

Abstract
Ammonia Borane (AB) has high hydrogen density (19.6 wt. %), and can, in principle, release up to 3 equivalents of H2 under mild catalytic conditions. A limited number of catalysts are capable of non-hydrolytic dehydrogenation of AB beyond 2 equivalents of H2 under mild conditions, but none of these is shown directly to derivatise borazine, the product formed after 2 equivalents of H2 are released. We present here a high productivity ruthenium-based catalyst for non-hydrolytic AB dehydrogenation that is capable of borazine dehydrogenation, and thus exhibits among the highest H2 productivity reported to date for anhydrous AB dehydrogenation. At 1 mol% loading, (phen)Ru(OAc)2(CO)2 (1) effects AB dehydrogenation through 2.7 equivalents of H2 at 70°C, is robust through multiple charges of AB, and is water and air stable. We further demonstrate that catalyst 1 has the ability both to dehydrogenate borazine in isolation and dehydrogenate AB itself. This is important, both because borazine derivatisation is productivity-limiting in AB dehydrogenation and because borazine is a fuel cell poison that is commonly released in H2 production from this medium.
Graphic Abstract
Introduction
Dehydrogenation of ammonia borane (AB, H3N-BH3) has been studied extensively as an approach for high-capacity hydrogen storage, because AB has high hydrogen density (19.6 wt%) and the ability to release H2 under mild conditions.1 Although catalytic hydrolysis is well known and efficient for H2 production from AB,2 non-hydrolytic dehydrogenation is a more desirable approach, because it (1) enables more facile re-reduction of dehydrogenated spent fuel3 and (2) minimizes evolution of ammonia, a hydrogen fuel cell poison, in the eluent gas stream.4, 1b Several transition metal catalysts are active for non-hydrolytic AB dehydrogenation, including complexes of iron,5 molybdenum,6 iridium,7 rhodium,8 nickel,9 palladium,10 and ruthenium.11, 12 Catalyst systems reported to date fit one of two classes:13 (1) those that release 1 equivalent of hydrogen quickly14, 7 and (2) those that release 2 or more equivalents slowly.9, 10a, 12, 15 The latter are known to proceed through (or stop at) borazine, N3B3H6, as an intermediate with its subsequent conversion to polyborazylene as a slow step in the overall mechanism of hydrogen evolution.16 This is problematic because (1) slow borazine derivatization limits H2 productivity,12 (2) borazine, which boils at 55 ºC, is poisonous to fuel cells, and (3) borazine is known to coordinate metals17 and deactivate some AB dehydrogenation catalysts.16
Maximizing the H2 release efficiency is highly desirable, and only a few catalysts achieved a high release extent of 2.5 equivalents or greater (Figure 1). Currently, Baker’s nickel-based system supported by Ender’s carbene is the highest extent of H2 release from AB reported to date. The Guan Fe-POCOP catalyst uses inexpensive iron but dehydrogenates fairly slowly and reacts through 2.5 equiv. H2. The Agapie Mo catalyst has the same productivity, and is limited by air and water sensitivity. Further, Wegner has recently presented a metal-free catalyst that is capable of releasing ca. 2.5 equiv. of H2. Despite high extent of H2 release observed with these catalytic systems, no study of direct borazine consumption was reported with any of them, and borazine remains a significant constituent of the reactive medium at the end of the AB dehydrogenation reaction for each.
Fig. 1.

Catalysts that dehydrogenate AB over 2.5 equiv.
In the absence of a catalyst, borazine undergoes slow dehydrogenative BN cross-linking (48–60 hours at 70 °C under reduced pressure with periodic degassing) to polyborazylene.18 This rate is insufficient to prevent borazine accumulation under catalytic hydrogen evolution conditions. Thus, there is a desire to find a catalyst system that will dehydrogenate borazine at a rate faster than (or commensurate with) the rate at which it reacts with ammonia borane itself. No such system has yet to be reported. We report here the reactivity of ruthenium complex 1 (Figure 2) with AB to produce H2 gas with high efficiency. 1 and its isomers are known to catalyse hydrogenation of ketones, olefins, and alkynes19 and hydroformylate olefins,20 but its reactivity with AB has not been studied. 1 not only dehydrogenates AB, but also reacts with borazine to form polyborazylene, therefore exceeding the extent of dehydrogenation available with other systems and decreasing the amount of fuel cell poison in the gaseous eluent stream as less volatile polyborazylene. In addition to decreasing the accumulation of borazine in the dehydrogenation reactor, conditions based on 1 yield unusually high productivity, 2.7 equivalents of H2 at mild reaction temperature, and are air and water tolerant.
