

Abstract
The scientific community has recently come to appreciate that, rather than existing as independent organisms, multicellular hosts and their microbiota comprise a complex evolving superorganism or metaorganism, termed a holobiont. This point of view leads to a re-evaluation of our understanding of different physiological processes and diseases. In this paper we focus on experimental and computational approaches which, when combined in one study, allowed us to dissect mechanisms (traditionally named host-microbiota interactions) regulating holobiont physiology. Specifically, we discuss several approaches for microbiota perturbation, such as use of antibiotics and germ-free animals, including advantages and potential caveats of their usage. We briefly review computational approaches to characterize the microbiota and, more importantly, methods to infer specific components of microbiota (such as microbes or their genes) affecting host functions. One such approach called transkingdom network analysis has been recently developed and applied in our study.1 Finally, we also discuss common methods used to validate the computational predictions of host-microbiota interactions using in vitro and in vivo experimental systems.
Keywords: antibiotics, bipartite betweenness centrality, germfree mice, holobiont, microbiome, microbiota, metagenomics, systems biology, transkingdom networks
Introduction
In addition to our own cells and genomes, humans are hosts to a vast community of microbes. This microbiota are not neutral neighbors, rather they are indispensable contributors to host physiology, acting in a symbiotic relationship with the host. The concept that we are holobionts, being comprised of hologenomes (our genomes and our microbial genomes) with complex interactions between our cells and our microbes, has been discussed for many years.2-9 More recently, the development of new tools to interrogate these hologenomic interactions has greatly expanded our ability to understand and define ourselves in terms of both our own human genome and that of our resident microbial partners.10 The holobiont view helps to explain the dramatic increase in the number of chronic inflammatory and autoimmune diseases with significant genetic component which occurred too rapidly to be attributed to the host genome alone.11,12 In contrast, our microbial genomes have a tremendous capacity for rapid adaptations which can influence health and disease.13 Therefore, in order to better understand the basis of many modern diseases a deeper insight into our interactions with our microbes is required.
Perturbing the microbiota: germ-free and antibiotic-treated mice
Microbiota contribution to specific host phenotypes, including diseases, is frequently determined through broad perturbation of the microbial community (Fig. 1). One extreme, but common method of microbiota perturbation is generation of animals, most commonly mice,14 devoid of microbes (i.e. germfree or axenic). Axenic and gnotobiotic insect and zebrafish models can also be informative in understanding fundamentals of host-microbe interactions;15-20 however, the mouse is the most widely used and powerful tool for understanding the connections between microbiota and disease.14,15 Study of germ free versus conventional organisms has revealed myriad roles for microbiota in host immunity and metabolism, among many other systems. For example, it has been long known that in the absence of microbiota, development of various immune cells is impaired and the ability to respond to infections is reduced.21,22 Germ free mice have also revealed that microbiota have profound influence on metabolism, for example, regulating bile acids pools23 and worsening glucose tolerance.24 Furthermore, it has been shown that the gut microbiota can mediate molecular cross-talk between host immune and metabolic functions.25,26 Additionally, derivation of specific genetically modified mouse models under germ free conditions has been used to investigate gene-specific interactions between host and microbiota.27 Although gnotobiotic technologies for germfree animals have existed since the 1950s,28 our ability to derive and work with germ-free mice is still a limiting factor for investigation. Therefore, an economically more feasible approach was introduced - using antibiotic treatments to alter microbiota composition ideally to the point of eliminating nearly all microbes with cocktails of antibiotics. One protocol using a cocktail of 4 antibiotics for 4–5 weeks has been exceptionally popular among investigators and allowed to demonstrate the involvement of microbiota in host functions and disease pathogenesis in a variety of models.29-32 Furthermore, a few studies compared both germfree and antibiotic-treated animals and detected concordant results.31,33 However, concordant results are not always observed between these 2 models. For example, in an animal model of common variable immunodeficiency (CVID) associated enteropathy (in B lymphocyte deficient mice), we found that derivation of B cell knockout (BcKO) and control mice as germfree abolished differences in the host phenotype between the 2 genotypes observed in conventional mice (Fig. 2A).25 This result demonstrated the essential role of microbiota in BcKO phenotype. However, the antibiotics cocktail protocol that has successfully mimicked germfree mice in other studies did not revert host alterations in the intestine of B cell deficient mice (Fig. 2B).
Figure 1.
