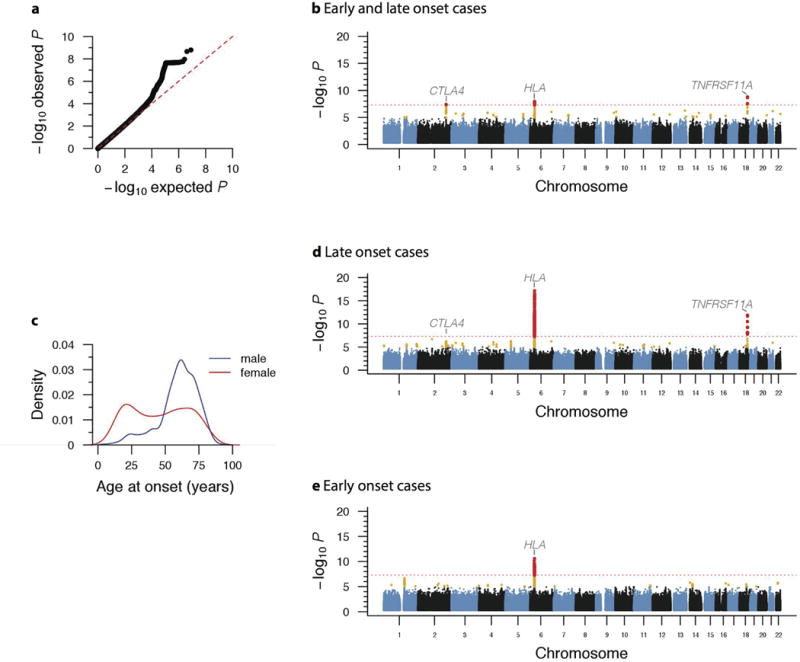
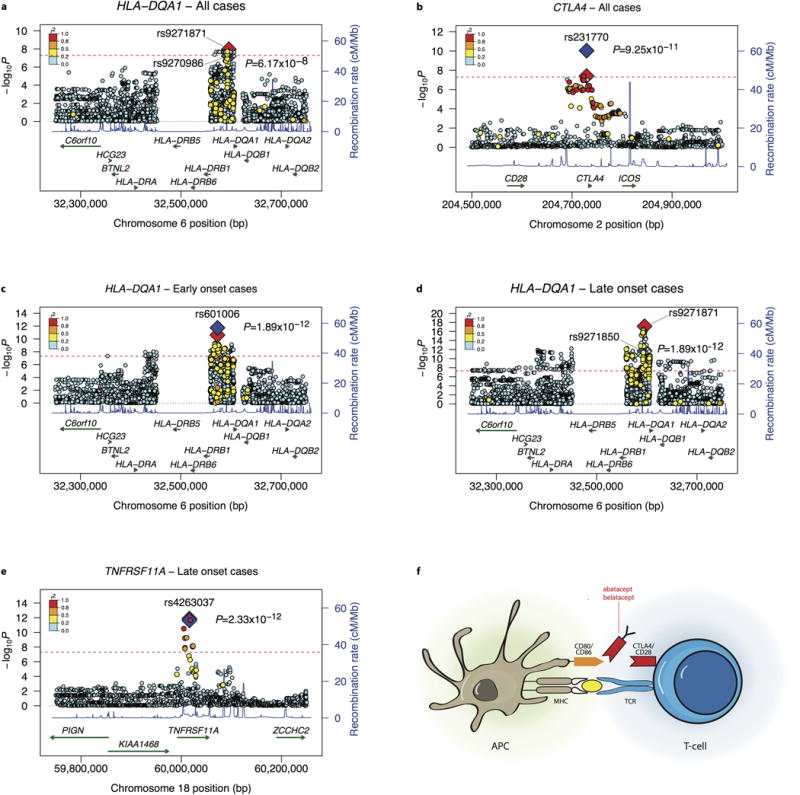
Alan E Renton
Alan E Renton, PhD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Hannah A Pliner
Hannah A Pliner, BS
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Carlo Provenzano
Carlo Provenzano, MD, PhD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Amelia Evoli
Amelia Evoli, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Roberta Ricciardi
Roberta Ricciardi, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Michael A Nalls
Michael A Nalls, PhD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Giuseppe Marangi
Giuseppe Marangi, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Yevgeniya Abramzon
Yevgeniya Abramzon
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Sampath Arepalli
Sampath Arepalli, MS
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Sean Chong
Sean Chong, BS
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Dena G Hernandez
Dena G Hernandez, MS
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Janel O Johnson
Janel O Johnson, PhD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Emanuela Bartoccioni
Emanuela Bartoccioni, PhD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Flavia Scuderi
Flavia Scuderi
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Michelangelo Maestri
Michelangelo Maestri, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
J Raphael Gibbs
J Raphael Gibbs, MS
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Edoardo Errichiello
Edoardo Errichiello
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Adriano Chiò
Adriano Chiò, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Gabriella Restagno
Gabriella Restagno, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Mario Sabatelli
Mario Sabatelli, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Mark Macek
Mark Macek, MS
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Sonja W Scholz
Sonja W Scholz, MD, PhD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Andrea Corse
Andrea Corse, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Vinay Chaudhry
Vinay Chaudhry, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Michael Benatar
Michael Benatar, MD, PhD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Richard J Barohn
Richard J Barohn, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
April McVey
April McVey, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Mamatha Pasnoor
Mamatha Pasnoor, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Mazen M Dimachkie
Mazen M Dimachkie, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Julie Rowin
Julie Rowin, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
John Kissel
John Kissel, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Miriam Freimer
Miriam Freimer, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Henry J Kaminski
Henry J Kaminski, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Donald B Sanders
Donald B Sanders, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Bernadette Lipscomb
Bernadette Lipscomb, RN
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Janice M Massey
Janice M Massey, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Manisha Chopra
Manisha Chopra, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
James F Howard Jr
James F Howard Jr, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Wilma J Koopman
Wilma J Koopman, RN
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Michael W Nicolle
Michael W Nicolle, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Robert M Pascuzzi
Robert M Pascuzzi, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Alan Pestronk