Fig. 2.

Ru catalysts for AB dehydrogenation. (phen)Ru(OAc)2(CO)2 (1), (phen)RuCl2(CO)2 (2), Shvo’s catalyst (3), Shvo oxidised form dimer (4).
Results and Discussion
Catalyst Development
We have previously demonstrated that the Shvo catalyst can lose its tetraphenylcyclopentadienone (CPD) ligand from the metal centre in the presence of bidentate nitrogen ligands such as 1,10-phenanthroline (phen) shown in Scheme 1.21 We suspect that the species produced by the expulsion of the CPD ligand contributes to the increase in rate of AB dehydrogenation. This species potentially consists of a ruthenium centre supported by two carbonyl ligands and the bidentate bis(nitrogen) ligand. We therefore investigated ruthenium scaffolds composed of these constituents. Whereas these square planar ruthenium complexes are not stable to isolation, we generate analogous compounds in situ. We reasoned that treatment of the known complex (phen)RuCl2(CO)2 (2)22 with TlOTf and ammonia borane would generate a reactive (phen)Ru(CO)2-based fragment.21 While 10% of 2 with 2 equivalents of TlOTf will liberate 2.5–2.7 equivalents of H2 from AB, the high loading of metals (both Ru and Tl) is undesirable. Further, this reaction is not as effective at lower catalyst loadings. We eliminated the usage of thallium by replacing the chlorine ligands with acetates (1, Figure 2)22b with the expectation that acetate groups should dissociate easily from the metal centre in the presence of an excess of boron with this dissociation driven by the strength of boron-acetate bonds.
Scheme 1.

Ruthenium dimer 4 loses CPD in the presence of bidentate nitrogen ligands such as phen.
Reactivity of 1 with AB
1 dehydrogenates AB efficiently at low catalyst loading, down to 1 mol%, producing 2.4–2.7 equiv. of hydrogen, as shown in Figure 3. The catalyst system is robust, capable of producing a similarly high extent of H2 release (2.6, 2.5, 2.4) in each of the multiple AB reloadings. Further, we compared the rates of catalytic reactions prepared in the glove box under nitrogen atmosphere and an analogous sample of 1 suspended in solvent and immersed in an ultrasonic bath in open air for 20 minutes before AB addition. We find that exposure of the catalyst to air and water in the atmosphere neither slows nor accelerates dehydrogenation; rather, these runs have analogous rates (Supplementary Information).
Fig. 3.

Left: H2 production from 1% of 1 at 70°C in diglyme releasing 2.7 equiv. Right: 11B kinetic profile of the consumption of AB catalyzed by 1% of 1 in 2:1 diglyme/benzene-d6.
Reactivity of 1 with Borazine
Treatment of AB with 1 results in the formation of a family of AB dehydrogenation products (Scheme 2) that has homology to those formed when the dehydrogenation is conducted with the Shvo catalyst (3),16 except that the reaction does not stop at borazine, but goes on to polyborazylene materials (9). Borazine (8) is the exclusive boron-nitrogen material formed when 3 is used as the catalyst, and it is the principal material that accumulates after two equivalents of H2 are produced. We expect that the Shvo system can not derivatise borazine because it is restricted to an outer sphere hydrogen transfer mechanism.
Scheme 2.

Typical BN byproducts seen in AB dehydrogenation reactions.
