

ABSTRACT
It is now widely recognized that a range of human diseases, including obesity, cancer and inflammatory bowel disease, is strongly linked to the microbiota. For decades, the microbiota has been proposed to contribute to the pathogenesis of colon cancer. Our recent work reveals that the organization of the mucosal microbiota into biofilms marks a subset of human colon cancer. Further, biofilm-positive colon mucosa in the colon cancer host yields an infrequently detected polyamine metabolite, N(1), N(12)-diacetylspermine, that deserves further study to determine its utility as a marker for colon neoplasia.
KEYWORDS: bacteroides, biofilm, colorectal cancer, escherichia coli, fusobacterium, microbiome, screening colonoscopy
The colonic microbiome has become increasingly recognized for roles in the initiation and progression of colorectal cancer and perhaps offers the best opportunity for proving the microbiota is causal in disease initiation. Our group has recently described the identification of polymicrobial bacterial biofilms and their metabolic contributions toward the colon cancer environment.1,2 Complex bacterial communities invading to colonize the mucus layer of the colonic mucosa—and therefore, encased in mucus–were identified on nearly all colorectal tumors (cancers and adenomas) proximal to the hepatic flexure, and a subset (12%) of tumors distal to the hepatic flexure in a United States population. The bacterial biofilms identified in our initial study1 were associated with epithelial changes relevant to oncogenic progression; including loss of the tumor suppressor, E-cadherin, increased levels of the angiogenic and pro-inflammatory cytokine, IL-6, as well as activation (by tyrosine phosphorylation) of the downstream-effector STAT3, and increased epithelial crypt cell proliferation. A follow-up metabolomic analysis of tissues with and without a biofilm from the cancer host revealed that biofilms contribute to enhanced N(1), N(12)-diacetylspermine, a polyamine regulator of cellular proliferation. Together these findings implicate colonic bacterial biofilms as contributors to a pro-oncogenic state.
A secreted network of densely organized mucin protects the epithelium of the healthy human colon from direct contact with luminal contents. A persistent breach and subsequent colonization of this mucus barrier by a dense consortium of luminal bacteria meets criteria for consideration as a colonic biofilm. Development of a colonic biofilm is a pathogenic state previously implicated in the inflammatory bowel diseases, ulcerative colitis and Crohn’s disease.3,4 Consistent with this association, we found biofilms from the sporadic colorectal cancer (CRC) host to be associated with a pro-oncogenic altered metabolome and epithelial cell biology with perhaps the most relevant association, in the context of cancer, being increased epithelial cell proliferation. The altered organization of bacteria in direct contact with the colonic epithelial cell membrane raises questions about the delivery and mechanisms by which microbial metabolic products secreted in such close proximity to host cells may facilitate carcinogenesis. An untargeted global metabolomics analysis revealed that an elevated N(1), N(12)-diacetylspermine pool, a polyamine relevant to oncogenesis, was in part due to contribution(s) from the bacterial biofilms. While excess levels of various polyamines have been recognized in association with colon cancer, including one study in which N(1), N(12)-diacetylspermine was detected in the urine of CRC patients,5 this is the first direct link of this polyamine metabolite to the microbiome and, specifically, biofilm formation. A decreasing gradient from the proximal to distal colon in both healthy and disease states has been reported for polyamine levels and ornithine decarboxylase (ODC) prior to our study.6,7 Our nearly universal detection of biofilms on CRCs in the proximal colon provides additional insight and an explanation for why this gradient might be playing a role in CRC. The idea that a microbial biofilm community, rather than individual organisms, may be primed to differentially produce metabolites that influence carcinogenesis is a new concept that warrants further investigation. These expression patterns, differing in geographic regions of the colon suggest that in situ metabolic analysis may be necessary to truly understand the microbial contributions to physiology and pathophysiology throughout the colon. Further, geographic metabolomic differences in the colon, driven by the microbiome, may contribute to the already established differences between the left and right colon.