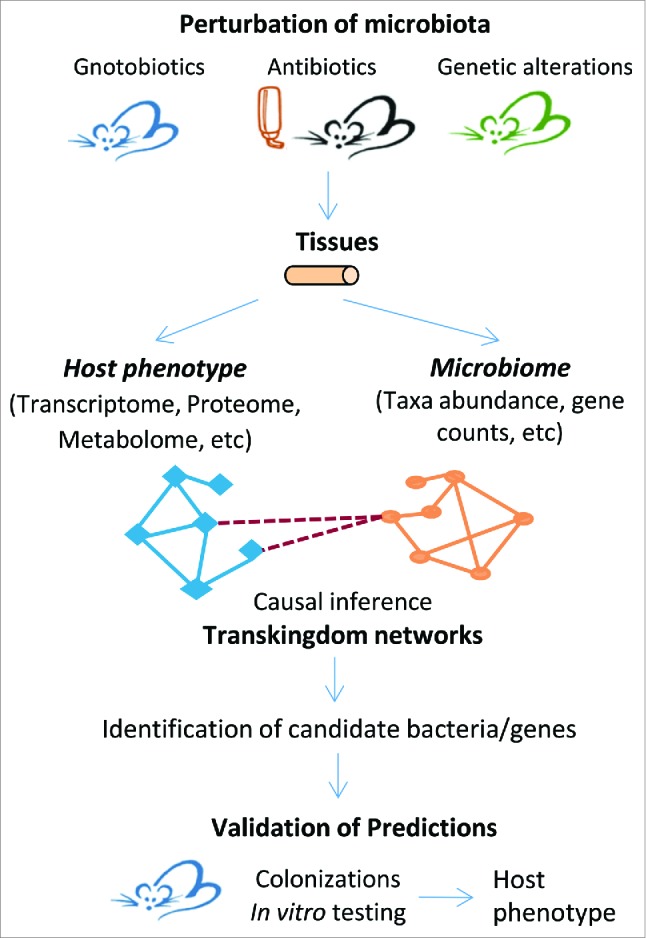
An approach to predict and test for members of microbiota affecting specific host functions using transkingdom network analysis.
Figure 2.

Different effects of the absence of microbiota (A) and of antibiotics (B) on the B cell knockout (BcKO) gene expression phenotype in the intestine. Each dot on graphs represents ratios of gene expression between BcKO and control mice from BcKO signature (Shulzhenko & Morgun et al., 2011). Similarity between gene expression alterations induced by B cell deficiency in conventional and germ-free (A) or conventional and antibiotic-treated (B) mice is estimated using correlation analysis and represented on the graphs.
Intrigued by this difference between germfree and antibiotic-treated mice, we sought to comprehensively evaluate the effects of antibiotics on the intestine. Surprisingly, we found that only about one third of gene expression changes in the gut of antibiotic-treated mice could be attributed to the depletion of normal microbiota verified by comparison to germfree mice.1 This effect was primarily manifested by decreased expression of genes related to many aspects of innate and adaptive immunity. Interestingly, antibiotics had much more pronounced effect on T cell numbers than on IgA-producing cells.1 This may be one of the reasons for the discrepancy between germfree and antibiotic-treated BcKO mice (Fig. 2) as their phenotype was IgA-dependent.25 An additional, non-mutually exclusive, explanation is that microbiota members insensitive to antibiotics (such as viruses) are responsible for alterations in intestines of B cell deficient mice. Indeed, this hypothesis is supported by other studies demonstrating outgrowth of diverse viruses in immunodeficient animals.34,35
Thus, since each type of microbiota perturbation presents some drawbacks, the selection of appropriate models and interpretation of results must be performed with consideration to the potential of model-specific effects. The results of our study that evaluated global intestinal transcriptome after treatment with antibiotics cocktail can aid in these decisions by providing a thorough characterization of changes in host gene expression that can be specifically attributed to 3 factors, namely, the lack of microbiota, the effect of antibiotics-resistant microbes, and direct effects of antibiotics.1
Microbiota characterization
Broad perturbations of microbial communities discussed above can provide evidence of causal roles of microbiota in host phenotypes. However, these experiments have to be followed by several questions to pinpoint the exact mechanisms: What microbes live in the particular host? Which of them are responsible for control of specific host phenotypes? What molecular mechanisms do microbiota use to influence the host? Advances in sequencing technologies over the past decade have greatly enhanced our ability to answer some of these questions. Herein, we briefly described culture-free methods, leaving out cultivation approaches reviewed elsewhere (Fig. 1).36,37
One approach to identify the taxonomic profile of a microbial community is with 16S rRNA amplicon sequencing, in which highly variable regions of 16S ribosomal rRNA from all microbes in a sample are amplified by PCR and subject to high throughput sequencing38 or hybridization to probes on a special microarray chip called Phylochip.1,39 In the case of sequencing, relative abundances of taxa are calculated from copy number of amplicons from the corresponding taxa. Abundance profiles for different taxonomic ranks are commonly generated (i.e., strain, species, genus, family etc.) and then can be used for further analyses.40 Taxonomic composition 16S amplicon sequencing can be also used to predict functional variables such as KEGG gene/pathway.41 In this case, gene composition of detected taxa is predicted from phylogenetic relationship of taxa with known annotated reference genome.
An alternative and/or complementary approach to identification of taxonomic composition by 16S is shotgun metagenomics sequencing, in which the whole genomes of all microbes (microbiome) in the sample are sequenced. With this method, gene/function abundance can be directly derived from shotgun metagenomics sequencing by assigning reads to protein sequence or protein families that have functional annotation in KEGG, SEED and COG database.42,43 This method can be very informative, as it could be not a single species that affects host phenotypes, but a bacterial function present across multiple species. In this case, the same function carried out by similar genes from different species is investigated as a single variable, providing potential molecular mechanisms regardless of taxonomy information. In addition, genome sequence reads can be compared to reference genomes in order to be assigned to specific taxa. Then the number of reads assigned to each taxon can be normalized to adjust for genome size and sequencing depth and generate abundance profiles for microbes.44
Systems approaches to infer microbial contributors to host physiology
Thanks to new sequencing technologies and new culturing techniques, we have made big advances in characterization of microbiota. Currently, the key challenge in the field is a transition from merely observational or descriptive studies to inference and testing of causal relationships between specific microbes and host biology. In other words, identification which specific taxa and/or bacterial functions are actually responsible for control of specific host function is now an area of active investigation. One recently proposed approach to this question is to systematically test libraries of randomly selected microbes through large scale colonization of germ free mice employed by Faith et al.45 Although the approach uses an analytical method that infers microbes specifically affecting host phenotype, this strategy is still highly labor and resource intensive, thus limiting its utilization to centers that have large gnotobiotic facilities.