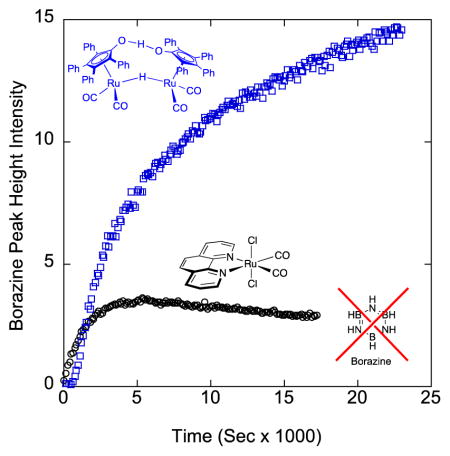
Unlike 3 and its derivatives,21 1 reacts with isolated borazine at 70°C, yielding polyborazylene as the major byproduct. We can observe this directly by 11B NMR (Figure 4). In this experiment, borazine was isolated by a vacuum transfer of the volatiles in a spent 3-catalysed AB dehydrogenation reaction.12a After the transfer of borazine and solvents into a J-Young NMR tube preloaded with 1, a 11B NMR spectrum was taken, and the solution was heated to 70 °C (see experimental section for further details). After heating for 24 hours, borazine (31 ppm) was converted, ca. 70%, relative to the external 11B standard (0 ppm) to give signals consistent with cross-linked polyborazylene and other unsaturated B-N byproducts. These are indicated by broad signals in the 11B NMR spectra from 24–34 ppm. Visual inspection of the NMR tube after the reaction shows insoluble, gelatine-like, white material, which is consistent with the formation of polymeric B-N species and accounts for the loss in integration of 11B products observed by NMR. In contrast, the background thermal decomposition reaction under these conditions gives only ca. 15% after 24 hours (see Supplementary Information).
Fig. 4.

Reaction of 1 with borazine (8) in 2:1 diglyme/benzene-d6. Bottom: initial 11B spectrum of 1 and borazine at room temperature. Top: 11B spectrum of 1 and borazine after 24 hrs at 70°C. Boron external standard BF3-OEt2 is at 0 ppm, and borazine is the only boron species present in the bottom spectrum at 31 ppm. In the top spectrum, borazine peak height has decreased and other broad boron species that are indicative of polyborazylene appear after 24 hrs. Areas under the peaks are integrated relative to the external standard BF3-OEt2 peak set to 1.00.
Reaction Intermediates
We observe by 11B NMR that reactions catalysed by 1 generate boron intermediates common in AB dehydrogenation reactions with catalysts outlined in Figure 1, which include μ-aminodiborane (5), cyclotriborazane (6), aminoborane cyclic tetramer (7), and borazine (8) (Scheme 2). However, at 1% loading of 1, we observe the appearance of polyborazylene (9) as early as the first 20 minutes into AB consumption, along with intermediates 6–8. This suggests that as borazine is produced, another mechanism is concurrently dehydrogenating it into polyborazylene. The borazine concentration builds steadily until approximately 50% of AB is consumed, and then the growth of the borazine peak tapers off followed by its conversion to polyborazylene. When we compare 11B NMR spectra of 1-catalysed AB dehydrogenation (10 mol% Ru atom loading) with our previously-reported reaction catalysed by the same loading of 3,12a we observe less borazine (31 ppm) build-up and more polyborazylene (24–34 ppm) in the reaction catalysed by 1 (Figure 5a). Plotting normalized peak height of borazine against time for reactions catalysed by 10 mol % of 1 and 3 (Figure 5b), we observe the decrease of borazine over time with 1, while the borazine peak rises in the reaction catalysed by 3.
Fig. 5.

a. Comparison of end-of-reaction 11B NMR spectra under representative conditions. Top: AB dehydrogenation catalysed by 10% Ru atom of 1 after 1.75 hours in 2:1 diglyme/benzene-d6 at 70°C. Bottom: AB dehydrogenation catalysed by 10% Ru atom of 3 after 1.75 hours in 2:1 diglyme/benzene-d6 at 70°C. Note the larger proportion of polyborazylene in the top spectra (24–34 ppm) compared to the bottom spectra. Boron standard (BF3-OEt2 inset tube) is at 0 ppm. Areas under the peaks are integrated relative to the external standard BF3-OEt2 peak set to 1.00. b. Peak height of borazine over time catalysed by 10% Ru atom of 1 (left) and 3 (right).
Homogeneity
In order to probe the homogeneity or heterogeneity of the dehydrogenation catalysis, we conducted several tests. We added ca. 100 μL of elemental mercury to the reaction conducted at 10% catalyst loading and monitored AB consumption via 11B kinetics in a mercury drop test of homogeneity. While there was a small drop in rate, the reaction proceeded similarly to the parent reaction, completing in approximately the same amount of time (Figure 6). This result suggests homogeneous catalysis, even though there are observable heterogeneous materials in the reaction. We therefore suspect that we have a homogeneous active species working in a heterogeneous suspension.
