggested that this assay was appropriate for screening compounds that inhibited the binding of Aβ oligomers to EphB2.
From a library of 840 drugs approved for clinical use, we screened for compounds that inhibited the binding of Aβ oligomers to EphB2. Each compound was applied to the cell‐free binding assay simultaneously with Aβ oligomers, and the amount of bound biotin‐Aβ was determined (Fig. 1C). As an assay quality parameter, we calculated Z′‐factor to be 0.89, supporting the quality of this assay. Among these 840 compounds, 52 compounds inhibited more than 30% of the Aβ oligomer binding to EphB2. We examined the effect of these compounds on the binding in additional two independent experiments. We selected 22 compounds that inhibited more than 30% of the Aβ oligomer binding to EphB2 in both experiments (Table 1, red dots in Fig. 1C).
Table 1.
Inhibitory effect of each compound on the binding of Aβ oligomers to EphB2 or PrPC
| Compound | Aβ binding (% of control) | |||
|---|---|---|---|---|
| EphB2 | ||||
| Screening | Reproducibility | PrPC | ||
| Montelukast sodium | 3.5 | 0.0 | 6.0 ± 0.4 | 0.4 ± 0.8 |
| Pirarubicin hydrochloride | 4.6 | 19.7 ± 1.8 | 33.1 ± 3.6 | 55.8 ± 9.3 |
| Minocycline hydrochloride | 8.8 | 3.9 ± 1.4 | 4.7 ± 2.1 | 4.2 ± 5.9 |
| Tosufloxacin tosilate | 12.2 | 11.2 ± 0.7 | 19.1 ± 2.6 | 42.2 ± 1.7 |
| Diclazuril | 13.3 | 11.3 ± 1.7 | 20.8 ± 5.1 | 12.4 ± 4.8 |
| Suramin sodium | 14.7 | 16.4 ± 1.9 | 21.2 ± 0.6 | 8.6 ± 4.0 |
| Miltefosine | 16.0 | 50.1 ± 1.9 | 53.0 ± 4.4 | 43.4 ± 2.3 |
| Cytochrome c | 16.4 | 27.1 ± 1.9 | 32.0 ± 2.0 | 53.4 ± 2.9 |
| l‐thyroxine | 28.1 | 53.8 ± 7.0 | 60.1 ± 3.7 | 61.0 ± 9.3 |
| Abamectin | 29.8 | 40.1 ± 2.1 | 45.4 ± 5.7 | 35.4 ± 12.5 |
| Nystatin | 35.6 | 33.2 ± 4.7 | 57.5 ± 0.8 | 41.6 ± 5.0 |
| Lysozyme hydrochloride | 40.8 | 48.7 ± 3.8 | 60.6 ± 2.0 | 48.3 ± 1.2 |
| Bexarotene | 48.5 | 32.7 ± 1.7 | 46.2 ± 1.2 | 23.3 ± 5.9 |
| Dihydroergotamine mesilate | 48.6 | 62.7 ± 6.7 | 65.3 ± 5.6 | 82.7 ± 5.8 |
| Retinoic acid | 48.9 | 39.5 ± 13.9 | 62.8 ± 2.3 | 27.8 ± 1.5 |
| Mifepristone | 53.3 | 49.1 ± 8.8 | 55.7 ± 2.2 | 20.7 ± 2.0 |
| Bromocriptine mesilate | 53.9 | 57.9 ± 6.3 | 69.5 ± 5.8 | 99.4 ± 9.3 |
| Toltrazuril | 58.6 | 55.8 ± 1.6 | 61.2 ± 8.7 | 35.9 ± 4.5 |
| Cepharanthine | 60.2 | 50.1 ± 4.0 | 66.3 ± 7.4 | 113.4 ± 4.5 |
| Indigocarmine | 62.2 | 55.9 ± 2.4 | 64.1 ± 8.5 | 47.4 ± 4.2 |
| Levonorgestrel | 62.6 | 64.1 ± 2.0 | 65.2 ± 4.2 | 95.6 ± 6.3 |
| Oxytetracycline hydrochloride | 63.2 | 53.7 ± 3.2 | 65.7 ± 4.8 | 58.0 ± 3.7 |
Among 52 compounds that inhibited more than 30% of the Aβ oligomer binding to EphB2 in the screening, 22 compounds that inhibited more than 30% of the Aβ oligomer binding to EphB2 in additional two independent experiments are listed. The effects of these 22 compounds on the binding of Aβ oligomers to EphB2 in the screening and in the additional two independent experiments (reproducibility) and on the binding of Aβ oligomers to PrPC were shown. The four compounds that inhibited less than 30% of the Aβ oligomer binding to PrPC are highlighted in gray. Values are mean (n = 2, ‘Screening’ column) or mean ± SD (n = 3). The concentration of nystatin or retinoic acid was 50 μm in the screening or 100 μm in other experiments. The concentration of cytochrome c or lysozyme hydrochloride was 10 μg·mL−1 in all experiments. The concentration of other all compounds was 100 μm in all experiments.