Therefore, a more desirable approach would be to first computationally predict the most likely important players or causal factors (microbes and/or microbial genes) among hundreds of measured variables and then to experimentally test the most promising candidates. This type of question is not unique to the field of holobiont biology. For instance, cancer researchers have been facing a similar problem attempting identify driver mutations responsible for progression of tumors. Application of novel systems approaches to genomic and transcriptomic data from tumors has recently offered an efficient solution to infer a short list of candidate drivers. Indeed, reconstruction and analysis of gene regulatory networks has proven to be an excellent tool for causal inference, allowing researchers to uncover gene-drivers of carcinogenesis of different tumors.46-48 Moreover, these networks can be built from different types of variables using a variety of available tools49 as long as all parameters are all measured in the same sample.
In our work studying the effect of antibiotics on intestine by colonizing germ-free mice with antibiotic-resistant microbiota, we demonstrated that these microbes had profound effect on intestinal transcriptome, in particular, decreasing expression of mitochondria and proliferation related genes.1 Indeed, we have faced the common problem that there is evidence of a microbiota effect on a host phenotype, but it was unclear which microbe was responsible. Building on our previous experience with gene networks,25,48,50 we developed a new approach that models host-microbiota interaction that we called transkingdom network.1,49 To build this model, microbial gene abundances and mouse transcriptome data were integrated into one network. Interrogating the microbial part of this network revealed 2 unexpected microbes (Pseudomonas aeruginosa and Escherichia coli) as candidates to drive mitochondrial depression and cell death in epithelium. Although these microbes are minor players in the healthy mouse microbiota, they are resistant to many antibiotics1 and therefore antibiotic ablation of the microbiota provides them with a prime opportunity to interact with the host. Furthermore, in order to use this network to infer which bacterial genes are responsible for microbiota effect on host, we adapted a specific topological metric, called betweenness centrality (Fig. 3). Instead of using standard betweenness centrality we employed the bipartite betweenness centrality. Although bipartite betweenness centrality had been originally proposed for human communication studies,51 our work provides first evidence that this topological metric can be useful to investigate biological networks and interrogate transkingdom communications. This approach allowed us to search for microbial genes that would represent “bottleneck” nodes in communication between microbiota and host. Among top predicted genes we found LasR, a well-known transcription factor of P. aeruginosa responsible for regulation of quorum sensing and secretion of virulence factors by this microbe. Importantly, we could experimentally validate not only the effect of P. aeruginosa on mammalian cells, but also the key role of LasR in this process. It is important to note, that while in this study we have used bacterial gene copy-number data, bipartite betweenness centrality can also be applied to taxonomic abundances derived from 16S sequencing (unpublished data). Thus, the reconstruction and interrogation of transkingdom network represents a promising approach for inference of causal players of host-microbiota interaction and it is the first one used to infer not only specific taxa, but also specific bacterial genes responsible for the effect of microbe on specific host phenotype.
Figure 3.
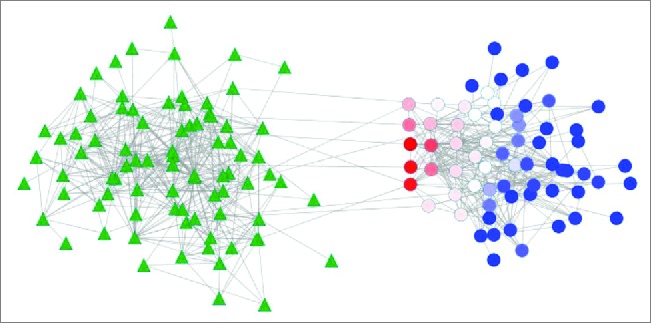
Bipartite betweenness centrality in transkingdom network. Microbial genes (circle nodes) and host genes (triangle nodes). Bi-partite betweenness centrality is calculated for each microbial gene based on the number of times it is present in the shortest paths connecting microbial genes and human genes (Dong et al., 2015). Microbial genes with high bipartite betweenness centrality (red) are more likely to be key regulators of host gene expression than genes with small values of bipartite betweenness centrality (blue).
Other tools have been also recently developed attempting to connect microbiome data with host phenotype.52-58 Although most of them do not go beyond simple association tests, 2 approaches should be specifically mentioned as similarly to our method they attempt to pinpoint microbes that have potential causal effect on unrelated phenotype.52,59 The first one, LEfSe, is a popular tool that has been already utilized by several groups.60-62 More importantly a few studies went beyond analyses and successfully validated computational predictions of this approach. For example, LEfSe has been applied on 16S and shotgun metagenomics data to identify Prevotella copri to be related to Rheumatoid arthritis and experimentally proved to be able to increase inflammatory response in the gut.61 The other approach,59 although it does not have specific tool, relies on extended generalized Lotka–Volterra dynamics on time series metagenomics data analysis. It was successfully applied to predict bacteria that would efficiently compete with C. difficile.57 Importantly, in this case the researchers could also confirm that all 3 microbes predicted could out-compete C. difficile in mouse colonization experiments.
