

Abstract
Addiction to psychostimulants such as Methamphetamine (MA) is a significant public health issue in the United States and currently, there are no FDA approved pharmacological interventions. Previously, using short term-selected mouse lines for high and low MA sensitivity that were derived from an F2 cross between C57BL/6J (B6) and DBA/2J (D2) strains, we identified a quantitative trait locus (QTL) on chromosome (chr) 11 that influenced sensitivity to MA-induced locomotor activity (D2 < B6). Using interval-specific murine congenic lines containing various D2 allelic segments on a B6 background, we fine mapped the QTL to a 206 kb critical interval on chromosome 11. To investigate the neurobiological mechanism by which this QTL decreases MA sensitivity, we conducted transcriptome analysis in a 10 Mb congenic mouse (chromosome 11: 50–60 Mb) on whole-striatum brain tissue punches compared to wild-type B6 littermate controls [1]. The data from this study can be found in the NCBI Gene Expression Omnibus (GSE66366).
| Specifications | |
|---|---|
| Organism/cell line/tissue | Mus musculus, C57BL/6J (B6) and DBA/2J (D2) |
| Sex | Male (CB-1), male (CB-2), female (CB-3), female (CB-4), male (CB-5), male (CB-6), female (CB-7), female (CB-8), female (CB-9), male (CB-10), female (CB-11), female (CB-12), male (CB-13), male (CB-14), male (CB-15), male (CB-16) |
| Sequencer or array type | Illumina HiSeq2500 |
| Data format | Raw (fastq.gz) and processed (TXT) |
| Experimental factors | N/A |
| Experimental features | A total of sixteen drug-naïve B6xD2 congenics (Line 4a) and wild-type B6 mice were used for this study. Whole-striatum tissue was dissected, RNA-extracted, and used for 50 bp single-end read library generation, sequencing and subsequent transcriptome analysis. |
| Consent | N/A |
| Sample source location | Chicago, Illinois USA |
1. Direct link to deposited data
2. Unix and R codes used for transcriptome analysis are provided on github
3. Experimental design, materials and methods
3.1. Experimental design
We conducted transcriptome analysis of total RNA extracted from striatum punches from eight drug-naïve Line 4a congenics heterozygous for a 10 Mb interval from the DBA/2J (D2) strain on chr.11 (Line 4a) and eight wild-type B6 controls [1]. A complete description of our samples and files that are available on GEO can be found in Table 1. A detailed experimental pipeline is illustrated in Fig. 1.
Table 1.
Sample information. For each replicate sample file used in transcriptome analysis, we list the file types available in GEO (Fastq and/or TXT) as well as the corresponding mouse genotype, sex, and cage. We also include the total number of reads for each replicate that was generated using the Illumina HiSeq2500 for sequencing.