Through this screening procedure, we also found five compounds that enhanced the binding of Aβ oligomers to EphB2 to more than 200% (Table 2).
Table 2.
Compounds that enhance the binding of Aβ oligomers to EphB2
| Compound | Aβ binding (% of control) |
|---|---|
| Domiphen bromide | 386.8 |
| Dopamine hydrochloride | 262.8 |
| Pentamidine isethionate | 238.6 |
| Levodopa | 214.9 |
| Fluvoxamine maleate | 206.6 |
Five compounds that increased the Aβ oligomer binding to EphB2 to more than 200% in the screening are listed. Values are mean (n = 2). The concentration of drugs was 100 μm.
To select compounds that selectively inhibited the Aβ oligomer binding to EphB2, we next examined the effects of these 22 compounds on the binding of Aβ oligomers to PrPC. We found four compounds (DIH, BRO, CEP, and LEV; Fig. 2A) that inhibited less than 30% of the Aβ oligomer binding to PrPC (Table 1). As shown in Fig. 2B, each of these four compounds inhibited the binding of Aβ oligomers to EphB2 in a concentration‐dependent manner. Additionally, under conditions in which an antibody against PrPC (ICSM35) 7 inhibited the binding of Aβ oligomers to PrPC, none of these four compounds inhibited the binding (Fig. 2C). These results suggested that these four compounds selectively inhibited the binding of Aβ oligomers to EphB2.
Figure 2.
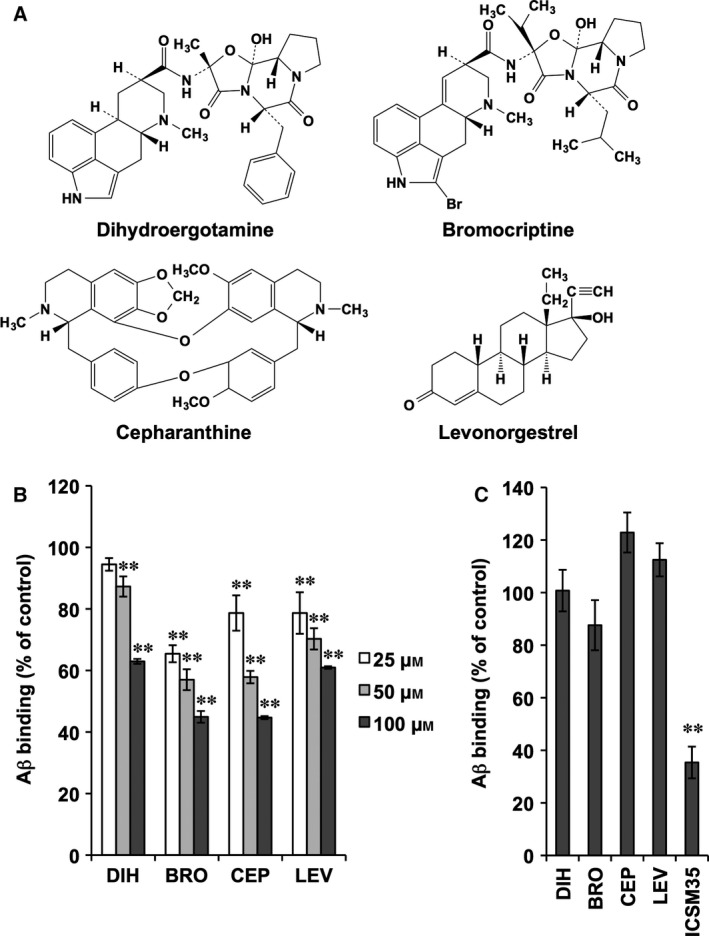
Selective inhibition of the binding of Aβ oligomer to EphB2 by selected compounds. (A) Structures of the four selected compounds are shown. (B) The indicated concentration of each compound was added simultaneously with Aβ oligomers to EphB2‐coated wells, and the amount of bound biotin‐Aβ was determined. (C) Each compound (100 μm) or ICSM35 (2 μg·mL−1) was added simultaneously with Aβ oligomer to PrPC‐coated wells, and the amount of bound biotin‐Aβ was determined. Values represent the mean ± SD (n = 3). **P < 0.01. Basically similar results were obtained in another independent experiment.
