

Graphical abstract
SEM images of F6 ethosomal formulation

Abbreviations: DEE, drug entrapment efficiency; PEG, polyethylene glycol; PI, polydispersity index; TH, Terbinafine Hydrochloride; MR, marketed cream
Keywords: Ethosomes, In-vitro drug diffusion, Terbinafine Hydrochloride, Transdermal flux
Abstract
The present study investigates the entrapment of terbinafine hydrochloride (TH) in ethosomal vesicles via unsonicated and sonication method. Carbopol 934P was incorporated in the best formulation, F6, obtained by sonication method. The formulated ethosomal gel obtained as such i.e. F6∗ was exploited to achieve a zero order release profile of TH. The composition includes phospholipid, ethanol and propylene glycol. Drug entrapment efficiency (DEE), in-vitro and ex-vivo drug diffusion studies, FT-IR and stability studies of the prepared ethosomes were investigated. The size and shape of F6 ethosomes vesicles were characterized by SEM. In-vitro drug release studies were performed using sigma dialysis membrane in phosphate buffer, pH 7.4 for 12 h while drug content was determined by HPLC. DEE was ranked from 55.33 ± 1.32% to 69.11 ± 2.11%. Highest DEE was seen with F6 ethosomal formulation with a vesicle size of 248 ± 1.02 nm. FT-IR studies confirmed that there was no chemical interaction between drug and excipients used in the formulation. Ex-vivo result suggested that drug diffusion observed after 12 h from F6∗ and marketed cream (MR) formulations was 74.01 ± 0.62% and 61.45 ± 0.86%, respectively. The results of similarity factor (f2 values) for MR and F6∗ ethosomal gel were 85.14 and 42.63, respectively. It revealed that F6∗ showed dissimilar dissolution profiles. Transdermal flux value for F6∗ and MR was found to be 144.61 ± 1.28 μg/cm2/h and 121.6 ± 1.16 μg/cm2/h, respectively. This study disclosed that F6∗ resides at targeted site for a relatively longer period of time thereby signifying the improved patient compliance.
Introduction
Ethosomes are soft malleable vesicles constituting phospholipids, ethanol (relatively high concentration) and water [1]. They act as non-invasive delivery carriers for targeting drugs to deep skin layers. Hence, when integrated into a vesicles membrane, ethosomes promote the vesicle to penetrate the stratum corneum [2]. The utility of ethosomes as a carrier of antiviral drug acyclovir was previously tested by Essa and coworkers for the topical treatment of herpetic infection. They demonstrated significant improvement of ethosomal 5% acyclovir system as compared to a 5% acyclovir cream by performing two-armed, double-blinded, randomized clinical trial [3]. In another study, enhanced drug delivery via ethosomal carrier was observed by an increase in depth and fluorescent activity [4].
A synergistic mechanism was suggested between ethanol, vesicles and skin lipids by Touitou and coworkers for elucidating the role of ethosomes in promoting enhanced drug delivery [5]. It was proposed that “ethanol effect” resulted in an interaction of ethanol with the lipid molecules in the polar head group region and exhibits reduction in the transition temperature of lipids in the stratum corneum, which ultimately increases their fluidity and decreases the density of lipid multilayer. This effect is followed by the “ethosomal effect” which involves the penetration and permeation of lipids due to the malleability and fusion of ethosomes with skin lipids. This step resulted in the release of drug into the deep layers of skin. It should be noted that since ethanol imparted flexible characteristics to vesicles, it allowed the ethosomal vesicles easier and deeper penetration into the deeper layers of the skin. The release of the drug in the deep layers of the skin and its transdermal absorption could then be the result of a fusion of ethosomes with skin lipids, and drug release at various points along the penetration pathway [6].
The pharmaceutical technology in recent years has witnessed the formulation of modified liposomes for skin mediated drug delivery, and in this regard, considerable attention has been paid to vesicular approaches involving transfersomes and ethosomes. These approaches utilize non-toxic and biodegradable chemicals which prolong half-life of a drug in order to provide a sustained drug delivery release effect [7], [8]. Ethosomal systems are very efficient in delivering substances in terms of quantity and depth by increasing cell permeability/lipid fluidity [9], [10], [11].