Validating predictions: microbiota transplants and in vitro testing
Similar to other fields of biology, the results of computational inferences of host microbiota interactions are much more valuable if confirmed by ‘wet-lab’ experiments (Fig. 1). Given our primary focus here in determining how microbes affect host biology, approaches taken in validation are reminiscent of widely used infection models using commensal microbes instead of pathogens. However, whereas in the infection models such microbes are normally absent in healthy hosts, commensal microorganisms can be present in normal microbiota. Therefore, monocolonization of germfree mice with a microbe of interest is seemingly the most straightforward solution to circumvent this problem and is still widely employed in experimental studies.63-66 Though it might look to be the “cleanest” way, concerns arise about the environment into which the selected microbes are introduced. For example, germfree mice have an undeveloped immune system and altered metabolism that could influence behavior of the tested species. Additionally, a given bacterium in monoassociation might not behave in the same way as it would in a community and might not induce same immune responses.45,67-69 For example, bacteria comprising a standard mix (Altered Schaedler Flora) were insufficient to induce colonic T regulatory cells in germfree mice when used individually.67 In another study, a common mouse pathogen Citrobacter rodentium did not trigger colitis when it was administered alone in contrast to when it was given in the presence of another bacterium.70 Another recently proposed approach would be to use “standardised” microbial communities14 which can be then supplemented with the microbe of interest. In addition, aforementioned antibiotic-treated mice can serve as another type of host. There are recent examples in the literature utilizing one of these or a combination of approaches to validate their predictions. For example, Iida and Dzutsev et al. predicted Alistipes shahii as a bacterium affecting the host's ability to respond to chemotherapy.71 To test this, antibiotic-treated mice that otherwise have poor response were given A. shahii, which improved response by inducing expression of inflammatory cytokines.71 In another study, the genus Sutterella was identified by LEfSe to be associated with low fecal IgA levels.62 This prediction was tested not by monocolonization, but by administration of an enriched culture of Sutterella to mice, which converted high fecal IgA mice to a low IgA phenotype.62
Finally, besides validation of effects of microbes predicted from animal models, there is a growing need for validating predictions generated from analyses of human microbiota associated with disease states. Therefore, humanized gnotobiotic mice (i.e. germfree mice colonized with human microbiota) have become a popular experimental system.72 Despite some concerns that this system does not fully recapitulate the effects human microbiota on immune system,73 some effects of microbiota on metabolism could be confirmed in humanized mice.72,74-76
Classical in vitro approaches can be also used for evaluation the effects of microbes on host cells (Fig. 1). This strategy has been very useful for investigation of effects of pathogens77 and probiotics.78-81 Furthermore, if candidates are not just particular bacteria but specific bacterial genes, genetically modified microbes have to be tested alongside with wild-type bacteria, as was done in our study on the effects of antibiotics and antibiotic-insensitive bacteria on intestinal phenotypes.1 In this study, besides identifying P. aeruginosa as a candidate microbe as a regulator of a specific host phenotype (mitochondrial depression and cell death), we also identified LasR as a likely bacterial gene regulator of this phenotype. We anticipated that, as LasR is a primary regulator of quorum sensing, soluble factors secreted by bacteria should play in the effect on host. To validate these predictions we treated an intestinal cell line with wild-type and knockout P. aeruginosa conditioned growth medium. Indeed, our analytical predictions were confirmed as medium from wild type bacterium led to mitochondrial repression and cell death, while LasR-deficient bacteria were unable to produce this effect.1
In conclusion, the field of host-microbiota interactions is rapidly transforming into a new discipline bringing biology and medicine into new era. While hologenome theory makes peace between Darwin's evolution and Lamarck's theory of Inheritance of acquired characteristics, new systems biology approaches armed with metagenomics and gnotobiotic techniques revolutionize our understanding of health and disease. Transkingdom networks put in practice the ‘hologenome theory of evolution’ by offering a robust framework for interrogation of hosts and their microbes as a whole. The insights from these networks provide a unique understanding of cellular and molecular mechanisms that govern social affairs between macro- and micro-species that altogether make up a holobiont.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
AM and NS are funded by U01 AI109695 and R01 DK103761.