| Sample_lane | Raw | Analyzed | Genotype | Sex | Cage | Reads |
|---|---|---|---|---|---|---|
| CB-1_L005 | .fastq.gz | .txt | B6 | M | 11PN7-89 | 22,610,234 |
| CB-1_L006 | .fastq.gz | .txt | B6 | M | 11PN7-89 | 25,543,567 |
| CB-2_L005 | .fastq.gz | .txt | Line 4a | M | 11PN7-87 | 22,157,350 |
| CB-2_L006 | .fastq.gz | .txt | Line 4a | M | 11PN7-87 | 25,262,056 |
| CB-3_L005 | .fastq.gz | .txt | B6 | F | 11PN7-81 | 22,947,115 |
| CB-3_L006 | .fastq.gz | .txt | B6 | F | 11PN7-81 | 25,854,797 |
| CB-4_L005 | .fastq.gz | .txt | Line 4a | F | 11PN7-80 | 18,723,353 |
| CB-4_L006 | .fastq.gz | .txt | Line 4a | F | 11PN7-80 | 21,157,403 |
| CB-5_L005 | .fastq.gz | .txt | B6 | M | 11PN7-89 | 19,515,383 |
| CB-5_L006 | .fastq.gz | .txt | B6 | M | 11PN7-89 | 21,956,561 |
| CB-6_L005 | .fastq.gz | .txt | Line 4a | M | 11PN7-87 | 16,942,883 |
| CB-6_L006 | .fastq.gz | .txt | Line 4a | M | 11PN7-87 | 19,532,150 |
| CB-7_L005 | .fastq.gz | .txt | B6 | F | 11PN7-86 | 22,179,855 |
| CB-7_L006 | .fastq.gz | .txt | B6 | F | 11PN7-86 | 25,134,998 |
| CB-8_L005 | .fastq.gz | .txt | Line 4a | F | 11PN7-90 | 19,457,924 |
| CB-8_L006 | .fastq.gz | .txt | Line 4a | F | 11PN7-90 | 22,174,685 |
| CB-9_L007 | .fastq.gz | .txt | B6 | F | 11PN7-90 | 19,596,836 |
| CB-9_L008 | .fastq.gz | .txt | B6 | F | 11PN7-90 | 21,221,174 |
| CB-10_L007 | .fastq.gz | .txt | Line 4a | M | 11PN7-85 | 21,101,927 |
| CB-10_L008 | .fastq.gz | .txt | Line 4a | M | 11PN7-85 | 22,813,084 |
| CB-11_L007 | .fastq.gz | .txt | B6 | F | 11PN7-90 | 18,491,655 |
| CB-11_L008 | .fastq.gz | .txt | B6 | F | 11PN7-90 | 19,918,292 |
| CB-12_L007 | .fastq.gz | .txt | Line 4a | F | 11PN7-84 | 20,899,297 |
| CB-12_L008 | .fastq.gz | .txt | Line 4a | F | 11PN7-84 | 22,566,381 |
| CB-13_L007 | .fastq.gz | N/A | B6 | M | 11PN7-83 | 25,351,760 |
| CB-13_L008 | .fastq.gz | .txt | B6 | M | 11PN7-83 | 27,435,087 |
| CB-14_L007 | .fastq.gz | .txt | Line 4a | M | 11PN7-82 | 20,096,221 |
| CB-14_L008 | .fastq.gz | .txt | Line 4a | M | 11PN7-82 | 21,735,984 |
| CB-15_L007 | .fastq.gz | .txt | B6 | M | 11PN7-83 | 20,676,230 |
| CB-15_L008 | .fastq.gz | .txt | B6 | M | 11PN7-83 | 22,347,388 |
| CB-16_L007 | .fastq.gz | .txt | Line 4a | M | 11PN7-82 | 22,164,119 |
| CB-16_L008 | .fastq.gz | .txt | Line 4a | M | 11PN7-82 | 23,901,507 |
Fig. 1.
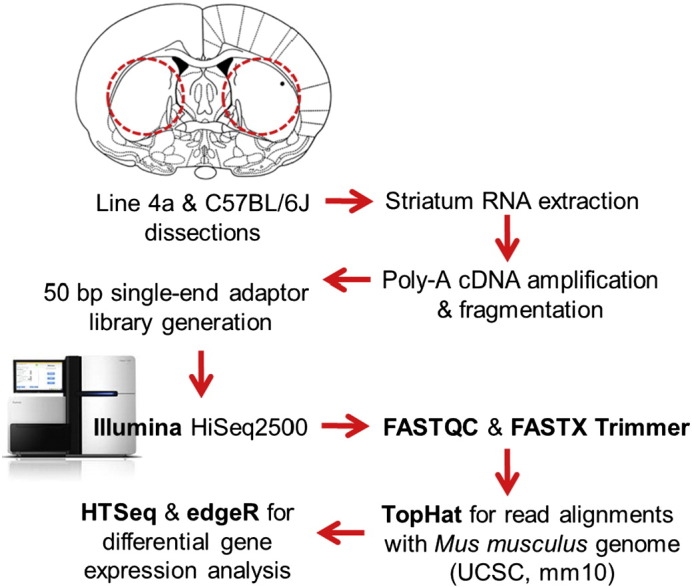
Experimental design. Bilateral striatum dissections are marked with red circles on the caudal end of the brain slice (Bregma − 0.10 mm). RNA extraction, cDNA library generation, sequencing followed by transcriptome analysis steps are included.