Effects of selected compounds on the binding of ephrinB2 to EphB2
The results in Fig. 2 suggested that the four selected compounds interacted with EphB2 rather than with Aβ oligomers to inhibit their binding. Thus, it is possible that these compounds may inhibit the binding between EphB2 and its endogenous ligands, which may cause adverse clinical effects. Thus, we examined the effects of these four compounds on the binding of one of the endogenous ligands, ephrinB2, to EphB2. As shown in Fig. 3, none of these four compounds inhibited the binding of ephrinB2 to EphB2. We confirmed the inhibitory effect of lithocholic acid, which interferes with Eph–ephrin interactions 18, on the binding of ephrinB2 to EphB2 (Fig. 3).
Figure 3.
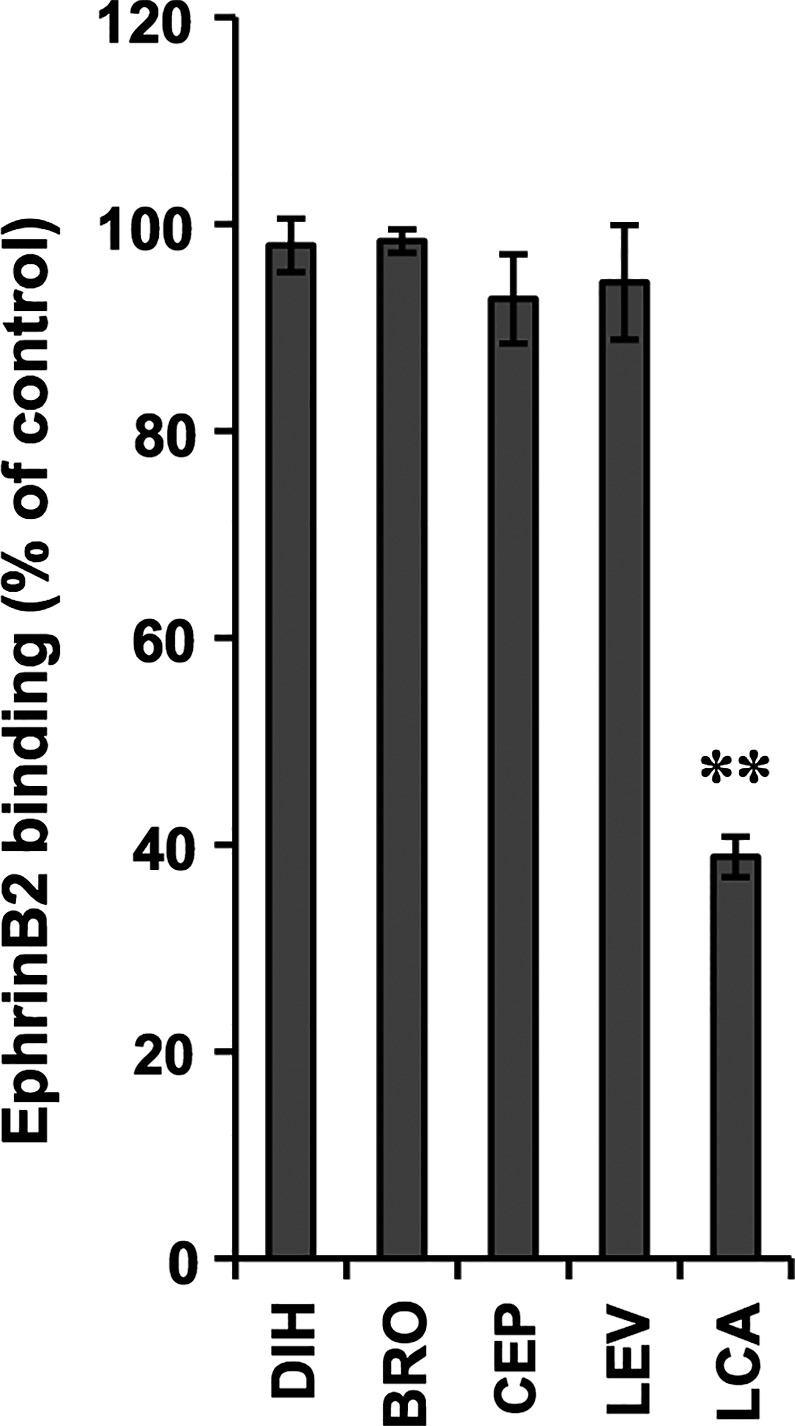
Effects of selected compounds on the binding of ephrinB2 to EphB2. Each compound (100 μm) was added simultaneously with biotinylated ephrinB2‐Fc to EphB2‐coated wells, and the amount of bound biotinylated ephrinB2‐Fc was determined. Values represent the mean ± SD (n = 3). **P < 0.01. LCA, lithocholic acid. Basically similar results were obtained in another independent experiment.