In some of the comparative studies, favorable attributes of ethosomes over liposomes, in terms of skin penetration and therapeutic effects as solution and cream have been outlined. These results are encouraging in relation to transdermal delivery of therapeutic agents via ethosomal vesicles [12]. Moreover, terbinafine hydrochloride (TH) is an allyl amine class derivative used for treating local and systemic infections. It is highly effective against dermatophytes and Aspergillus species for superficial and systemic fungal infections [13], [14], [15].
Hence, the objective of the present study involves the formulation of ethosomes containing Carbopol 934P and phospholipids as vesicle forming agent along with TH to observe its effect at targeted site for a relatively longer period of time with a zero order release profile.
Material and methods
Materials
Terbinafine hydrochloride and propylene glycol were obtained from Orchid Pharmaceuticals, India, and Sandoz Chemicals Ltd., India, respectively. Soya phosphatidylcholine was purchased from Sigma-Aldrich Chem, Germany. Carbopol 934P was procured from Correl Pharma Ltd., Mumbai, India. Cellophane membrane grade 110 was purchased from Hi Media Laboratories, Mumbai, India. Methyl paraben, Propyl paraben, Triethanolamine, methanol and high purity 99.9% Ethanol Omnis Grade were obtained from SD Fine chemicals, Mumbai, India. All other materials were of analytical grade.
Preparation of unsonicated ethosomes and sonicated ethosomes
Ethosomes were prepared by dissolving 10 mg TH in different concentration of ethanol and water in required quantity along with 2 mL of propylene glycol by maintaining the concentration of phospholipids (10 mg and 20 mg). The ingredients were formulated according to the procedure developed by Touitou and coworkers [9]. The ethanolic mixture in required quantity was taken separately and heated at 30 ± 2 °C on a water bath. The water was slowly added to ethanol mixture dropwise in the center of the vessel. The resulting mixture was stirred at 700 rpm for 10 min to obtain the ethosomal vesicles. In order to obtain the sonicated ethosomes, ethosomes obtained by the above procedure were subjected to sonication at 4 °C by probe sonicator (SH 70G, Bandelin Sonopuls UW 2070, Berlin, Germany) by stirring at 700 rpm for 15 min (3 cycles at a gap of 5 min) (see Table 1).
Table 1.
Formulation of ethosomes containing Terbinafine Hydrochloride.
| Ingredients | Unsonicated |
Sonicated |
||||||
|---|---|---|---|---|---|---|---|---|
| F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | |
| Terbinafine HCl (mg) | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 |
| Phospholipids (mg) | 10 | 10 | 10 | 20 | 10 | 10 | 10 | 20 |
| Ethanol (mL) | 4 | 5 | 6 | 5 | 4 | 5 | 6 | 5 |
| Propylene glycol (mL) | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Distilled water | q.s | q.s | q.s | q.s | q.s | q.s | q.s | q.s |
F6∗: formulation of ethosomes as gel.
q.s: quantity sufficient.
Preparation of ethosomes gel based formulations
The best available formulation, F6 was selected for in vitro and ex vivo observations. For this purpose, Carbopol 934P (1% w/w) was first dispersed in 8 mL ethanol followed by adding distilled water, 0.2% methyl paraben and 0.02% propyl paraben as preservatives. To this, triethanolamine (1% w/v) was added with continuous stirring (20 min) until a transparent alkaline gel was obtained finally [18]. The gel base so prepared was added to F6 formulation by continuous stirring which leads to ethosomal gel base formulations (denoted as F6∗).
