

Abstract
Introduction
Venous thromboembolism (VTE) is common in sickle cell disease (SCD); however, the risk factors associated with VTE in patients with sickle variant syndromes are not known. The primary aim of this study was to determine hematologic and clinical risk factors for VTE in adults with hemoglobin SC or Sβ+ thalassemia genotypes.
Materials and Methods
We conducted a retrospective cross-sectional analysis of patients with hemoglobin SC and Sβ+ thalassemia genotypes followed at the Sickle Cell Center for Adults from 2008 to 2012. Data on baseline hematologic parameters and SCD-specific comorbidities were collected from review of electronic records.
Results
A total of 116 patients, 85 (73%) with hemoglobin SC disease and 31 (27%) with Sβ+-thalassemia, were included for analysis. Thirty-two (28%) patients had a verified history of non-catheter related VTE. Mean baseline hemoglobin levels were higher among individuals with a history of VTE compared to those without (11.7 g/dL vs. 11.0 g/dL, p=0.003). In addition, the prevalence of surgical splenectomy was higher among patients with VTE compared to those without (25.0% vs. 4.8%, p=0.001). On multivariate analysis, elevated baseline hemoglobin (odds ratio [OR] 2.45 (95% confidence interval [CI] 1.42–4.23) and history of surgical splenectomy (OR 5.76 [CI 1.43–23.22) were independently associated with VTE risk.
Conclusions
Higher baseline hemoglobin is a risk factor for non-catheter-related VTE in patients with hemoglobin SC or Sβ+ thalassemia genotypes. Surgical splenectomy, which is a known risk factor for VTE in other hemoglobinopathies such as β-thalassemia intermedia, is also associated with VTE in sickle variant syndromes. Future studies are needed to validate these findings and to investigate the mechanisms of hypercoagulability observed in patients with hemoglobin SC and Sβ+ thalassemia.
Introduction
Venous thromboembolism (VTE) is a common complication in adults with sickle cell disease (SCD),(1–3) occurring in over 10–15% of individuals with SCD by age 40.(1,2) As with other complications of SCD, genotype appears to modify the risk of VTE; however studies evaluating this genotypic variation in VTE have demonstrated conflicting results. Although patients with hemoglobin SC disease or Sβ+ thalassemia were found to have a lower incidence of VTE compared to those with SS or Sβ0 thalassemia in adolescence and early adulthood using data from the Cooperative Study of Sickle Cell Disease,(2) the prevalence of VTE was paradoxically higher among SC and Sβ+ thalassemia patients using a more recent SCD cohort of older adult patients.(1) This age-related phenotypic variation is similar to that observed with osteonecrosis of the femoral head,(4) and, in the case of VTE, may reflect a complex interplay between risk factors such as catheter use, hemolysis, endothelial damage, hypercoagulability, and viscosity for venous thrombotic events in SCD.(5,6)
Few studies have evaluated risk factors for complications in adults with sickle cell variant genotypes. High baseline hemoglobin is associated with viscosity-related sequelae such as proliferative retinopathy and multi-organ failure in SC disease;(7–9) whereas hemoglobin levels do not appear to be associated with other common complications such as sensorineural hearing loss in SC patients.(8) To date, however, the risk factors for VTE in patients with sickle cell disease variants have not been investigated.
We hypothesized that VTE in patients with hemoglobin SC disease or Sβ+ thalassemia is associated with characteristic baseline laboratory and clinical risk factors. In order to address this question, we performed a retrospective analysis of a large cohort of patients with sickle variant syndromes followed at the Sickle Cell Center for Adults at Johns Hopkins.
Methods
We conducted a retrospective cross-sectional analysis of patients with either hemoglobin SC or Sβ+ thalassemia cared for at the Sickle Cell Center for Adults at Johns Hopkins between August 2008 and January 2012. Inclusion criteria were age ≥ 18 years and known genotype. The study was approved by the Institutional Review Board and was deemed to be exempt from informed consent.
Relevant comorbidities for each patient were collected via review of electronic records. Demographic information including age, sex, and genotype were recorded for all patients, as were sickle-cell specific comorbidities including VTE, avascular necrosis, retinopathy, stroke, leg ulcer, and history of surgical splenectomy. Baseline hematologic parameters, including white blood cell count (WBC), hemoglobin, platelet count, and absolute reticulocyte count (ARC), were also collected and were defined as a steady-state, non-pregnancy-related outpatient value taken at least one month after hospitalization and at least three months after a transfusion.