Alan Pestronk, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Charlie Wulf
Charlie Wulf
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Julaine Florence
Julaine Florence, PhD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Derrick Blackmore
Derrick Blackmore, BSc
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Aimee Soloway
Aimee Soloway, RN
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Zaeem Siddiqi
Zaeem Siddiqi, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Srikanth Muppidi
Srikanth Muppidi, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Gil Wolfe
Gil Wolfe, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
David Richman
David Richman, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Michelle M Mezei
Michelle M Mezei, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Theresa Jiwa
Theresa Jiwa
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Joel Oger
Joel Oger, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Daniel B Drachman
Daniel B Drachman, MD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1,
Bryan J Traynor
Bryan J Traynor, MD, PhD
1Neuromuscular Diseases Research Unit, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Renton, Pliner, Marangi, Abramzon, Johnson, Errichiello, Traynor); Institute of General Pathology, Catholic University, Rome, Italy (Provenzano, Bartoccioni, Scuderi); Institute of Neurology, Catholic University, Rome, Italy (Evoli, Sabatelli); Department of Neuroscience, Cisanello Hospital, University of Pisa, Pisa, Italy (Ricciardi, Maestri); Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Nalls); Institute of Medical Genetics, Catholic University, Rome, Italy (Marangi); Genomics Technology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Arepalli, Chong, Hernandez); Computational Biology Core, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Porter Neuroscience Research Center, Bethesda, Maryland (Gibbs); Rita Levi Montalcini Department of Neuroscience, University of Turin, Turin, Italy (Errichiello, Chiò); Molecular Genetics Unit, Department of Clinical Pathology, ASO OIRM-S Anna, Turin, Italy (Restagno); Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland (Macek, Scholz, Corse, Chaudhry, Drachman, Traynor); Department of Neurology, University of Miami, Miami, Florida (Benatar); Department of Neurology, University of Kansas Medical Center, Kansas City (Barohn, McVey, Pasnoor, Dimachkie); Department of Neurology, University of Illinois College of Medicine, Chicago (Rowin); Department of Neurology, Ohio State University Medical Center, Columbus (Kissel, Freimer); Department of Neurology, George Washington University, Washington, DC (Kaminski); Department of Neurology, Duke University Medical Center, Durham, North Carolina (Sanders, Lipscomb, Massey); Department of Neurology, University of North Carolina, Chapel Hill (Chopra, Howard); Department of Clinical Neurosciences, London Health Sciences Centre, London, Ontario, Canada (Koopman, Nicolle); Department of Neurology, Indiana University–Purdue University, Indianapolis (Pascuzzi); Department of Neurology, Washington University School of Medicine, St Louis, Missouri (Pestronk, Wulf, Florence); Department of Medicine, University of Alberta Hospital, Edmonton, Alberta, Canada (Blackmore, Soloway, Siddiqi); Department of Neurology, University at Buffalo SMBS, State University of New York, Buffalo (Muppidi, Wolfe); Department of Neurology, University of California, Davis Medical Center (Richman); Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada (Mezei, Jiwa, Oger)
1