Fig. 6.
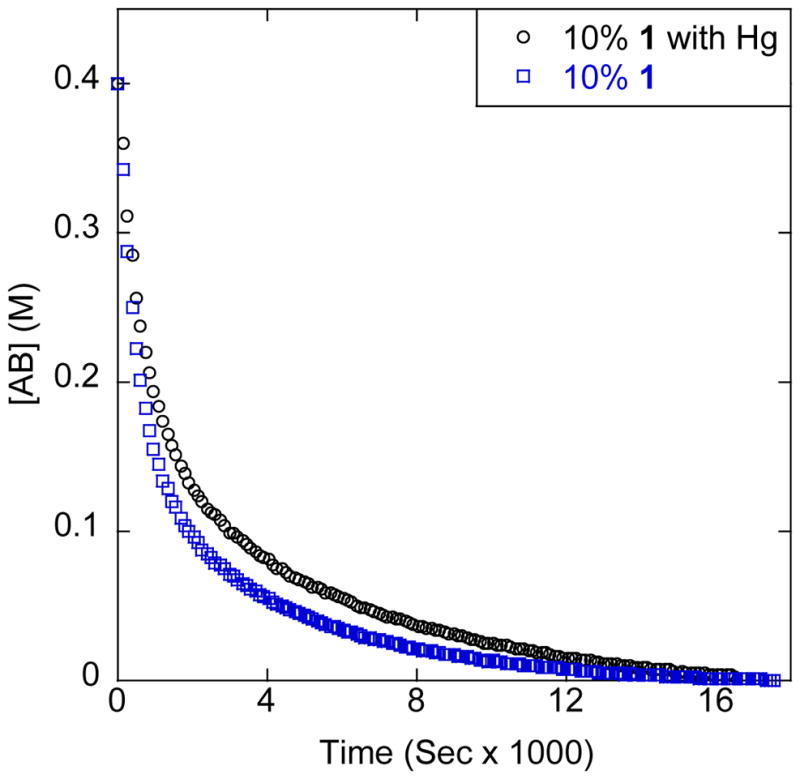
Hg addition homogeniety test in 2:1 diglyme/benzene-d6 at 70ºC. Black circles: 10 mol% of 1 with Hg. Blue squares: 10% of 1.
Conclusions
In summary we present here a high productivity non-hydrolytic ammonia borane dehydrogenation system releasing up to 2.7 equiv. of H2 at low catalyst loading. It has desirable kinetic properties, air and water tolerance, and reusability. We also show that this catalyst has reactivity with borazine itself, thus minimizing borazine concentration in the gas eluent stream of the H2 generation reactor.
Experimental
All air and water sensitive procedures were carried out either in a Vacuum Atmosphere glove box under nitrogen (0.5–10 ppm O2 for all manipulations) or using standard Schlenk techniques under nitrogen. Deuterated NMR solvents were purchased from Cambridge Isotopes Laboratories. Benzene-d6 and diethylene glycol dimethyl ether (diglyme, Alfa Aesar) were dried over sodium benzophenone ketyl and distilled prior to use. Ammonia borane (NH3BH3, AB) was purchased from Sigma Aldrich and used under N2 atmosphere only without further purifications. RuCl3•3H2O was purchased from Pressure Chemical Co. and used without further purifications. Shvo’s catalyst (3) was purchased from Strem Chemicals and used without further purifications. Acetic acid was purchased from EMD Millipore and used without further purification. Formic acid, 1,10-phenanthroline, and silver acetate were purchased from Alfa Aesar and used without further purification. [RuCl2(CO)2]n, (phen)RuCl2(CO)2 (2) and (phen)Ru(OAc)2(CO)2 (1) were synthesized according to literature procedures (more details in the Supplementary Information)23.22 1H and 11B NMR spectra were obtained on Varian 600 MHz (600 MHz in 1H, 192 MHz in 11B), 500 MHz, and 400 MHz spectrometers with chemical shifts reported in units of ppm. All 1H chemical shifts were referenced to the residual 1H solvent (relative to TMS). All 11B chemical shifts were referenced to a BF3-OEt2 in diglyme in a co-axial external standard (0 ppm). 11B spectra are phased, baseline corrected, and backwards linear predicted 3–8 points. NMR spectra were taken in 8″ J-Young tubes (Wilmad) with Teflon valve plugs. FTIR were taken on KBr salt plate, or KBr pellet on a Bruker Vertex 80 FTIR.