HPLC determination of TH from dissolution sample
HPLC determination was performed according to the procedure described by Patel [19]. Briefly, the dissolution media consisted of phosphate buffer (pH 7.4). At a specific time interval, samples of TH were withdrawn, filtered and analyzed using a HPLC (Shimadzu HPLC, Class VP series) system. Chromatographic separation was performed on a RP C-18 column (250 mm 34.6 mm i.d., particle size 5 μm) at 25 °C. The mobile phase used was a mixture of methanol–acetonitrile (60:40 v/v) with (0.15% triethylamine and 0.15% phosphoric acid) at flow rate of 1.2 mL/min. Loop size: 20 μL stock solutions of TH were prepared in PBS (pH 7.4) as 1 mg/mL. Calibration curve was prepared for each of the analytes after appropriate dilution of stock solutions to obtain final concentrations of 0.1, 0.5, 1, 2, 5, 10, 15 and 30 μg/mL, and the detection was performed at 224 nm using a UV detector. The run time for the assay was 8 min. The retention time for TH was 6.11 ± 0.2 min. The calibration curve was prepared taking the peak area of the analytes (TH) versus the concentration (μg/mL) using a weighted (1/concentration2) linear least squares regression as the mathematical model. The regression equation of the calibration curve was then used to calculate the drug entrapment efficiency, in-vitro and ex-vivo drug release. The lowest limit of quantization for TH was determined from the peak signal to noise level (S/N) as 10.
Drug entrapment efficiency (DEE)
Drug entrapment efficiency (DEE) was determined by applying ultracentrifugation method. All the formulations were subjected to ultracentrifugation at 20,000 rpm for 3 h (Remi Elektrotechnik Limited, Mumbai, India) by using centrisort tubes [16], [17]. The supernatant liquid after centrifugation was diluted with 1:1 water–ethanol solution. The amount of TH present in the formulation was analyzed by using HPLC method as mentioned above. The drug entrapment percentage was calculated using the given equation:
where Qt = total amount of drug added and Qs = amount of drug obtained in supernatant.
Vesicle size and shape
Dynamic Light Scattering method was used to measure the size of prepared formulation. The particle size was measured using a Malvern Zetasizer 300 HSA (Malvern Instruments, UK) at 25 °C. The samples were diluted with 1:1 ethanol:water solution. The authors are aware that the dilution may change the microstructure of the vesicles, but the dilution was required to allow sufficient amount of light to pass through the solution. Charges on drug loaded ethosomes vesicles and zeta potential measurement were determined by analyzing the formulation for 60 s. Polydispersity (PI) index was also determined as a measurement of particle size homogeneity for the prepared ethosomal formulations.
Scanning electron microscopy
Scanning electron microscopy was conducted to characterize the surface morphology of ethosomal formulation (F6) for which a drop of ethosomal formulation was mounted on clear glass stub, air-dried and coated with Polaron E 5100 Sputter coater (Polaron, UK) and visualized under SEM (JEOL-JSM-6510, Japan).
In vitro diffusion study
In vitro drug diffusion study was carried out to select the formulation for ex vivo release studies by using a Franz diffusion cell with Cellophane dialysis membrane of grade ‘110’. Firstly, the studies were carried out with ethosomal formulations (F1-F8), gel based ethosomal formulations (F6∗) and the marketed cream (1% w/v Archicare Pharma, India). The dialysis membrane was mounted on Franz diffusion cell with 3 mg TH applied through donor compartment on the dialysis membrane and reservoir compartment was filled with 10 mL methanol and 40 mL phosphate buffer solution (pH 7.4). The reservoir compartment was replenished with same quantity of PBS and the study was carried out at 37 ± 1 °C at a speed of 400 rpm/min for 12 h. HPLC was used to obtain the amount of drug released.
Ex-vivo diffusion study of ethosomes gel
Male albino rats weighing 120–150 g were sacrificed for abdominal skin. Formulation (F6∗) was incorporated into the gel base to get ethosomal gel. 1.5 g of ethosomal gel which was equivalent to 3 mg of drug was applied through donor compartment on the skin. Marketed cream (MR) of same drug (0.3 g) was taken and applied through the donor compartment on other diffusion cell. In both the diffusion cells, reservoir compartment was filled with 10 mL of methanol and 40 mL PBS (pH 7.4) at 37 ± 1 °C at 400 rpm/min for 12 h. Samples were withdrawn from reservoir compartment at 1,2,4,6,8,10 and 12 h time intervals and the amount of drug release was determined by HPLC method. Each time the reservoir compartment was replenished with the same quantity of PBS buffer. Transdermal flux (F = slope/area) values for ethosomal gel and MR cream were calculated and reported.