VTE events were verified by duplex ultrasound, ventilation-perfusion scan or computed tomography angiography when available. In situations where radiology reports were not available, only patients who had been prescribed treatment doses of anticoagulation were included as cases. Catheter-related VTE events were excluded from this study. Only patients who had complete information on all steady-state hematologic parameters and comorbidities were included. The present report represents a secondary analysis of a previously published study of VTE using all SCD genotypes from this cohort.(1)
Bivariate analyses were performed using t-test and chi-squared statistics. Logistic regression was used to identify independent risk factors for VTE among patients with sickle cell variants. All statistics were performed using STATA Data Analysis and Statistical Software (Version 12; College Station, TX). Statistical significance was defined as a p-value <0.05.
Results
Of the 158 patients with electrophoresis-verified SC or Sβ+-thalassemia in our cohort, 39 were missing steady-state or transfusion-free hematologic values and 3 experienced a catheter related VTE. We therefore included 116 patients with sickle cell variant genotypes in the present study. Demographic, hematologic, and clinical characteristics according to history of VTE are summarized in Table 1. In the cohort, 85 (73%) patients had SC disease, and 31 (27%) had Sβ+-thalassemia disease. A history of non-catheter-related VTE was recorded in 32 (28%) patients. Of the 32 VTE events, 15 were pulmonary emboli (PE), 7 were isolated deep venous thromboses (DVT), 8 were both PE and DVT, 1 was a cerebral vein thrombosis and 1 was an abdominal vein thrombosis. Additionally, 1 patient with DVT/PE also experienced a cerebral vein thrombosis, and 1 patient with PE had catheterization-proven chronic thromboembolic pulmonary hypertension. Females comprised 57% of the cohort, and the median age of the cohort was 43 years (range 21–72 years). There were no significant differences for age, sex, or sickle variant genotype between patients with and without VTE.
Table 1.
Baseline characteristics by VTE status for patients with SC and Sβ+-thalassemia genotypes
| No VTE (n=84) |
VTE (n=32) |
p-value | |
|---|---|---|---|
| Demographics: | |||
| Age (years) | 41.4 (21–72) | 47.3 (23–70) | 0.032 |
| Female | 49 (58.3%) | 17 (53.1%) | 0.613 |
| Hemoglobin SC disease | 63 (75.0%) | 22 (68.8%) | 0.497 |
| Hemoglobin Sβ+-thalassemia | 21 (25%) | 10 (31.2%) | 0.497 |
| Laboratory Values: | |||
| Hemoglobin (g/dL) | 11.0 (7.6–14.2) | 11.7 (10.0–13.5) | 0.003 |
| Platelets (K/cu mm) | 276 (72–648) | 316 (123–521) | 0.157 |
| Reticulocyte count (%) | 3.7 (1.1–15.3) | 3.2 (1.5–6.4) | 0.236 |
| Absolute reticulocyte count | 148 (45–534) | 138 (60–255) | 0.452 |
| White blood cell count (K/cu mm) | 8.2 (4.0–14.9) | 8.3 (4.0–13.9) | 0.742 |
| Comorbidities: | |||
| Avascular necrosis | 28 (33.3%) | 13 (40.6%) | 0.463 |
| Retinopathy | 52 (61.9%) | 16 (50.0%) | 0.245 |
| Stroke | 7 (8.3%) | 4 (12.5%) | 0.494 |
| Surgical splenectomy | 4 (4.8%) | 8 (25.0%) | 0.001 |
| Leg ulcer | 3 (3.6%) | 2 (6.2%) | 0.525 |
VTE = venous thromboembolism. Results reported as mean (range) or number (%).
For the hematologic parameters, the VTE group demonstrated a higher mean baseline hemoglobin level compared to the non-VTE group (11.7 g/dL vs. 11.0 g/dL, p=0.003) (Figure 1). Of the VTE events, 26/32 (81%) occurred in patients with baseline hemoglobin ≥ 11 g/dL (p=0.002) and 20/32 (63%) occurred with baseline hemoglobin ≥ 11.5 g/dL (p=0.006). On subgroup analysis by genotype, the mean baseline hemoglobin levels for the VTE group compared to the non-VTE group for patients with hemoglobin SC disease was 11.7 g/dL vs. 11.2 g/dL (p=0.038) and for Sβ+-thalassemia patients was 11.6 g/dL vs. 10.5 g/dL (p=0.012). Mean WBC, platelet counts, and reticulocyte counts did not differ between the groups. In addition, the prevalence of prior splenectomy was significantly higher on bivariate analysis comparing patients with VTE to those without (25.0% vs. 4.8%, p=0.001). No differences between groups were found for the other recorded comorbidities.