Hydrogen Quantification
In a typical reaction, 7.7 mg AB (0.25 mmol) was combined with catalyst (1–10 mol%) in a 2 mL Schlenk bomb equipped with a Teflon stir bar while in a glove box under nitrogen. Diglyme (0.6 mL) was added to the flask to create a light yellow mixture. The eudiometer was constructed as follows: The side arm of the valve was connected to a piece of Tygon tubing, which was connected to a 3-way valve. Center of the 3-way valve was connected to the Schlenk line to allow vacuum and nitrogen purge of the apparatus. The last valve was connected to Tygon tubing that was adapted to 20 gauge (0.03″) Teflon tubing with a needle. The tubing was threaded through open end of a burette that was sealed with a Teflon stopcock on the other end. The burette was filled with water. The entire apparatus was then inverted into a 500 mL Erlenmeyer filled with water and clamped onto a metal ring stand. Opening the 3-way-valve to the Schlenk line and the bomb (reaction still closed and wrapped in foil at room temperature), the space in the 3-way valve and tubing was vacuumed and refilled with N2 for 5 minutes twice and then opening all 3 valves to the line, N2 purge was conducted through out the burette for 10 minutes. Then the 3-way valve was turned so that only the bomb and the side connected to the burette were open, and N2 was turned off. The water in the burette was pulled upwards by a pipette filler bulb and the initial volume was recorded. The bomb was then submerged into a 70 ºC oil bath for 2 minutes to allow the temperature of the system to equilibrate. The reactor’s valve was opened to release gas from the reactor headspace while heating in a regulated oil bath. The volume of liberated gas was recorded periodically until gas evolution ceased. Liberated hydrogen was quantified by recording its volume displacement in the eudiometer. Reaction proceeds from initial yellow to gray, to black suspension.
Kinetic Profiles of AB Dehydrogenation Recorded via 11B NMR
In a typical reaction, 7.7 mg of AB was combined with catalyst (1.0 – 10.0 mol%) in a J-Young NMR tube while in a glove box under nitrogen. The AB concentration and catalyst concentrations may be varied. Diglyme (0.4 mL) and benzened6 (0.2 mL) were added to the tube. The sample tube was immediately inserted into a temperature-equilibrated (70 °C), pre-shimmed, and pre-locked NMR, and the kinetic monitoring commenced. Disappearance of AB in the solution was monitored by the relative integration of its characteristic peak in the 11B spectrum (−22 ppm) and the BF3-OEt2 (0 ppm) standard. The acquisition involved a 1.84 s pulse sequence in which 16,384 complex points were recorded, followed by 1 s relaxation delay. To eliminate B–O peaks from the borosilicate NMR tube and probe, the 11B FIDs were processed with backward linear prediction. Safety note: caution should be used when carrying out these reactions, as the release of hydrogen can lead to sudden pressurization of reaction vessels.
Reactivity of 1 with Borazine
Borazine was generated in a J. Young NMR tube by the reaction of Shvo’s catalyst (6.7 mg, 2.5%, 0.0062 mmol) with AB (7.7 mg, 0.25 mmol) in 2:1 diglyme/benzene-d6 (0.4 mL and 0.2 mL respectively) in a 70ºC oil bath for 16–20 hours.12a,16 The NMR tube was connected via a flame-dried U-tube to another J. Young NMR tube containing 1 (1.2 mg, 0.0025 mmol), diglyme (0.4 mL), and 11B external standard. The volatiles (H2, borazine, benzene-d6) were transferred under static vacuum while the borazine-containing tube was heated gently with a heat gun while the receiving tube was submerged in liquid N2 until all of the ca. 0.2 mL of benzene was transferred to the receiving tube. The J. Young valves were closed and the U-tube system was flushed with N2. While the N2 line was closed and the U-tube is under N2, the receiving tube was opened briefly to return it to atmospheric pressure. 1H and 11B NMR spectra were taken, and then the borazine-containing tube was submerged in a 70 ºC oil bath for 24 hours. Peak heights relative to the external standard (BF3-OEt2 in diglyme (0 ppm)) were compared since polyborazylene and borazine peaks overlap in the 11B spectrum and integrations could not be done accurately. The procedure for the borazine background reaction was the same, except for the absence of 1 in the receiving tube (See Supplementary Information for graphical representation).