Characterization of release data
The description of dissolution profiles has been attempted using different release models [20]. The data were evaluated according to the following equations:
where Mt is the amount of drug dissolved at time t, Mo the initial amount of drug, K1 is the first order release constant, K0 the zero order release constant, KH the Higuchi rate constant, Kk the release constant and n is the diffusional release exponent indicative of the operating release mechanism. The correlation coefficient (r2) was used as an indicator of the best fitting, for each of the models considered.
Similarity factor
The similarities between two dissolution profiles were assessed by a pairwise model independent procedure and similarity factor (f2) was applied for the F6∗ ethosomal formulation and marketed cream.
where n is the sampling number, Rt and Tt are the percent dissolved of the reference and test products at each time point t, and Wt is the optional weight factor (normally taken as 1). It should be noted that f2 values higher than 50 exhibited the similarity of dissolution profiles.
FT-IR spectroscopy
Infrared spectrum (FT-IR, Spectrum RX1, Perkin Elmer Ltd., Switzerland) was obtained to monitor the functional groups present in the compound by scanning the sample in potassium bromide disks.
Stability studies
In order to analyze the efficacy of prepared formulation at room temperature (25 ± 1 °C) and under refrigerated conditions (4 °C), different formulations were kept in a borosilicate vial for 2 months [21], [22], [23]. The sample was analyzed for entrapment efficiency, vesicle size and ex vivo diffusion studies.
Statistical analysis
All the data are represented as Mean ± SD (n = 3). Drug entrapment efficiency for F2 and F6 formulation, ex-vivo diffusion study of F6∗ formulation and marketed cream were analyzed by applying Newman–Keuls multiple comparison tests by using Graph pad prism version 5 (Graph pad prism Software, Inc.). The values were confirmed to be significant with p value less than 0.05 (P < 0.05).
Results and discussion
Preparation of unsonicated ethosomes and sonicated ethosomes
The high concentration of ethanol in ethosomal formulations makes the vesicle membrane leaky thereby decreasing the drug entrapment. Hence, propylene glycol (stabilizing agent) was added as a permeation enhancer to increase drug retention in skin. In the ethosomal formulations, the ethanol concentration is varied by keeping the concentration of PEG constant. It should be noted that if we increase the concentration of PEG, there is a decrease in the entrapment efficiency of TH [25]. Our results suggested that PEG showed synergistic effect with ethanol at higher concentration. Hence PEG was kept constant i.e. 2% and the effect of ethanol concentration was studied at this concentration of PEG.
Selection of carbopol to prepare ethosomes gel based formulation
A hydrophilic polymer carbopol 934P was incorporated in the gel to control the drug release. It is an acid polymer which disperses readily in water, exhibits excellent compatibility and yields an acid solution with low viscosity [24].
Characterization of ethosomes entrapped with Terbinafine Hydrochloride (TH)
Drug entrapment efficiency
Table 2 shows DEE of ethosomes containing TH. The results indicated that in unsonicated ethosomes, the entrapment efficiency was increased from formulation F1 to F2 (40% w/v ethanol and 50% w/v ethanol). However, when the concentration of ethanol was increased from 50% w/v to 60% w/v for formulation F3, a decrease in entrapment efficiency was observed from 64.61 ± 2.13% to 55.33 ± 1.32%. This can be explained due to greater fluidity effect of ethanol which leads to drug leakage. The maximum amount of drug was loaded in sonicated ethosomal formulations from F5 to F8. The drug entrapment efficiency for the formulation F6 was found to be 69.11 ± 2.11% containing 50% w/v ethanol (Table 2). Nevertheless, the relatively high entrapment of TH within the ethosomal vesicles is explained by multilamerality and the presence of ethanol content. This shows that sonicated ethosomes of TH have more potential than unsonicated ethosomes. The result revealed that less soluble TH shows significant enhancement in the DEE by sonication method.