Figure 1.
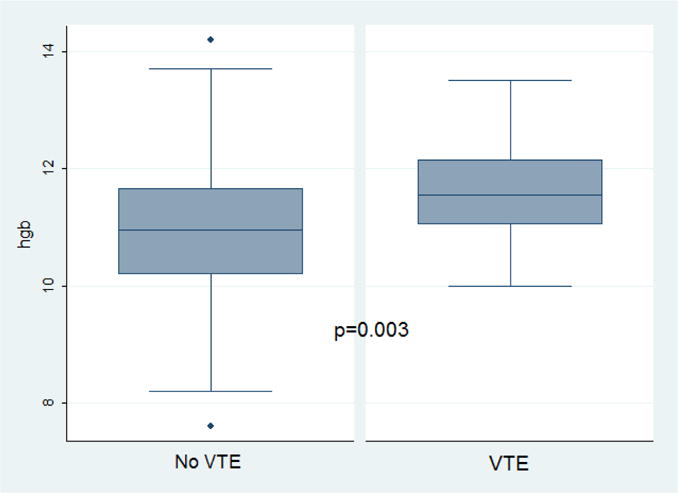
Hemoglobin values by venous thromboembolism (VTE) status for patients with SC or Sβ+ thalassemia genotypes
The logistic regression model for VTE is shown in Table 2. Individuals with sickle variant syndromes were found to have a 2.5 fold (95% confidence interval (CI) 1.4–4.2) increased risk of VTE for every 1 mg/dL increase in hemoglobin level. On subset analysis by genotype, patients with hemoglobin SC disease had a 2.1 fold (CI 1.1–3.8) increased risk of VTE per 1 mg/dL of hemoglobin, and those with Sβ+-thalassemia had a 6.3 fold (CI 1.19–33.25) increased risk of VTE per unit of hemoglobin. History of surgical splenectomy was also associated with VTE, with an OR of 5.8 (CI 1.4–23.2). Subgroup analysis by VTE type revealed that prior splenectomy was primarily associated with an increased risk of PE or unusual vein thrombosis (OR 5.6 [CI 1.4–21.5]) rather than isolated DVT (OR 1.2 [CI 0.1–13.2]), although these analyses were based on a small number of events. A similar differential risk between PE and DVT was not observed with hemoglobin level.
Table 2.
Logistic regression risk factor model for VTE
| Variable | OR | 95% CI |
|---|---|---|
| Age | 1.05† | 1.01–1.09 |
| Female | 1.97 | 0.66–5.85 |
| Hemoglobin SC genotype | 0.62 | 0.21–1.80 |
| Hemoglobin | 2.45† | 1.42–4.23 |
| Surgical splenectomy | 5.76* | 1.43–23.22 |
VTE = venous thromboembolism.
p < 0.05,
p < 0.01
Discussion
VTE is now recognized as a common complication of SCD; however, a comprehensive evaluation of risk factors for VTE in sickle variant genotypes has not yet been performed. In this cohort of over 100 patients with SC or Sβ+ thalassemia, we found that elevated baseline hemoglobin and history of surgical splenectomy were significantly associated with VTE. This relationship was independent of age and sex, which have been shown to influence hemoglobin levels in SCD.(7,10) These findings suggest that high hemoglobin levels may influence VTE risk in SCD and that surgical splenectomy may contribute to hypercoagulability in SCD patients.
Increased whole blood viscosity has long been hypothesized to underlie certain sequelae of SCD, including proliferative retinopathy and avascular necrosis (AVN). Proliferative retinopathy is more prevalent in individuals with SC compared to SS genotype,(11) and baseline hemoglobin levels have been shown to be higher among both SS and SC patients with retinopathy compared to those without. (7,12) A similar association with higher hemoglobin levels has also been demonstrated for femoral head AVN, and individuals with coinheritance of hemoglobin SS and α-thalassemia genotypes have a higher incidence of osteonecrosis of the femoral head compared to those with SS alone.(4) In light of these observations, several clinical groups routinely perform therapeutic phlebotomy for patients with sickle cell variant syndromes who experience clinical complications.(8,13,14) The role of viscosity in the pathophysiology of VTE and role of phlebotomy for treatment or prophylaxis for VTE in patients with SC or Sβ+ thalassemia, however, requires more formal investigation.