Catalyst Reuse Reactions
To test the reusability of catalyst 1, we studied the production of H2 gas by eudiometry for successive runs with 1.0 mol% catalyst. For run 1, we added 7.7 mg AB, 1.2 mg 1, and 0.6 mL diglyme to a 2 mL Schlenk flask equipped with a small stir bar. The reaction was heated at 70 °C in a regulated oil bath and reaction progress monitored by displacement of water by H2 gas in an inverted 50 mL burette. For successive runs, we added 7.7 mg AB and 0.2 mL diglyme and repeated the reaction at 70 °C. Pseudo-first order rate constants for H2 productions over these runs are 12.1, 10.8, and 7.0 × 10−5 s−1.
Supplementary Material
Acknowledgments
This work was sponsored by the National Science Foundation (CHE-1054910) and the Hydrocarbon Research Foundation. We are grateful to the National Science Foundation (DBI-0821671, CHE-0840366), the National Institutes of Health (1 S10 RR25432), and the University of Southern California for sponsorship of NMR spectrometers. Fellowship assistance from The Sonosky Foundation of the USC Wrigley Institute (X.Z.), Dornsife College (L.K.), and USC undergraduate research fellowship program (L.K.) is gratefully acknowledged.
Footnotes
Electronic Supplementary Information (ESI) available: Details of experimental and spectral data. See DOI: 10.1039/x0xx00000x
No competing financial interests have been declared.
References
- 1.(a) Staubitz A, Robertson APM, Manners I. Chem Rev. 2010;110:4079–4124. doi: 10.1021/cr100088b. [DOI] [PubMed] [Google Scholar]; (b) Stephens FH, Pons V, Baker RT. Dalton Trans. 2007:2613–2626. doi: 10.1039/b703053c. [DOI] [PubMed] [Google Scholar]; (c) Marder TB. Angew Chem Int Ed. 2007;46:8116–8118. doi: 10.1002/anie.200703150. [DOI] [PubMed] [Google Scholar]; (d) Hamilton CW, Baker RT, Staubitz A, Manners I. Chem Soc Rev. 2009;38:279–293. doi: 10.1039/b800312m. [DOI] [PubMed] [Google Scholar]; (e) Baitalow F, Bauman J, Wolf G, Jaenicke-Rossler K, Leitner G. Thermochim Acta. 2002;391:159–168. [Google Scholar]; (f) Wolf G, Baumann J, Baitalow F, Hoffmann FP. Thermochim Acta. 2000;343:19–25. [Google Scholar]; (g) Wang JS, Geanangel RA. Inorg Chim Acta. 1988;148:185–190. [Google Scholar]; (h) Bluhm ME, Bradley MG, Butterick R, III, Kusari U, Sneddon LG. J Am Chem Soc. 2006;128:7748–7749. doi: 10.1021/ja062085v. [DOI] [PubMed] [Google Scholar]; (i) Rassat SG, Aardahl CL, Autrey T, Smith RS. Energy Fuels. 2010;24:2596–2606. [Google Scholar]; (j) Kalviri HA, Gartner F, Ye G, Korobkov I, Baker RT. Chem Sci. 2015;6:618–624. doi: 10.1039/c4sc02710h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Yan JM, Zhang XB, Akita T, Haruta M, Xu Q. J Am Chem Soc. 2010;132:5326–5327. doi: 10.1021/ja910513h. [DOI] [PubMed] [Google Scholar]; (b) Jiang HL, Umegaki T, Akita T, Zhang XB, Haruta M, Xu Q. Chem Eur J. 2010;16:3132–3137. doi: 10.1002/chem.200902829. [DOI] [PubMed] [Google Scholar]; (c) Ramachandran PV, Gagare PD. Inorg Chem. 2007;46:7810–7817. doi: 10.1021/ic700772a. [DOI] [PubMed] [Google Scholar]; Nelson DJ, Truscott BJ, Egbert JD, Nolan SP. Organometallics. 2013;32:3769–3772. [Google Scholar]