Table 2.
In-vitro diffusion kinetics, EE and VS of TH released from ethosomes.
| Formulation code | Drug entrapment efficiency (%) ± S.D | Zeta potential (mV) | Vesicle size (nm) | PI | Zero order |
First order |
Higuchi model |
Korsmeyer Peppas model |
|
|---|---|---|---|---|---|---|---|---|---|
| R2 | R2 | R2 | n | k | |||||
| F1 | 60.59 ± 1.92 | −7.10 ± 0.51 | 712 ± 1.32 | 0.317 ± 0.01 | 0.998 | 0.882 | 0.892 | 0.621 | 1.301 |
| F2 | 62.61 ± 2.13 | −7.21 ± 0.16 | 681 ± 1.56 | 0.216 ± 0.02 | 0.997 | 0.809 | 0.893 | 0.711 | 1.457 |
| F3 | 55.33 ± 1.32 | −7.19 ± 0.18 | 539 ± 1.41 | 0.208 ± 0.01 | 0.999 | 0.770 | 0.908 | 0.786 | 1.605 |
| F4 | 63.38 ± 1.18 | −7.16 ± 0.22 | 812 ± 2.21 | 0.389 ± 0.03 | 0.995 | 0.845 | 0.916 | 0.636 | 1.354 |
| F5 | 66.83 ± 1.52 | −7.61 ± 0.38 | 332 ± 1.12 | 0.214 ± 0.02 | 0.997 | 0.767 | 0.889 | 0.791 | 1.582 |
| F6 | 69.11 ± 2.11 | −7.96 ± 0.65 | 248 ± 1.02 | 0.206 ± 0.10 | 0.999 | 0.732 | 0.928 | 0.812 | 1.669 |
| F7 | 56.83 ± 1.09 | −7.41 ± 0.46 | 227 ± 1.11 | 0.212 ± 0.04 | 0.997 | 0.685 | 0.947 | 0.793 | 1.585 |
| F8 | 63.21 ± 2.62 | −7.13 ± 0.25 | 695 ± 1.25 | 0.312 ± 0.02 | 0.998 | 0.800 | 0.908 | 0.652 | 1.398 |
Vesicle size and shape
The vesicle size of the formulated sonicated and unsonicated ethosomes was found to be in the range of 539–812 nm and 227–695 nm, respectively. It was observed that vesicles size decreases with increase in the concentration of ethanol. The data obtained in Table 2 clearly indicate that as the quantity of phospholipids was increased, sizes of vesicles increased. The smallest vesicle for unsonicated ethosomes was observed with formulation F3 (539 ± 1.41 nm) and highest vesicle size with formulation F1 (712 ± 1.32 nm). The sonicated ethosomes formulation F7 shows smallest vesicle size of 227 ± 18 nm and the formulation F8 shows highest vesicle size 695 ± 1.25 nm. Significant difference in the vesicle size was observed for sonicated (F8) and unsonicated (F4) formulations. Moreover, sonicated ethosomes exhibited uniformity in the vesicular size and shape as compared to that of unsonicated ethosomes. The results showed that sonicated ethosomal formulation F6 exhibited vesicle size of 248 ± 11 nm. Verma et al. [7] view that vehicles up to 300 nm are able to release the medicament in deep layers of skin. Polydispersity index (PI) of various formulations is shown in Table 2. Compare to all formulations, F6 formulation showed a less PI value of 0.206 ± 0.10. It indicates the formation of homogeneous ethosomal vesicle that may finally lead to decrease in mean particle size.