Recently, some compounds were reported to inhibit the binding of Aβ to its receptors. For example, Deane et al. 19 identified FPS‐ZM1 as an inhibitor for the binding of Aβ to the receptor for advanced glycation end products, and Risse et al. identified Chicago Sky Blue 6B as an inhibitor for the binding of Aβ to PrPC 20. Furthermore, Fu et al. reported that rhynchophylline binds to EphA4 and stimulated LTP in the presence of Aβ oligomer 21.
As described above, Aβ oligomers directly bind to EphB2 and induce its degradation, resulting in LTP impairment and memory dysfunction 5. Because artificial induction of EphB2 expression in the dentate gyrus rescues LTP and memory function in a mouse model of AD 5, induction of EphB2 expression is a potential drug target for treatment of AD. However, EphB2 is also involved in the development of cancer 22, 23 and may stimulate cancer progression. Thus, inhibiting the binding between Aβ oligomers and EphB2 is a better target for developing drugs aimed at AD treatment. Here, we found 22 compounds with such activity. Among them, 18 compounds also inhibited the binding of Aβ oligomers to PrPC, whereas four compounds did not. Although we focused on the latter group for development of AD therapeutics, the former group is also interesting. In addition to EphB2, other receptors for Aβ oligomers, such as PrPC 6, 7, 24, leukocyte immunoglobulin‐like receptor B2 25, and IgG Fcγ receptor II‐b 26, are involved in the inhibitory effect on LTP and in neurotoxicity. Therefore, compounds that inhibit the binding of Aβ oligomers to various receptors may have beneficial effects for treatment of patients with AD. Indeed, among the 18 compounds identified here, montelukast 27, minocycline 28, 29, and bexarotene 30 reportedly have beneficial effects in animal models of AD.
The four compounds we identified that selectively inhibited the binding of Aβ oligomers to EphB2 (DIH, BRO, CEP, and LEV) are approved for use in humans as treatments for migraine, Parkinson's disease, and leucopenia, as well as for use as a contraceptive, respectively. Among these compounds, DIH, BRO, and CEP penetrate the blood–brain barrier 31, 32, 33. Thus, our results suggest that these compounds, especially DIH, BRO, and CEP, may be safe and effective drugs for treatment of AD. On the other hand, these compounds required relatively high concentration to show their inhibitory effect on the binding of Aβ oligomers to EphB2. Therefore, we also consider the drug modification, using these approved medicines as leads.
As described above, the compounds identified in this study are expected to ameliorate Aβ oligomer‐induced LTP impairment. EphB2 overexpression suppresses Aβ oligomer‐induced neurotoxicity in cultured hippocampal neurons 34, and activation of EphB2 attenuates tau phosphorylation in a mouse model of AD 35. Because both Aβ oligomer‐induced neurotoxicity and tau phosphorylation are important factors in AD pathology, the four compounds identified here may also prove useful for AD treatment through these mechanisms.
In conclusion, we screened for compounds that selectively inhibit the binding of Aβ oligomers to EphB2 from a library of approved drugs already in clinical use, and identified dihydroergotamine mesilate, bromocriptine mesilate, cepharanthine, and levonorgestrel. We propose further analysis of these compounds as candidates for AD treatment.
Author contribution
KS, TI, and TM conceived and designed the project; KS and TA acquired the data; KS analyzed and interpreted the data; and KS, TI, and TM wrote the paper.
Acknowledgements
This work was supported by JSPS KAKENHI Grant Number 15J02654, 24659037 and the Center of Innovation Program from Japan Science and Technology Agency.