References
- [1].Morgun A, Dzutsev A, Dong X, Greer RL, Sexton DJ, Ravel J, Schuster M, Hsiao W, Matzinger P, Shulzhenko N. Uncovering effects of antibiotics on the host and microbiota using transkingdom gene networks. Gut 2015; 64(11):1732-43; PMID:25614621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Waldor MK, Bordenstein SR, Theis KR. Host Biology in Light of the Microbiome: Ten Principles of Holobionts and Hologenomes. PLOS Biol 2015; 13:e1002226; PMID:26284777; http://dx.doi.org/ 10.1371/journal.pbio.1002050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gordon J, Youle M, Knowlton N, Rohwer F, Relman DA. Superorganisms and Holobionts. Microbe Magazine 2013; 8:152-3; http://dx.doi.org/ 10.1128/microbe.8.152.1 [DOI] [Google Scholar]
- [4].Biagi E, Candela M, Fairweather-Tait S, Franceschi C, Brigidi P. Ageing of the human metaorganism: the microbial counterpart. Age 2011; 34:247-67; PMID:21347607; http://dx.doi.org/ 10.1007/s11357-011-9217-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gilbert SF. A holobiont birth narrative: the epigenetic transmission of the human microbiome. Front Genet 2014; 5:282; PMID:25191338; http://dx.doi.org/ 10.3389/fgene.2014.00282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dheilly NM. Holobiont–Holobiont Interactions: Redefining Host–Parasite Interactions. PLoS Pathog 2014; 10:e1004093; PMID:24992663; http://dx.doi.org/ 10.1371/journal.ppat.1004093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Mindell DP. Phylogenetic consequences of symbioses: Eukarya and Eubacteria are not monophyletic taxa. Biosystems 1992; 27:53-62; PMID:1391691; http://dx.doi.org/ 10.1016/0303-2647(92)90046-2 [DOI] [PubMed] [Google Scholar]
- [8].Rosenberg E, Koren O, Reshef L, Efrony R, Zilber-Rosenberg I. The role of microorganisms in coral health, disease and evolution. Nat Rev Microbiol 2007; 5:355-62; PMID:17384666; http://dx.doi.org/ 10.1038/nrmicro1635 [DOI] [PubMed] [Google Scholar]
- [9].Margulis L, Fester R. Symbiosis as a source of evolutionary innovation : speciation and morphogenesis. Cambridge, Mass: MIT Press, 1991. [PubMed] [Google Scholar]
- [10].van Opstal EJ, Bordenstein SR. MICROBIOME. Rethinking heritability of the microbiome. Science 2015; 349:1172-3; PMID:26359393; http://dx.doi.org/ 10.1126/science.aab3958 [DOI] [PubMed] [Google Scholar]
- [11].Okada H, Kuhn C, Feillet H, Bach JF. The ‘hygiene hypothesis’ for autoimmune and allergic diseases: an update. Clin Exp Immunol 2010; 160:1-9; PMID:20415844; http://dx.doi.org/ 10.1111/j.1365-2249.2010.04139.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, Benchimol EI, Panaccione R, Ghosh S, Barkema HW, et al.. Increasing Incidence and Prevalence of the Inflammatory Bowel Diseases With Time, Based on Systematic Review. Gastroenterology 2012; 142:46-54.e42; PMID:22001864; http://dx.doi.org/ 10.1053/j.gastro.2011.10.001 [DOI] [PubMed] [Google Scholar]
- [13].Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nat Rev Genet 2012; 13:260-70; PMID:22411464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Macpherson AJ, McCoy KD. Standardised animal models of host microbial mutualism. Mucosal Immunol 2015; 8:476-86; PMID:25492472; http://dx.doi.org/ 10.1038/mi.2014.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Milligan-Myhre K, Charette JR, Phennicie RT, Stephens WZ, Rawls JF, Guillemin K, Kim CH. Study of host-microbe interactions in zebrafish. Methods Cell Biol 2011; 105:87-116; PMID:21951527; http://dx.doi.org/ 10.1016/B978-0-12-381320-6.00004-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bae YS, Choi MK, Lee W-J. Dual oxidase in mucosal immunity and host–microbe homeostasis. Trends Immunol 2010; 31:278-87; PMID:20579935; http://dx.doi.org/ 10.1016/j.it.2010.05.003 [DOI] [PubMed] [Google Scholar]
- [17].Buchon N, Broderick NA, Chakrabarti S, Lemaitre B. Invasive and indigenous microbiota impact intestinal stem cell activity through multiple pathways in Drosophila. Genes Dev 2009; 23:2333-44; PMID:19797770; http://dx.doi.org/ 10.1101/gad.1827009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shin SC, Kim SH, You H, Kim B, Kim AC, Lee KA, Yoon JH, Ryu JH, Lee WJ. Drosophila Microbiome Modulates Host Developmental and Metabolic Homeostasis via Insulin Signaling. Science 2011; 334:670-4; PMID:22053049; http://dx.doi.org/ 10.1126/science.1212782 [DOI] [PubMed] [Google Scholar]
- [19].Stephens WZ, Wiles TJ, Martinez ES, Jemielita M, Burns AR, Parthasarathy R, Bohannan BJ, Guillemin K. Identification of Population Bottlenecks and Colonization Factors during Assembly of Bacterial Communities within the Zebrafish Intestine. mBio 2015; 6:e01163-15; PMID:26507229; http://dx.doi.org/ 10.1128/mBio.01163-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kanther M, Sun X, Mühlbauer M, Mackey LC, Flynn EJ 3rd, Bagnat M, Jobin C, Rawls JF. Microbial Colonization Induces Dynamic Temporal and Spatial Patterns of NF-κB Activation in the Zebrafish Digestive Tract. Gastroenterology 2011; 141:197-207; PMID:21439961; http://dx.doi.org/ 10.1053/j.gastro.2011.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 2009; 9:313-23; PMID:19343057; http://dx.doi.org/ 10.1038/nri2515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kamada N, Seo S-U, Chen GY, Núñez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol 2013; 13:321-35; PMID:23618829; http://dx.doi.org/ 10.1038/nri3430 [DOI] [PubMed] [Google Scholar]