The magnitude of the zeta potential gives an indication of the potential stability of the colloidal system. If all the particles in suspension have a large negative or positive zeta potential, then they will tend to repel each other and there will be no tendency for the particles to come together. However, if the particles have low zeta potential values, then there will be no force to prevent the particles to come together and flocculate. The zeta potential, for assessing the stability, showed that unsonicated and sonicated ethosomes were observed from 7.10 ± 0.51 mV to 7.96 ± 0.65 mV, meaning that the formulated ethosomes do not aggregate rapidly. The charge of vesicles is an important parameter that can influence both stability and skin vesicle interaction. The pH of the ethosomal formulation was found to be in the range of 3.8 ± 0.58 to 4.1 ± 0.11 (data not shown). The image of Scanning electron microscopy for the ethosomal formulation (F6) is shown in Fig. 6. It elucidates that the prepared ethosomal formulations show multilamellar vesicles.
Fig. 6.
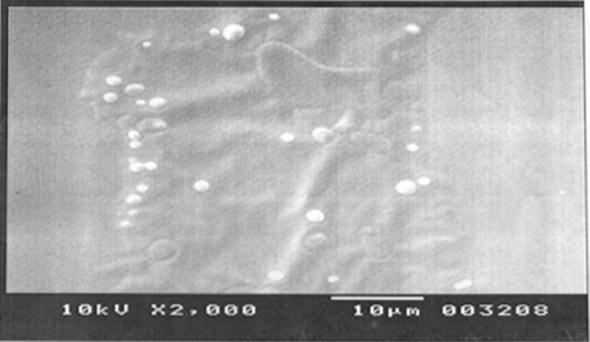
SEM images of ethosomal formulation (F6).
In vitro drug diffusion studies
The diffusion of entrapped TH molecules from the vesicles of sonicated and unsonicated ethosomal formulation was transferred to the surrounding media by diffusing through the cellophane membrane into the receptor medium. From Fig. 1a, the amount of TH diffused through a membrane from unsonicated formulations F1 and F3 was 70.74 ± 1.60% and 78.78 ± 0.36% after 12 h, and the enhancement in the diffusion of the drug was 8.7 ± 0.11% which might be due to the “penetration enhancer” effect of ethanol. In addition, when the concentration of phospholipids was increased from 1% to 2%, the amount of TH diffused for unsonicated formulation F2 was 75.72 ± 0.91% and that of formulation F4 showed 66.06 ± 1.80% after 12 h. It might be due to the effect of phospholipids which will allow the vesicles to fuse easily thereby leading to larger vesicle size. From our findings, it is clear that the release of TH was reduced with increasing phospholipids concentration, whereas increase in the alcohol content increases the drug release from the formulation F2 (75.72 ± 0.91% after 12 h). When the alcohol concentration was raised from 40% w/v to 60% w/v in formulation F1 to F3, a significant difference in the diffusion of the drug was observed. It might be due to increased fluidity of the bilayer membranes with increasing concentration of ethanol. Similarly, the amount of drug diffused through a dialysis membrane from the sonicated formulations F5 and F7 was 79.47 ± 1.08% and 88.31 ± 0.91% after 12 h. The amount of drug diffused from formulation F7 was higher among the sonicated ethosomal formulations. Moreover, the amount of drug diffused from formulation F7 was also higher than all unsonicated ethosomal formulations but at the same time drug entrapment efficiency was found to be low when compared to the same sonicated ethosomes formulation. Among the sonicated ethosomes formulation, the higher drug entrapment efficiency was found with formulation F6 i.e. 69.11 ± 2.11% with small vesicle size of 248 nm, when compared with other sonicated ethosomal formulations. Amounts of drug diffused from sonicated ethosomal formulations F6 and F7 were 84.28 ± 1.03% and 88.31 ± 0.91% respectively as shown in Fig. 1b, which showed insignificant difference in the amount of drug diffused from both the formulations.
Fig. 1.