References
- 1. Haass C and Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta‐peptide. Nat Rev Mol Cell Biol 8, 101–112. [DOI] [PubMed] [Google Scholar]
- 2. Shankar GM, Li S, Mehta TH, Garcia‐Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA et al (2008) Amyloid‐beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med 14, 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ and Selkoe DJ (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long‐term potentiation in vivo. Nature 416, 535–539. [DOI] [PubMed] [Google Scholar]
- 4. Malenka RC and Bear MF (2004) LTP and LTD: an embarrassment of riches. Neuron 44, 5–21. [DOI] [PubMed] [Google Scholar]
- 5. Cisse M, Halabisky B, Harris J, Devidze N, Dubal DB, Sun B, Orr A, Lotz G, Kim DH, Hamto P et al (2011) Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature 469, 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lauren J, Gimbel DA, Nygaard HB, Gilbert JW and Strittmatter SM (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid‐beta oligomers. Nature 457, 1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Freir DB, Nicoll AJ, Klyubin I, Panico S, Mc Donald JM, Risse E, Asante EA, Farrow MA, Sessions RB, Saibil HR et al (2011) Interaction between prion protein and toxic amyloid beta assemblies can be therapeutically targeted at multiple sites. Nat Commun 2, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kullander K and Klein R (2002) Mechanisms and functions of Eph and ephrin signalling. Nat Rev Mol Cell Biol 3, 475–486. [DOI] [PubMed] [Google Scholar]
- 9. Sheffler‐Collins SI and Dalva MB (2012) EphBs: an integral link between synaptic function and synaptopathies. Trends Neurosci 35, 293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dalva MB, Takasu MA, Lin MZ, Shamah SM, Hu L, Gale NW and Greenberg ME (2000) EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell 103, 945–956. [DOI] [PubMed] [Google Scholar]
- 11. Grunwald IC, Korte M, Wolfer D, Wilkinson GA, Unsicker K, Lipp HP, Bonhoeffer T and Klein R (2001) Kinase‐independent requirement of EphB2 receptors in hippocampal synaptic plasticity. Neuron 32, 1027–1040. [DOI] [PubMed] [Google Scholar]
- 12. Henderson JT, Georgiou J, Jia Z, Robertson J, Elowe S, Roder JC and Pawson T (2001) The receptor tyrosine kinase EphB2 regulates NMDA‐dependent synaptic function. Neuron 32, 1041–1056. [DOI] [PubMed] [Google Scholar]
- 13. Simon AM, de Maturana RL, Ricobaraza A, Escribano L, Schiapparelli L, Cuadrado‐Tejedor M, Perez‐Mediavilla A, Avila J, Del Rio J and Frechilla D (2009) Early changes in hippocampal Eph receptors precede the onset of memory decline in mouse models of Alzheimer's disease. J Alzheimers Dis 17, 773–786. [DOI] [PubMed] [Google Scholar]
- 14. Qu M, Jiang J, Liu XP, Tian Q, Chen LM, Yin G, Liu D, Wang JZ and Zhu LQ (2013) Reduction and the intracellular translocation of EphB2 in Tg2576 mice and the effects of beta‐amyloid. Neuropathol Appl Neurobiol 39, 612–622. [DOI] [PubMed] [Google Scholar]
- 15. Mizushima T (2011) Drug discovery and development focusing on existing medicines: drug re‐profiling strategy. J Biochem 149, 499–505. [DOI] [PubMed] [Google Scholar]
- 16. Aimi T, Suzuki K, Hoshino T and Mizushima T (2015) Dextran sulfate sodium inhibits amyloid‐beta oligomer binding to cellular prion protein. J Neurochem 134, 611–617. [DOI] [PubMed] [Google Scholar]
- 17. Zhang JH, Chung TD and Oldenburg KR (1999) A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 4, 67–73. [DOI] [PubMed] [Google Scholar]