- [23].Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, Bäckhed F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-β-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 2013; 17:225-35; PMID:23395169; http://dx.doi.org/ 10.1016/j.cmet.2013.01.003 [DOI] [PubMed] [Google Scholar]
- [24].Caesar R, Reigstad CS, Backhed HK, Reinhardt C, Ketonen M, Lunden GO, Cani PD, Bäckhed F. Gut-derived lipopolysaccharide augments adipose macrophage accumulation but is not essential for impaired glucose or insulin tolerance in mice. Gut 2012; 61:1701-7; PMID:22535377; http://dx.doi.org/ 10.1136/gutjnl-2011-301689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Shulzhenko N, Morgun A, Hsiao W, Battle M, Yao M, Gavrilova O, Orandle M, Mayer L, Macpherson AJ, McCoy KD, et al.. Crosstalk between B lymphocytes, microbiota and the intestinal epithelium governs immunity vs. metabolism in the gut. Nat Med 2011; 17:1585-93; PMID:22101768; http://dx.doi.org/ 10.1038/nm.2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Greer RL, Morgun A, Shulzhenko N. Bridging immunity and lipid metabolism by gut microbiota. J Allergy Clin Immunol 2013; 132:253-62; PMID:23905915; http://dx.doi.org/ 10.1016/j.jaci.2013.06.025 [DOI] [PubMed] [Google Scholar]
- [27].Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, Rennick DM, Sartor RB. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun 1998; 66:5224-31; PMID:9784526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Orland FJ, Blayney JR, Harrison RW, Reyniers JA, Trexler PC, Ervin RF, Gordon Ha, Wagner M. Experimental caries in germfree rats inoculated with enterococci. J Am Dent Assoc 1955; 50:259-72; PMID:13232956; http://dx.doi.org/ 10.14219/jada.archive.1955.0061 [DOI] [PubMed] [Google Scholar]
- [29].Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004; 118:229-41; PMID:15260992; http://dx.doi.org/ 10.1016/j.cell.2004.07.002 [DOI] [PubMed] [Google Scholar]
- [30].Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, Littman DR. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 2008; 4:337-49; PMID:18854238; http://dx.doi.org/ 10.1016/j.chom.2008.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hill DA, Siracusa MC, Abt MC, Kim BS, Kobuley D, Kubo M, Kambayashi T, Larosa DF, Renner ED, Orange JS, et al.. Commensal bacteria-derived signals regulate basophil hematopoiesis and allergic inflammation. Nat Med 2012; 18:538-46; PMID:22447074; http://dx.doi.org/ 10.1038/nm.2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, Kastenmuller W, Deming C, Quinones M, Koo L, Conlan S, et al.. Compartmentalized control of skin immunity by resident commensals. Science 2012; 337:1115-9; PMID:22837383; http://dx.doi.org/ 10.1126/science.1225152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Reikvam DH, Erofeev A, Sandvik A, Grcic V, Jahnsen FL, Gaustad P, McCoy KD, Macpherson AJ, Meza-Zepeda LA, Johansen FE. Depletion of Murine Intestinal Microbiota: Effects on Gut Mucosa and Epithelial Gene Expression. PLoS ONE 2011; 6:e17996; PMID:21445311; http://dx.doi.org/ 10.1371/journal.pone.0017996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Young GR, Eksmond U, Salcedo R, Alexopoulou L, Stoye JP, Kassiotis G. Resurrection of endogenous retroviruses in antibody-deficient mice. Nature 2012; 491:774-8; PMID:23103862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Handley SA, Thackray LB, Zhao G, Presti R, Miller AD, Droit L, Abbink P, Maxfield LF, Kambal A, Duan E, et al.. Pathogenic simian immunodeficiency virus infection is associated with expansion of the enteric virome. Cell 2012; 151:253-66; PMID:23063120; http://dx.doi.org/ 10.1016/j.cell.2012.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Allen-Vercoe E. Bringing the gut microbiota into focus through microbial culture: recent progress and future perspective. Curr Opin Microbiol 2013; 16:625-9; PMID:24148301; http://dx.doi.org/ 10.1016/j.mib.2013.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Stewart EJ. Growing unculturable bacteria. J Bacteriol 2012; 194:4151-60; PMID:22661685; http://dx.doi.org/ 10.1128/JB.00345-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al.. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010; 7:335-6; PMID:20383131; http://dx.doi.org/ 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Vujkovic-Cvijin I, Dunham RM, Iwai S, Maher MC, Albright RG, Broadhurst MJ, Hernandez RD, Lederman MM, Huang Y, Somsouk M, et al.. Dysbiosis of the gut microbiota is associated with HIV disease progression and tryptophan catabolism. Sci Transl Med 2013; 5:193ra91; PMID:23843452; http://dx.doi.org/ 10.1126/scitranslmed.3006438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kuczynski J, Stombaugh J, Walters WA, Gonzalez A, Caporaso JG, Knight R. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr Protoc Microbiol 2012; 27:E:1E.5:1E.5.1 -1E.5.20. doi: 10.1002/9780471729259.mc01e05s27; PMID:23184592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, et al.. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 2013; 31:814-21; PMID:23975157; http://dx.doi.org/ 10.1038/nbt.2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Glass EM, Wilkening J, Wilke A, Antonopoulos D, Meyer F. Using the metagenomics RAST server (MG-RAST) for analyzing shotgun metagenomes. Cold Spring Harb Protoc 2010; 2010:pdb prot5368; PMID:20150127 [DOI] [PubMed] [Google Scholar]