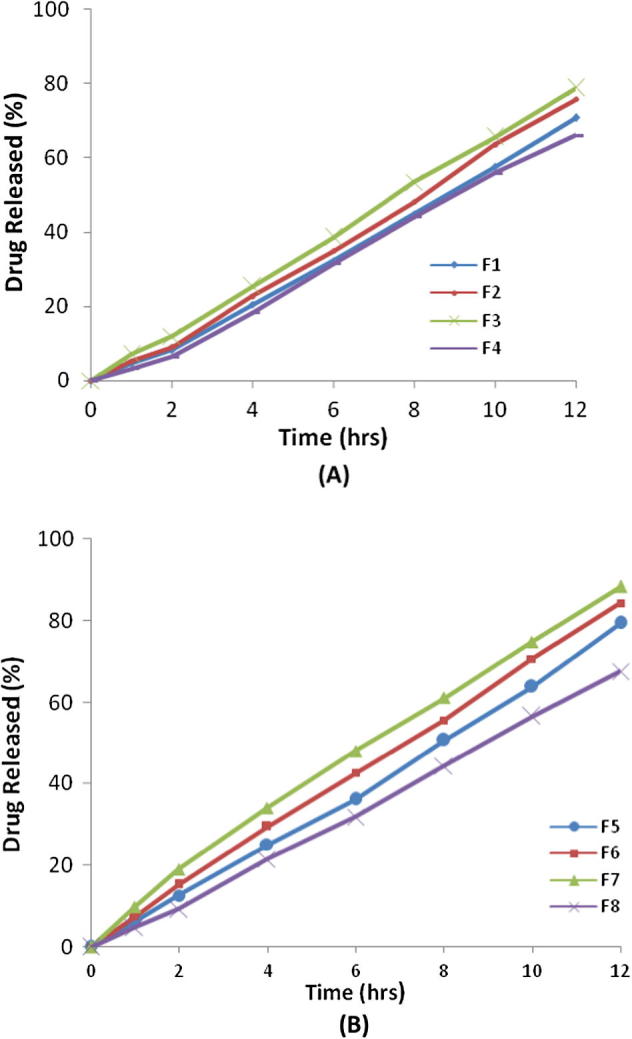
In-vitro drug release profiles of unsonicated formulations (A) and sonicated ethosomes formulations (B).
Kinetic analysis of dissolution data
The results suggested that the developed unsonicated and sonicated ethosomes followed zero order kinetics. The release pattern based on the diffusional exponents (n) values for the unsonicated ethosomal formulation ranges from 0.621 to 0.786 and that of sonicated ethosomal formulation ranges from 0.652 to 0.812. However, in case of F6 sonicated ethosomes formulation, it showed higher diffusional exponents which might be attributed to lower concentration of phospholipids. Similar results were also reported with soya lecithin concentration which resulted in an increase in mean particle size of ethosomal vesicles [9], [25]. This optimized amount of ethanol enables the drug to dissolve completely in the vesicle which facilitated the drug to pass through the membrane at a constant rate. Thus taking into consideration all the parameters, F6 was selected as the most satisfactory formulation and was further evaluated for the stability studies. The results of f2 values showed similar dissolution profiles for marketed cream (f2 = 85.14) and F6∗ ethosomal formulation (f2 = 42.63). Hence F6∗ ethosomal formulation showed dissimilar dissolution profiles.
Ex-vivo diffusion study of Ethosomes Gel
The in vitro studies show that the percentage of drug released from the sonicated ethosomes formulation F6 was 84.28 ± 1.03%, whereas sonicated ethosomes gel based formulation F6∗ showed 74.01 ± 0.62% drug release after 12 h. The study revealed significant difference in the drug release. No difference was observed in the percentage drug content of F6 sonicated ethosomes formulation and F6∗ sonicated ethosomes gel based formulation.
Fig. 4 shows the ethosomal gel based formulation when compared with marketed cream containing 1% w/w TH. The amount of drug diffused from marketed cream was 61.45 ± 0.86% which was lesser than the drug diffused from ethosomal gel i.e. 74.01 ± 0.62%. Transdermal flux value for ethosomal gel and marketed cream was found to be 144.61 ± 1.28 μg/cm2/h and 121.6 ± 1.16 μg/cm2/h, respectively. This data reveals that ethosomal gel (F6∗) resides at targeted site for a relatively longer period of time and signifies improved patient compliance.