- 3.(a) Davis BL, Dixon DA, Garner EB, Gordon JC, Matus MH, Scott B. Angew Chem Int Ed. 2009;48:6812–6816. doi: 10.1002/anie.200900680. [DOI] [PubMed] [Google Scholar]; (b) Sutton AD, Burrell AK, Dixon DA, Garner EB, III, Gordon JC, Nakagawa T, Ott KC, Robinson JP, Vasiliu M. Science. 2011;331:1426–1429. doi: 10.1126/science.1199003. [DOI] [PubMed] [Google Scholar]
- 4.(a) Smythe NC, Gordon JC. Eur J Inorg Chem. 2010:509–521. [Google Scholar]; (b) Sutton AD, Burrell AK, Dixon DA, Gardner EB, Gordon JC, Nakagawa T, Ott KC, Robinson JP, Vasiliu M. Science. 2011;331:1426–1429. doi: 10.1126/science.1199003. [DOI] [PubMed] [Google Scholar]
- 5.(a) Vance JR, Robertson APM, Lee K, Manners I. Chem Eur J. 2011;17:4099–4103. doi: 10.1002/chem.201003397. [DOI] [PubMed] [Google Scholar]; (b) Baker RT, Gordon JC, Hamilton CW, Henson NJ, Lin PH, Maguire S, Murugesu M, Scott BL, Smythe NC. J Am Chem Soc. 2012;134:5598–5609. doi: 10.1021/ja210542r. [DOI] [PubMed] [Google Scholar]; (c) Bhattacharya P, Krause JA, Guan H. J Am Chem Soc. 2014;136:11153–11161. doi: 10.1021/ja5058423. [DOI] [PubMed] [Google Scholar]; (d) Glüer A, Förster M, Celinski VR, Schmedt auf der Günne J, Holthausen MC, Schneider S. ACS Catal. 2015;5:7214–7217. [Google Scholar]
- 6.Buss JA, Edouard GA, Cheng C, Shi J, Agapie T. J Am Chem Soc. 2014;136:11272–11275. doi: 10.1021/ja5059923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denny MC, Pons V, Hebdon TJ, Heinekey M, Goldberg KI. J Am Chem Soc. 2006;128:12048–12049. doi: 10.1021/ja062419g. [DOI] [PubMed] [Google Scholar]
- 8.Jaska CA, Temple K, Lough AJ, Manners I. J Am Chem Soc. 2003;125:9424–9434. doi: 10.1021/ja030160l. [DOI] [PubMed] [Google Scholar]; (b) Jaska CA, Manners I. J Am Chem Soc. 2004;126:1334–1335. doi: 10.1021/ja039162w. [DOI] [PubMed] [Google Scholar]; (c) Shrestha RP, Diyabalanage HVK, Semelsberger TA, Ott KC, Burrell AK. Int J Hydrogen Energy. 2009;34:2616–2621. [Google Scholar]; (d) Douglas TM, Chaplin AB, Weller AS. J Am Chem Soc. 2008;130:14432–14433. doi: 10.1021/ja806582n. [DOI] [PubMed] [Google Scholar]; (e) Alcaraz G, Sabo-Etienne S. Angew Chem Int Ed. 2010;49:7170–7179. doi: 10.1002/anie.201000898. [DOI] [PubMed] [Google Scholar]
- 9.Keaton RJ, Blacquiere JM, Baker RT. J Am Chem Soc. 2007;129:1844–1845. doi: 10.1021/ja066860i. [DOI] [PubMed] [Google Scholar]
- 10.(a) Kim SK, Han WS, Kim TJ, Kim TY, Nam SW, Mitoraj M, Piecoś Ł, Michalak A, Hwang SJ, Kang SO. J Am Chem Soc. 2010;132:9954–9955. doi: 10.1021/ja101685u. [DOI] [PubMed] [Google Scholar]; (b) Kim S-K, Hong S-A, Son H-J, Han W-SA, Hwang S-J, Kang SO. Dalton Trans. 2015;44:7373–7381. doi: 10.1039/c5dt00599j. [DOI] [PubMed] [Google Scholar]; (c) Kim SK, Hong SA, Son HJ, Han WS, Michalak A, Hwang SJ, Kang SO. Dalton Trans. 2015;44:7373–7381. doi: 10.1039/c5dt00599j. [DOI] [PubMed] [Google Scholar]