- 18. Giorgio C, Hassan Mohamed I, Flammini L, Barocelli E, Incerti M, Lodola A and Tognolini M (2011) Lithocholic acid is an Eph‐ephrin ligand interfering with Eph‐kinase activation. PLoS ONE 6, e18128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deane R, Singh I, Sagare AP, Bell RD, Ross NT, LaRue B, Love R, Perry S, Paquette N, Deane RJ et al (2012) A multimodal RAGE‐specific inhibitor reduces amyloid beta‐mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest 122, 1377–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Risse E, Nicoll AJ, Taylor WA, Wright D, Badoni M, Yang X, Farrow MA and Collinge J (2015) Identification of a compound that disrupts binding of amyloid‐beta to the prion protein using a novel fluorescence‐based assay. J Biol Chem 290, 17020–17028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fu AK, Hung KW, Huang H, Gu S, Shen Y, Cheng EY, Ip FC, Huang X, Fu WY and Ip NY (2014) Blockade of EphA4 signaling ameliorates hippocampal synaptic dysfunctions in mouse models of Alzheimer's disease. Proc Natl Acad Sci USA 111, 9959–9964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang SD, Rath P, Lal B, Richard JP, Li Y, Goodwin CR, Laterra J and Xia S (2012) EphB2 receptor controls proliferation/migration dichotomy of glioblastoma by interacting with focal adhesion kinase. Oncogene 31, 5132–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Farshchian M, Nissinen L, Siljamaki E, Riihila P, Toriseva M, Kivisaari A, Ala‐Aho R, Kallajoki M, Verajankorva E, Honkanen HK et al (2015) EphB2 promotes progression of cutaneous squamous cell carcinoma. J Invest Dermatol 135, 1882–1892. [DOI] [PubMed] [Google Scholar]
- 24. Kudo W, Lee HP, Zou WQ, Wang X, Perry G, Zhu X, Smith MA, Petersen RB and Lee HG (2012) Cellular prion protein is essential for oligomeric amyloid‐beta‐induced neuronal cell death. Hum Mol Genet 21, 1138–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim T, Vidal GS, Djurisic M, William CM, Birnbaum ME, Garcia KC, Hyman BT and Shatz CJ (2013) Human LilrB2 is a beta‐amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer's model. Science 341, 1399–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kam TI, Song S, Gwon Y, Park H, Yan JJ, Im I, Choi JW, Choi TY, Kim J, Song DK et al (2013) FcgammaRIIb mediates amyloid‐beta neurotoxicity and memory impairment in Alzheimer's disease. J Clin Invest 123, 2791–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lai J, Hu M, Wang H, Long Y, Miao MX, Li JC, Wang XB, Kong LY and Hong H (2014) Montelukast targeting the cysteinyl leukotriene receptor 1 ameliorates Abeta1‐42‐induced memory impairment and neuroinflammatory and apoptotic responses in mice. Neuropharmacology 79, 707–714. [DOI] [PubMed] [Google Scholar]
- 28. Choi Y, Kim HS, Shin KY, Kim EM, Kim M, Park CH, Jeong YH, Yoo J, Lee JP, Chang KA et al (2007) Minocycline attenuates neuronal cell death and improves cognitive impairment in Alzheimer's disease models. Neuropsychopharmacology 32, 2393–2404. [DOI] [PubMed] [Google Scholar]
- 29. Ryu JK, Franciosi S, Sattayaprasert P, Kim SU and McLarnon JG (2004) Minocycline inhibits neuronal death and glial activation induced by beta‐amyloid peptide in rat hippocampus. Glia 48, 85–90. [DOI] [PubMed] [Google Scholar]
- 30. Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ et al (2012) ApoE‐directed therapeutics rapidly clear beta‐amyloid and reverse deficits in AD mouse models. Science 335, 1503–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang Y, Aun R and Tse FL (1998) Brain uptake of dihydroergotamine after intravenous and nasal administration in the rat. Biopharm Drug Dispos 19, 571–575. [DOI] [PubMed] [Google Scholar]
- 32. Friis ML, Paulson OB and Hertz MM (1979) Transfer of bromocriptine across the blood‐brain barrier in man. Acta Neurol Scand 59, 88–95. [DOI] [PubMed] [Google Scholar]
- 33. Okamoto M, Ono M and Baba M (2001) Suppression of cytokine production and neural cell death by the anti‐inflammatory alkaloid cepharanthine: a potential agent against HIV‐1 encephalopathy. Biochem Pharmacol 62, 747–753. [DOI] [PubMed] [Google Scholar]
- 34. Geng D, Kang L, Su Y, Jia J, Ma J, Li S, Du J and Cui H (2013) Protective effects of EphB2 on Abeta1‐42 oligomer‐induced neurotoxicity and synaptic NMDA receptor signaling in hippocampal neurons. Neurochem Int 63, 283–290. [DOI] [PubMed] [Google Scholar]
- 35. Jiang J, Wang ZH, Qu M, Gao D, Liu XP, Zhu LQ and Wang JZ (2015) Stimulation of EphB2 attenuates tau phosphorylation through PI3K/Akt‐mediated inactivation of glycogen synthase kinase‐3beta. Sci Rep 5, 11765. [DOI] [PMC free article] [PubMed] [Google Scholar]