The vast majority of biofilm-covered tissues were identified on individuals with right-sided colon cancer, proximal to the hepatic flexure, raising questions about the relationship between the microbiome and site-specific cancer. While the division between the right and left colon has traditionally been at the splenic flexure based on embryonic derivation, differences in incidence, mutation profile, MSI status, and outcome have been noted between the proximal and distal colon for decades.8 Distinct epidemiological, phenotypic, and molecular pathological differences based on tumor anatomical location suggest different risk factors, susceptibilities, and pathways of transformation associated with right vs. left colon carcinogenesis. These differences have been thoroughly studied and largely attributed to the following explanations: (1) inherently distinct biological characteristics acquired during development exist between proximal and distal colon cells, which, in turn, determine different responses to common environmental insults; or (2) there are different procarcinogenic exposures in proximal vs. distal colon (such as bacterial biofilms and their metabolites as in our study); or (3) most likely, there exists a combination of both unique environmental insults and differences in the innate susceptibility of the target cells.9 Our 2 studies lend further support to this observation and we would propose that bacterial biofilms could, in part, be contributing to this difference, specifically in the etiology of colon cancers proximal to the hepatic flexure. The task of determining causation requires a solid epidemiological link of exposure to biofilms before disease onset, a measureable host response, an experimental model to test the hypothesis and finally evidence that removal of the putative causative bacterium (or downstream metabolite(s)) or mechanism prevents disease.
While biofilms of CRC were detected on both tumors and paired normal tissues, we focused on analyzing epithelial changes of the paired normal tissue to begin to discern what effect biofilms may have on cells that have not undergone oncogenic transformation. Biofilms in the cancer host were determined to be expansive, spanning at least the length of the resected specimen. Thus, we speculate that the entire colon is biofilm-covered in some CRC patients. While the paired normal tissue from the cancer host revealed biological changes relevant to oncogenic transformation, the finding that biofilm-covered mucosa identified in a subset (~10–20%) of healthy individuals undergoing routine screening colonoscopy also exhibits loss of E-cadherin from the apical zonula adherens, enhanced mucosal IL-6, and increased crypt cell proliferation was of particular interest to our group. Thus, we posit that colon mucosal biofilms may mark the individual at high risk for development of colon neoplasia, a risk potentially augmented by other established risk factors for colon cancer such as obesity, diabetes mellitus, smoking among others. To begin to address this hypothesis, a longitudinal prospective study has been designed to establish biofilm stability, progression and host:bacterial mechanisms while, in parallel, seeking to detect colon neoplasia in a healthy cohort over time.
From our study, we note that had we focused solely on the cancer microbiome using 16S rRNA sequence analysis, we would have missed all the critical observations of our study. However, by combining detailed microbiologic, biologic and principal coordinates analysis of 16s rRNA sequence data, we identified biofilms, procarcinogenic mucosal signaling and a progressive dysbiosis of microbial communities in the transition from tissues without a biofilm, to normal tissues with a biofilm, to the cancer microbiome. Based on the procarcinogenic biological changes associated with biofilm communities on normal tissues, we propose that increasing oncogenic potential accompanies this microbial dysbiosis (Fig. 1). An animal model of right (proximal) colon biofilm formation and carcinogenesis would be enormously helpful to assess the oncogenic potential of biofilm communities and to interrogate the mechanisms of carcinogenesis. However, no such murine model yet exists and development may be hampered by the reported variable and loosely-organized mucus layer in the proximal mouse colon that is penetrable by bacteria in the basal state.
Figure 1.
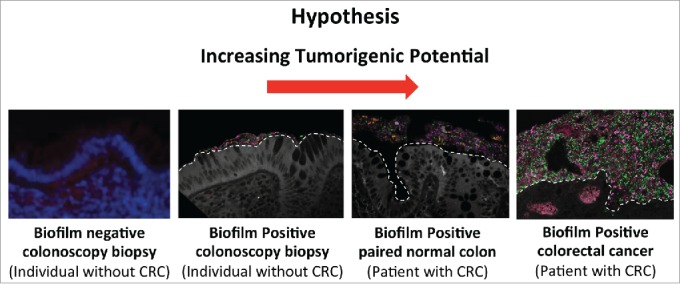
Working hypothesis for the pro-carcinogenic capacity of biofilms identified on the colon mucosa of control subjects undergoing screening colonoscopy and individuals with CRC.