- [43].Nayfach S, Fischbach MA, Pollard KS. MetaQuery: a web server for rapid annotation and quantitative analysis of specific genes in the human gut microbiome. Bioinformatics 2015; 31(20):3368-70; PMID:26104745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sharpton TJ. An introduction to the analysis of shotgun metagenomic data. Front Plant Sci 2014; 5:209; PMID:24982662; http://dx.doi.org/ 10.3389/fpls.2014.00209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Faith JJ, Ahern PP, Ridaura VK, Cheng J, Gordon JI. Identifying gut microbe-host phenotype relationships using combinatorial communities in gnotobiotic mice. Sci Transl Med 2014; 6:220ra11; PMID:24452263; http://dx.doi.org/ 10.1126/scitranslmed.3008051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao XD, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, et al.. The transcriptional network for mesenchymal transformation of brain tumours. Nature 2010; 463:318-U68; PMID:20032975; http://dx.doi.org/ 10.1038/nature08712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Akavia UD, Litvin O, Kim J, Sanchez-Garcia F, Kotliar D, Causton HC, Pochanard P, Mozes E, Garraway LA, Pe'er D. An Integrated Approach to Uncover Drivers of Cancer. Cell 2010; 143:1005-17; PMID:21129771; http://dx.doi.org/ 10.1016/j.cell.2010.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Mine KL, Shulzhenko N, Yambartsev A, Rochman M, Sanson GFO, Lando M, Varma S, Skinner J, Volfovsky N, Deng T, et al.. Gene network reconstruction reveals cell cycle and antiviral genes as major drivers of cervical cancer. Nat Commun 2013; 4:1806; PMID:23651994; http://dx.doi.org/ 10.1038/ncomms2693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Dong X, Yambartsev A, Ramsey S, Thomas L, Shulzhenko N, Morgun A. Reverse enGENEering of Regulatory Networks from Big Data: A Roadmap for Biologists. Bioinform Biol Insights 2015; 9:61-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Skinner J, Kotliarov Y, Varma S, Mine KL, Yambartsev A, Simon R, Huyen Y, Morgun A. Construct and Compare Gene Coexpression Networks with DAPfinder and DAPview. BMC Bioinformatics 2011; 12:286; PMID:21756334; http://dx.doi.org/ 10.1186/1471-2105-12-286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Borgatti SP, Everett MG. Network analysis of 2-mode data. Social Networks 1997; 19:243-69; http://dx.doi.org/ 10.1016/S0378-8733(96)00301-2 [DOI] [Google Scholar]
- [52].Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol 2011; 12:R60; PMID:21702898; http://dx.doi.org/ 10.1186/gb-2011-12-6-r60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lingner T, Asshauer KP, Schreiber F, Meinicke P. CoMet–a web server for comparative functional profiling of metagenomes. Nucleic Acids Res 2011; 39:W518-23; PMID:21622656; http://dx.doi.org/ 10.1093/nar/gkr388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Parks DH, Beiko RG. Identifying biologically relevant differences between metagenomic communities. Bioinformatics 2010; 26:715-21; PMID:20130030; http://dx.doi.org/ 10.1093/bioinformatics/btq041 [DOI] [PubMed] [Google Scholar]
- [55].Arndt D, Xia J, Liu Y, Zhou Y, Guo AC, Cruz JA, Sinelnikov I, Budwill K, Nesbø CL, Wishart DS. METAGENassist: a comprehensive web server for comparative metagenomics. Nucleic Acids Res 2012; 40:W88-95; PMID:22645318; http://dx.doi.org/ 10.1093/nar/gks497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].McMurdie PJ, Holmes S. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol 2014; 10:e1003531; PMID:24699258; http://dx.doi.org/ 10.1371/journal.pcbi.1003531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, et al.. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 2015; 517:205-8; PMID:25337874; http://dx.doi.org/ 10.1038/nature13828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, Wu X, Li J, Tang L, Li Y, et al.. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med 2015; 21:895-905; PMID:26214836; http://dx.doi.org/ 10.1038/nm.3914 [DOI] [PubMed] [Google Scholar]
- [59].Stein RR, Bucci V, Toussaint NC, Buffie CG, Ratsch G, Pamer EG, Sander C, Xavier JB. Ecological modeling from time-series inference: insight into dynamics and stability of intestinal microbiota. PLoS Comput Biol 2013; 9:e1003388; PMID:24348232; http://dx.doi.org/ 10.1371/journal.pcbi.1003388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, Codelli JA, Chow J, Reisman SE, Petrosino JF, et al.. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013; 155:1451-63; PMID:24315484; http://dx.doi.org/ 10.1016/j.cell.2013.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, Rostron T, Cerundolo V, Pamer EG, Abramson SB, et al.. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2013; 2:e01202; PMID:24192039; http://dx.doi.org/ 10.7554/eLife.01202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Moon C, Baldridge MT, Wallace MA, Burnham CA, Virgin HW, Stappenbeck TS. Vertically transmitted faecal IgA levels determine extra-chromosomal phenotypic variation. Nature 2015; 521:90-3; PMID:25686606; http://dx.doi.org/ 10.1038/nature14139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An Immunomodulatory Molecule of Symbiotic Bacteria Directs Maturation of the Host Immune System. Cell 2005; 122:107-18; PMID:16009137; http://dx.doi.org/ 10.1016/j.cell.2005.05.007 [DOI] [PubMed] [Google Scholar]