Our finding of surgical splenectomy as a risk factor for VTE in SC or Sβ+ thalassemia is consistent with the phenomenon observed in other hemolytic disorders, such as β-thalassemia intermedia and hereditary spherocytosis.(15,16) We were unable to verify the time from splenectomy to VTE in our cohort; however, previous reports have suggested that the increased risk of VTE after splenectomy persists for years to decades and is not solely attributable to the surgical procedure itself.(15–17) Although the pathophysiology of VTE risk with splenectomy remains unclear, potential theories include decreased clearance of abnormal erythrocytes, chronic intravascular hemolysis, and increased coagulability.(18–20)
In SCD, functional asplenia occurs in nearly 90% of infants with SS disease by age 1,(21) whereas splenic dysfunction in sickle variants syndromes can be delayed until adolescence and early adulthood.(10,22) Therefore, we would expect that a majority of patients in our cohort would have some degree of functional asplenia. However, in one study of individuals with SC disease, erythrocyte pit counts were highest among patients who had undergone surgical splenectomy, suggesting that surgical splenectomy results in a more profound degree of splenic dysfunction compared to functional asplenia alone.(22) This finding may relate to the increased risk of VTE that we specifically observed among splenectomized patients in our study. Further studies are needed to determine the role of splenic dysfunction in VTE risk in SCD.
The major strength of our study is our large cohort of adult patients with sickle variant genotypes and available sickle-cell specific phenotypic data. Limitations include the retrospective nature of our study, inability to document the date of splenectomy, and reliance on a single hemoglobin value to define baseline counts. We also did not have measures of viscosity and splenic function in the present study. In addition, a history of comorbidities may not have been recorded in electronic records of some patients.
Conclusions
In summary, elevated hemoglobin level and surgical splenectomy are significantly associated with VTE risk in patients with SC or Sβ+ thalassemia genotypes. Further studies are needed to validate these findings and to investigate the pathophysiology of increased coagulability in SCD.
Highlights.
Venous thromboembolism is common in adults with hemoglobin SC disease and Sβ+ thalassemia genotypes.
Higher baseline hemoglobin is significantly associated with VTE risk in patients with hemoglobin SC disease and Sβ+ thalassemia.
History of prior splenectomy is also significantly associated with VTE risk in these patients.
Acknowledgments
R.P. Naik was supported by National, Heart, Lung, and Blood Institute (NHLBI) award K08HL125100.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Addendum
R.P. Naik and S. Lanzkron designed the study and analyzed the results. T.T. Yu, J. Nelson, S. Lanzkron, and R.P. Naik collected the data. R.P. Naik, S. Lanzkron, and M.B. Streiff interpreted the findings. T.T. Yu, J. Nelson, M.B. Streiff, S. Lanzkron, and R.P. Naik wrote and critically revised the manuscript.
References
- 1.Naik RP, Streiff MB, Haywood C, Jr, Nelson JA, Lanzkron S. Venous Thromboembolism in Adults with Sickle Cell Disease: A Serious and Under-recognized Complication. Am J Med. 2013;126:443–449. doi: 10.1016/j.amjmed.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naik RP, Streiff MB, Haywood C, Jr, Segal JB, Lanzkron S. Venous thromboembolism incidence in the Cooperative Study of Sickle Cell Disease. J Thromb Haemost. 2014;12:2010–2016. doi: 10.1111/jth.12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stein PD, Beemath A, Meyers FA, Skaf E, Olson RE. Deep venous thrombosis and pulmonary embolism in hospitalized patients with sickle cell disease. Am J Med. 2006;119:897.e7–897.e11. doi: 10.1016/j.amjmed.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 4.Milner PF, Kraus AP, Sebes JI, Sleeper LA, Dukes KA, Embury SH, et al. Sickle cell disease as a cause of osteonecrosis of the femoral head. N Engl J Med. 1991;325:1476–1481. doi: 10.1056/NEJM199111213252104. [DOI] [PubMed] [Google Scholar]