3.2. Mice
All procedures in the mice were approved by the University of Chicago Institutional Animal Care and Use Committee and were conducted with strict adherence to the National Institute of Health guidelines for the care and use of laboratory animals. Colony rooms were maintained on a 12:12 h light–dark cycle (lights on at 600 h). Littermate mice derived from heterozygous congenics bred with B6 were housed in same-sex groups of two-five mice per cage with standard laboratory chow and water available ad libitum. Line 4a congenics were generated by several generations of backcrossing male or female mice that were heterozygous for the D2 allelic segment from the “11P” congenic strain (chr11: 0–90 Mb) to the B6 strain (see [1]).
3.3. Brain dissections
Drug-naïve, Line 4a congenics and wild-type B6 mice were habituated to a climate-controlled procedure room located next door to the vivarium for 2 h prior to brain dissections. For this particular study, samples were collected on a single day from 10:00 am–12:30 pm from eight Line 4a heterozygous congenics and eight wild-type B6 mice. Mice were sacrificed individually in a random cage order via live, rapid decapitation with sharpened shears. All subsequent procedures were performed with 70% ethanol and RNAse zap (Ambion)-sterilized tools and RNAse free consumables. Whole brains were carefully removed from the skull using tweezers and forceps and placed into an ice-cold brain matrix (Stoelting). Razor blades were simultaneously inserted into slots 2 and 5 corresponding to Bregma 2.90 to − 0.10 mm, and brain sections were immediately transferred to an ice-cold petri dish. Correct sectioning was confirmed by using the anterior commissure as the dorsal landmark and the cortex as the lateral and ventral landmarks. Bilateral 2.5 mm striatal punches were carefully harvested at a right degree angle relative to the surface of the petri dish (Fig. 1) and immediately submerged in 300 μL of RNAlater (Qiagen, Valencia, CA) in a 1.5 mL microfuge tube at room temperature. Within a few hours, the samples were transferred to 4 °C for 48 h. Afterwards, tissues were dry-blotted using Kim Wipes, moved into new microfuge tubes, and stored at − 80 °C until further processing. Left and right striatum were pooled for each sample for RNA extractions.
3.4. RNA-extraction & cDNA library preparation
For total RNA extractions [2], tissues were homogenized using a tissue homogenizer (PowerGen 125, Hampton, NH) in ice-cold Trizol (Invitrogen, Carlsbad, CA) for 1 min. Homogenates were then spun at 12,000 RPM for 10 min in a refrigerated centrifuge set to 4 °C to remove insoluble material. Supernatants were carefully transferred to phase-lock tubes (5 PRIME, Gaithersburg, MA) and warmed to room temperature and then mixed with equal volume Chloroform. Tubes were then vigorously shaken until a milky solution was formed, and then spun at 13,000 RPM for 5 min. The top clear aqueous layer was then removed and transferred to a new microfuge tube, mixed with one volume 70% molecular biological grade ethanol and mixed by pipetting. The RNA extracts were then further purified through RNeasy columns (RNeasy mini kit; Qiagen, Valencia, CA). Eluted RNA was quality checked via nanodrop (260/280 > 2) (Thermo Scientific, Waltham, MA) and Agilent 2100 Bioanalyzer (Santa Clara, CA). Samples were then diluted to 100 ng/μL, frozen on dry ice, and provided to the University of Chicago Genomics Core Facility for cDNA library preparation using the TruSeq oligo-dT kit, 50 bp single-end reads (Illumina, San Diego, CA).
3.5. Sequencing & transcriptome analysis
Samples CB-1 through CB-8 were multiplexed across lanes 5 and 6, while samples CB-9 through CB-16 were multiplexed over lanes 7 and 8 for sequencing on the Illumina HiSeq2500 platform at the University of Chicago Genomics Core Facility. Reads per lane ranged at 16,942,883–27,435,087 with an overall summed range of 36,475,033–52,786,847 per sample. Fastq files were quality checked via FASTQC (v0.10.1) [3] and all of the samples possessed Phred quality scores > 30 (i.e. < 0.1% sequencing error) (Fig. 2). Using the FastX-Trimmer from the FastX-Toolkit (v0.0.13.2), the 51st base was trimmed to enhance read quality and prevent misalignment. Fastq files were processed with TopHat (v2.0.0) [4] to align reads to the reference genome (mm10; UCSC Genome Browser). Due to errors with TopHat processing, sample 13 replicate CB-13_L007.fastq.gz was not included in the subsequent analyses. TopHat output SAM files that were generated were then processed with the Python package HTSeq (v0.6.1p1) [5] to compute read counts per gene. The HTSeq TXT file output for each replicate was then assembled into a matrix and analyzed in edgeR (v2.14) [6], a Bioconductor R package for differential gene expression analysis. EdgeR models read counts using a negative binomial distribution to account for variability in the number of reads via generalized linear models. In order to consider a gene expressed, we set the count-per-million (CPM) threshold to be > 1. Thus, for instance, the lowest number of reads in CB-6_L005 (Table 1) to consider a gene expressed would be 16. The distribution of p-values from the generalized linear model (GLM) likelihood ratio test for the effect of Line 4a versus wild-type B6 was used to calculate FDRs for differential expression. Covariates that were included in the model included “home cage” to account for cage effects on gene expression. Both the unix and R codes are publically available on github (https:/github.com/wevanjohnson/hnrnph1).