- [64].Bouskra D, Brézillon C, Bérard M, Werts C, Varona R, Boneca IG, Eberl G. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 2008; 456:507-10; PMID:18987631; http://dx.doi.org/ 10.1038/nature07450 [DOI] [PubMed] [Google Scholar]
- [65].Hooper LV. Molecular Analysis of Commensal Host-Microbial Relationships in the Intestine. Science 2001; 291:881-4; PMID:11157169; http://dx.doi.org/ 10.1126/science.291.5505.881 [DOI] [PubMed] [Google Scholar]
- [66].Fei N, Zhao L. An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice. ISME J 2012; 7:880-4; PMID:23235292; http://dx.doi.org/ 10.1038/ismej.2012.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Geuking MB, Cahenzli J, Lawson MA, Ng DC, Slack E, Hapfelmeier S, McCoy KD, Macpherson AJ. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity 2011; 34:794-806; PMID:21596591; http://dx.doi.org/ 10.1016/j.immuni.2011.03.021 [DOI] [PubMed] [Google Scholar]
- [68].Carvalho FA, Koren O, Goodrich JK, Johansson ME, Nalbantoglu I, Aitken JD, Su Y, Chassaing B, Walters WA, González A, et al.. Transient Inability to Manage Proteobacteria Promotes Chronic Gut Inflammation in TLR5-Deficient Mice. Cell Host Microbe 2012; 12:139-52; PMID:22863420; http://dx.doi.org/ 10.1016/j.chom.2012.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kamada N, Kim YG, Sham HP, Vallance BA, Puente JL, Martens EC, Núñez G. Regulated Virulence Controls the Ability of a Pathogen to Compete with the Gut Microbiota. Science 2012; 336:1325-9; PMID:22582016; http://dx.doi.org/ 10.1126/science.1222195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kamada N, Kim YG, Sham HP, Vallance BA, Puente JL, Martens EC, Núñez G. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science 2012; 336:1325-9; PMID:22582016; http://dx.doi.org/ 10.1126/science.1222195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, Molina DA, Salcedo R, Back T, Cramer S, et al.. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013; 342:967-70; PMID:24264989; http://dx.doi.org/ 10.1126/science.1240527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Nguyen TL, Vieira-Silva S, Liston A, Raes J. How informative is the mouse for human gut microbiota research? Dis Model Mech 2015; 8:1-16; PMID:25561744; http://dx.doi.org/ 10.1242/dmm.017400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Chung H, Pamp SJ, Hill JA, Surana NK, Edelman SM, Troy EB, Reading NC, Villablanca EJ, Wang S, Mora JR, et al.. Gut Immune Maturation Depends on Colonization with a Host-Specific Microbiota. Cell 2012; 149:1578-93; PMID:22726443; http://dx.doi.org/ 10.1016/j.cell.2012.04.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, et al.. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013; 341:1241214; PMID:24009397; http://dx.doi.org/ 10.1126/science.1241214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Smith MI, Yatsunenko T, Manary MJ, Trehan I, Mkakosya R, Cheng J, Kau AL, Rich SS, Concannon P, Mychaleckyj JC, et al.. Gut microbiomes of Malawian twin pairs discordant for kwashiorkor. Science 2013; 339:548-54; PMID:23363771; http://dx.doi.org/ 10.1126/science.1229000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Thaiss CA, Zeevi D, Levy M, Zilberman-Schapira G, Suez J, Tengeler AC, Abramson L, Katz MN, Korem T, Zmora N, et al.. Transkingdom control of microbiota diurnal oscillations promotes metabolic homeostasis. Cell 2014; 159:514-29; PMID:25417104; http://dx.doi.org/ 10.1016/j.cell.2014.09.048 [DOI] [PubMed] [Google Scholar]
- [77].Jenner RG, Young RA. Insights into host responses against pathogens from transcriptional profiling. Nat Rev Microbiol 2005; 3:281-94; PMID:15806094; http://dx.doi.org/ 10.1038/nrmicro1126 [DOI] [PubMed] [Google Scholar]
- [78].Mack DR, Michail S, Wei S, McDougall L, Hollingsworth MA. Probiotics inhibit enteropathogenic E. coli adherence in vitro by inducing intestinal mucin gene expression. Am J Physiol 1999; 276:G941-50; PMID:10198338 [DOI] [PubMed] [Google Scholar]
- [79].Duary RK, Batish VK, Grover S. Immunomodulatory activity of two potential probiotic strains in LPS-stimulated HT-29 cells. Genes Nutr 2014; 9:398; PMID:24682881; http://dx.doi.org/ 10.1007/s12263-014-0398-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Christensen HR, Frokiaer H, Pestka JJ. Lactobacilli Differentially Modulate Expression of Cytokines and Maturation Surface Markers in Murine Dendritic Cells. J Immunol 2002; 168:171-8; PMID:11751960; http://dx.doi.org/ 10.4049/jimmunol.168.1.171 [DOI] [PubMed] [Google Scholar]
- [81].Fink LN, Zeuthen LH, Christensen HR, Morandi B, Frokiaer H, Ferlazzo G. Distinct gut-derived lactic acid bacteria elicit divergent dendritic cell-mediated NK cell responses. Int Immunol 2007; 19:1319-27; PMID:17951600; http://dx.doi.org/ 10.1093/intimm/dxm103 [DOI] [PubMed] [Google Scholar]