- 5.Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007;21:37–47. doi: 10.1016/j.blre.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lim MY, Ataga KI, Key NS. Hemostatic abnormalities in sickle cell disease. Curr Opin Hematol. 2013 Sep;20(5):472–477. doi: 10.1097/MOH.0b013e328363442f. [DOI] [PubMed] [Google Scholar]
- 7.Hayes RJ, Condon PI, Serjeant GR. Haematological factors associated with proliferative retinopathy in sickle cell-haemoglobin C disease. Br J Ophthalmol. 1981;65:712–717. doi: 10.1136/bjo.65.10.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gualandro SF, Fonseca GH, Yokomizo IK, Gualandro DM, Suganuma LM. Cohort study of adult patients with haemoglobin SC disease: clinical characteristics and predictors of mortality. Br J Haematol. 2015 doi: 10.1111/bjh.13625. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 9.Hassell KL, Eckman JR, Lane PA. Acute multiorgan failure syndrome: a potentially catastrophic complication of severe sickle cell pain episodes. Am J Med. 1994;96:155–162. doi: 10.1016/0002-9343(94)90136-8. [DOI] [PubMed] [Google Scholar]
- 10.Ballas SK, Lewis CN, Noone AM, Krasnow SH, Kamarulzaman E, Burka ER. Clinical, hematological, and biochemical features of Hb SC disease. Am J Hematol. 13:37–51. doi: 10.1002/ajh.2830130106. [DOI] [PubMed] [Google Scholar]
- 11.Downes SM, Hambleton IR, Chuang EL, Lois N, Serjeant GR, Bird AC. Incidence and natural history of proliferative sickle cell retinopathy: observations from a cohort study. Ophthalmology. 2005;112:1869–1875. doi: 10.1016/j.ophtha.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 12.Hayes RJ, Condon PI, Serjeant GR. Haematological factors associated with proliferative retinopathy in homozygous sickle cell disease. Br J Ophthalmol. 1981;65:29–35. doi: 10.1136/bjo.65.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lionnet F, Hammoudi N, Stojanovic KS, Avellino V, Grateau G, Girot R, Haymann JP. Hemoglobin sickle cell disease complications: a clinical study of 179 cases. Haematologica. 2012;97:1136–1141. doi: 10.3324/haematol.2011.055202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Markham MJ, Lottenberg R, Zumberg M. Role of phlebotomy in the management of hemoglobin SC disease: case report and review of the literature. Am J Hematol. 2003;73:121–125. doi: 10.1002/ajh.10328. [DOI] [PubMed] [Google Scholar]
- 15.Taher A, Isma’eel H, Mehio G, Bignamini D, Kattamis A, Rachmilewitz EA, Cappellini MD. Prevalence of thromboembolic events among 8,860 patients with thalassaemia major and intermedia in the Mediterranean area and Iran. Thromb Haemost. 2006;96:488–491. [PubMed] [Google Scholar]
- 16.Schilling RF, Gangnon RE, Traver MI. Delayed adverse vascular events after splenectomy in hereditary spherocytosis. J Thromb Haemost. 2008;6:1289–1295. doi: 10.1111/j.1538-7836.2008.03024.x. [DOI] [PubMed] [Google Scholar]
- 17.Thomsen RW, Schoonen WM, Farkas DK, Riis A, Fryzek JP, Sorensen HT. Risk of venous thromboembolism in splenectomized patients compared with the general population and appendectomized patients: a 10-year nationwide cohort study. J Thromb Haemost. 2010;8:1413–1416. doi: 10.1111/j.1538-7836.2010.03849.x. [DOI] [PubMed] [Google Scholar]
- 18.Cappellini MD. Coagulation in the pathophysiology of hemolytic anemias. Hematology Am Soc Hematol Educ Program. 2007:74–78. doi: 10.1182/asheducation-2007.1.74. [DOI] [PubMed] [Google Scholar]
- 19.Cappellini MD, Robbiolo L, Bottasso BM, Coppola R, Fiorelli G, Mannucci AP. Venous thromboembolism and hypercoagulability in splenectomized patients with thalassaemia intermedia. Br J Haematol. 2000;111:467–473. doi: 10.1046/j.1365-2141.2000.02376.x. [DOI] [PubMed] [Google Scholar]
- 20.Ataga KI. Hypercoagulability and thrombotic complications in hemolytic anemias. Haematologica. 2009;94:1481–1484. doi: 10.3324/haematol.2009.013672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rogers ZR, Wang WC, Luo Z, Iyer RV, Shalaby-Rana E, Dertinger SD, Shulkin BL, Miller JH, Files B, Lane PA, Thompson BW, Miller ST, Ware RE. Biomarkers of splenic function in infants with sickle cell anemia: baseline data from the BABY HUG Trial. Blood. 2011;117:2614–2617. doi: 10.1182/blood-2010-04-278747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lane PA, O’Connell JL, Lear JL, Rogers ZR, Woods GM, Hassell KL, Wethers DL, Luckey DW, Buchanan GR. Functional asplenia in hemoglobin SC disease. Blood. 1995;85:2238–2244. [PubMed] [Google Scholar]