Fig. 2.

FASTQC output per sequencing lane. All four lanes 5–8, corresponding to panels A–D respectively, exhibited high Phred scores > 30 with the exception of the 51st base.
4. Discussion
Here we describe our transcriptome analysis pipeline that we used to generate a list of 91 differentially expressed genes in Line 4a relative to wild-type B6 (FDR < 0.05), when accounting for cage [1]. Total RNA that was extracted from striatum punches was used to generate 50 bp single-end cDNA libraries which were then sequenced on the Illumina HiSeq2500 platform. Transcriptome analysis was conducted using a variety of open source packages, mostly available through Bioconductor, using the Unix operating system and the R statistical computing interface.
To explore the importance of including “home cage” as a covariate in our edgeR analysis (cage-corrected), we also generated a gene list without the covariate (uncorrected). Interestingly, when we do not include “home cage”, we obtain a larger list of 1035 genes (FDR < 0.05). In comparison to our cage-corrected list, 51 genes are shared with an even distribution when we sort by p-value. Moreover, 9 out of 10 of our top genes on the cage-corrected gene list versus the uncorrected list are shared. As described in [1], ingenuity pathway analysis (IPA; Ingenuity Systems, Redwood city, CA) revealed the top network in our cage-corrected list to be “Cellular Development, Nervous System Development and Function, Behavior”. We also identified canonical pathways pertaining to neuronal function such a Glutamate Receptor Signaling, Gαq Signaling, and G-protein Coupled Receptor Signaling. Not surprisingly, IPA analysis of our uncorrected list provides a broader and ontological terms that were more general and did not have as obvious of relevance to the brain and behavior. Some of these terms included “Molecular transport, Nucleic Acid Metabolism, Small Molecule Biochemistry” and “Cancer, Organismal Injury and Abnormalities, Gastrointestinal Disease”. Our top canonical pathways were RhoGDI Signaling, and Molecular Mechanisms of Cancer, and Signaling by Rho Family GTPases. Because we examined brain tissue relevant to psychostimulant behavior, we would expect an overrepresentation of neurobiological terms, as we observed with our cage-corrected gene list [1]. Our analysis hence suggests “home cage” to be a crucial covariate for our analysis, and thus should be considered in future transcriptomic analyses of rodent brain tissue.
Funding
R01DA039168 (CBD), R00DA029635 (CBD), F31DA040324-01A1 (NY), R01HG005692 (WEJ).
References
- 1.Iakoubova O.A., Olsson C.L., Dains K.M., Ross D.A., Andalibi A., Lau K., Choi J., Kalcheva I., Cunanan M., Louie J., Nimon V., Machrus M., Bentley L.G., Beauheim C., Silvey S., Cavalcoli J., Lusis A.J., West D.B. Genome-tagged mice (GTM): two sets of genome-wide congenic strains. Genomics. 2001;74:89–104. doi: 10.1006/geno.2000.6497. [DOI] [PubMed] [Google Scholar]
- 2.Bryant C.D., Kole L.A., Guido M.A., Sokoloff G., Palmer A.A. Congenic dissection of a major QTL for methamphetamine sensitivity implicates epistasis. Genes Brain Behav. 2012;11:623–632. doi: 10.1111/j.1601-183X.2012.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data.http://www.bioinformatics.babraham.ac.uk/projects/fastqc Available online at. [Google Scholar]
- 4.Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anders S., Pyl P.T., Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robinson M.D., Mccarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]