Traditionally, efforts have focused on linking specific bacterial agents and their respective toxins to CRC. This has led to the identification of putative bacterial oncogenic drivers of CRC including, for example, enterotoxigenic Bacteroides fragilis expressing the BFT toxin, Escherichia coli harboring the pks virulence island encoding the genes required to make the colibactin genotoxin, and Fusobacterium nucleatum containing the FadA adhesin. Each of these organisms has been linked through epidemiological studies with human CRC and has been shown to induce colon tumors in genetically susceptible murine models of disease. This work yielded essential mechanistic details about the bacterial capacity to induce oncogenesis. However, in tackling the complexity of human disease, we think comprehensive epidemiologic and experimental approaches are necessary to identify ‘if, how and which’ microbial consortia or species are carcinogenic. The discovery that bacterial biofilm communities may harbor oncogenic risk provides a starting point for several new lines of investigation that may yield additional insight regarding the potential tumorigenic contribution of the microbiota to CRC initiation and progression.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the National Institutes of Health through R01 CA151393, a grant from the Institute Mérieux and a research agreement with Bristol- Myers Squibb Co -International Immuno-Oncology Network-IION Resource Model.
References
- 1.Dejea CM, Wick EC, Hechenbleikner EM, White JR, Mark Welch JL, Rossetti BJ, Peterson SN, Snesrud EC, Borisy GG, Lazarev M, et al.. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc Natl Acad Sci U S A 2014; 111:18321-6; PMID:25489084; http://dx.doi.org/ 10.1073/pnas.1406199111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson CH, Dejea CM, Edler D, Hoang LT, Santidrian AF, Felding BH, Ivanisevic J, Cho K, Wick EC, Hechenbleikner EM, et al.. Metabolism links bacterial biofilms and colon carcinogenesis. Cell Metab 2015; 21:891-7; PMID:25959674; http://dx.doi.org/ 10.1016/j.cmet.2015.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swidsinski A, Khilkin M, Kerjaschki D, Schreiber S, Ortner M, Weber J, Lochs H. Association between intraepithelial escherichia coli and colorectal cancer. Gastroenterology 1998; 115:281-6; PMID:9679033 [DOI] [PubMed] [Google Scholar]
- 4.Swidsinski A, Loening-Baucke V, Herber A. Mucosal flora in crohn's disease and ulcerative colitis - an overview. J Physiol Pharmacol 2009; 60 Suppl 6:61-71; PMID:20224153 [PubMed] [Google Scholar]
- 5.Hiramatsu K, Takahashi K, Yamaguchi T, Matsumoto H, Miyamoto H, Tanaka S, Tanaka C, Tamamori Y, Imajo M, Kawaguchi M, et al.. N(1),N(12)-diacetylspermine as a sensitive and specific novel marker for early- and late-stage colorectal and breast cancers. Clin Cancer Res 2005; 11:2986-90; PMID:15837752; http://dx.doi.org/11/8/2986 [DOI] [PubMed] [Google Scholar]
- 6.Zehnter E, Roisch U, Kruis W, Breuer C, Diehl V. Ornithine decarboxylase levels in patients with normal colonic mucosa. Eur J Clin Chem Clin Biochem 1996; 34:529-33; PMID:8864401 [DOI] [PubMed] [Google Scholar]
- 7.Allgayer H, Roisch U, Zehnter E, Ziegenhagen DJ, Dienes HP, Kruis W. Colonic ornithine decarboxylase in inflammatory bowel disease: Ileorectal activity gradient, guanosine triphosphate stimulation, and association with epithelial regeneration but not the degree of inflammation and clinical features. Dig Dis Sci 2007; 52:25-30; PMID:17171446; http://dx.doi.org/ 10.1007/s10620-006-9515-4 [DOI] [PubMed] [Google Scholar]
- 8.Bufill JA. Colorectal cancer: Evidence for distinct genetic categories based on proximal or distal tumor location. Ann Intern Med 1990; 113:779-88; PMID:2240880 [DOI] [PubMed] [Google Scholar]
- 9.Glebov OK, Rodriguez LM, Nakahara K, Jenkins J, Cliatt J, Humbyrd CJ, DeNobile J, Soballe P, Simon R, Wright G, et al.. Distinguishing right from left colon by the pattern of gene expression. Cancer Epidemiol Biomarkers Prev 2003; 12:755-62; PMID:12917207 [PubMed] [Google Scholar]