- 11.(a) Blaquiere N, Diallo-Garcia S, Gorelsky I, Black A, Fagnou K. J Am Chem Soc. 2008;130:14034–14035. doi: 10.1021/ja804235t. [DOI] [PubMed] [Google Scholar]; (b) Kaβ M, Fridrich A, Drees M, Schneider S. Angew Chem Int Ed. 2009;48:905–907. doi: 10.1002/anie.200805108. [DOI] [PubMed] [Google Scholar]; (c) Lu Z, Williams TJ. Chem Commun. 2014;50:5391–5393. doi: 10.1039/c3cc47384h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Conley BL, Williams TJ. Chem Commun. 2010;46:4815–4817. doi: 10.1039/c003157g. [DOI] [PubMed] [Google Scholar]; (b) Conley BL, Pennington-Boggio MK, Boz E, Williams TJ. Chem Rev. 2010;110:2294–2312. doi: 10.1021/cr9003133. [DOI] [PubMed] [Google Scholar]
- 13.Pons V, Baker RT, Szymczak NK, Heldebrant DJ, Linehan JC, Matus MH, Grant DJ, Dixon DA. Chem Commun. 2008;48:6597–5999. doi: 10.1039/b809190k. [DOI] [PubMed] [Google Scholar]
- 14.(a) Blaquiere N, Diallo-Garcia S, Gorelsky I, Black A, Fagnou K. J Am Chem Soc. 2008;130:14034–14035. doi: 10.1021/ja804235t. [DOI] [PubMed] [Google Scholar]; (b) Käβ M, Friedrich A, Drees M, Schneider S. Angew Chem Int Ed. 2009;48:905–907. doi: 10.1002/anie.200805108. [DOI] [PubMed] [Google Scholar]
- 15.(a) Chapman AM, Haddow MF, Wass DF. J Am Chem Soc. 2011;133:8826–8829. doi: 10.1021/ja201989c. [DOI] [PubMed] [Google Scholar]; (b) Wright WRH, Berkeley ER, Alden LR, Baker RT, Sneddon LG. Chem Commun. 2011;47:3177–3179. doi: 10.1039/c0cc05408a. [DOI] [PubMed] [Google Scholar]; (c) Lu Z, Schweighauser L, Hausmann H, Wegner HA. Angew Chem Int Ed. 2015;54:15556–15559. doi: 10.1002/anie.201508360. [DOI] [PubMed] [Google Scholar]
- 16.Lu Z, Conley BL, Williams TJ. Organometallics. 2012;31:6705–6714. doi: 10.1021/om300562d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carter TJ, Heiden ZM, Szymczak NK. Chem Sci. 2015;6:7258–7266. doi: 10.1039/c5sc02348c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Fazen PJ, Remsen EE, Beck JS, Carroll PJ, McGhie AR, Sneddon LG. Chem Mater. 1995;7:1942–1956. [Google Scholar]; (b) Lynch AT. PhD. University of Pennsylvania; 1989. [Google Scholar]
- 19.Frediani P, Bianchi M, Salvini A, Guarducci R, Carluccio LC, Piacenti F. J Organomet Chem. 1995;498:187. [Google Scholar]
- 20.Frediani P, Bianchi M, Salvini A, Carluccio LC, Rosi L. J Organomet Chem. 1997;547:35. [Google Scholar]
- 21.Zhang X, Lu Z, Foellmer LK, Williams TJ. Organometallics. 2015;34:3732. [Google Scholar]
- 22.(a) Krishnamurthy GN, Shashikala N. J Serb Chem Soc. 2009;74:1085–1096. [Google Scholar]; (b) Black D, Deacon G, Thomas N. Aust J Chem. 1982;35:2445–2453. [Google Scholar]; (c) Colton R, Farthing R. Aust J Chem. 1967;20:1283. [Google Scholar]; (d) Anderson PA, Deacon GB, Haarmann KH, Keene FR, Meyer TJ, Reitsma DA, Skelton BW, Strouse GF, Thomas NC. Inorg Chem. 1995;34:6145–6157. [Google Scholar]; (e) Frediani P, Bianchi M, Salvini A, Guarducci R, Carluccio LC, Piacenti F. J Organomet Chem. 1994;476:7. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.