Fig. 4.
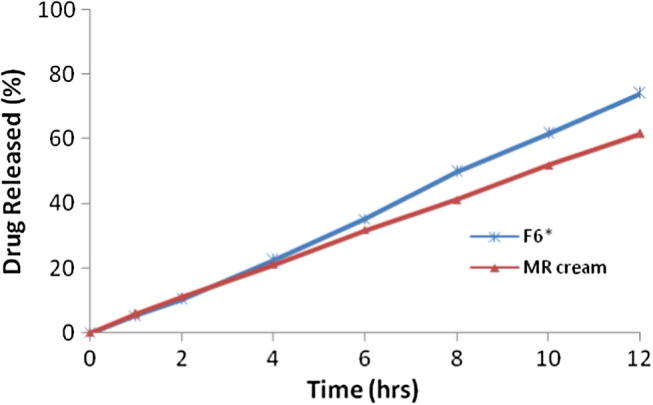
Comparison of ex-vivo diffusion study of formulation F6∗ and marketed cream.
The value of diffusion release exponent for ethosomal gel based formulations was found to be nearly 1, which indicates that absorption of drugs through the skin follows zero order release (Fig. 2). Systems that obey zero order release are ideal for transdermal drug delivery as they provide constant release of drug over an extended period of time and reflect improve in therapeutic index [26]. The kinetics of TH form the gel based formulation (F6∗) across skin shows zero order release profile, when compared with cellophane membrane (non-Fickian). This may be due to the fact that skin has complex structure and release profile of drugs from the ethosomal gel based formulation through skin cannot be exactly matched with cellophane membrane. It is suggested that other than the ethosomes, skin also modified the diffusion properties of TH which can be explained in a way that some component of the skin might be interacting with the ethosomes and altering its diffusion properties. The sonicated ethosomal gel based formulations can be prepared easily with high entrapment efficiency with TH as compared to solid lipid nanoparticle formulations by avoiding many steps such as co-processing of the lipids [27].
Fig. 2.
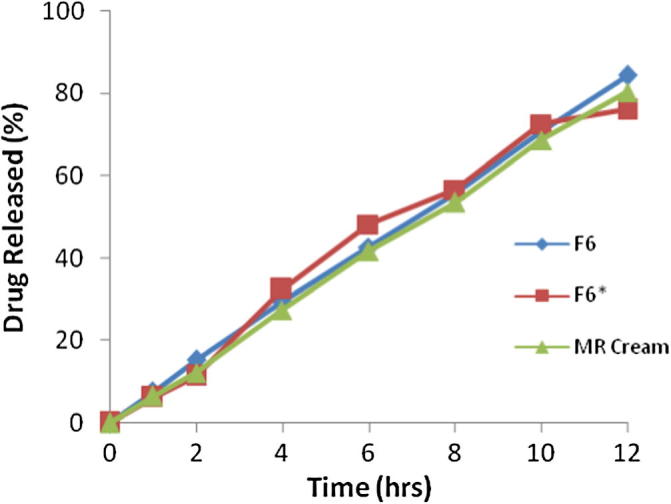
Comparison of in-vitro drug release profiles for the formulation F6, ethosomal gel (F6∗) and marketed cream.
The percentage of drug retention remained highest for ethosomes gel based formulations when compared to the marketed cream and the values obtained for ex vivo studies are nearly equal to the values of in vitro studies. Our results are in agreement with the findings obtained by Sakine and Ozgen for terbinafine loaded liposomal and ethosomal gels for nail drug delivery [16]. Hence, it can be concluded that TH ethosomal gel incorporated with hydrophilic carbopol could exhibit the required quantity of gel effectively for extended period of time with a zero order release profile.
FT-IR studies
FT-IR spectrum of pure TH shows C C stretching bands at 1517.17 cm−1, aromatic C C stretching bands at 2223.19 cm−1, aromatic C—H stretching bands at 3038.08 cm−1, aromatic alkenyl C C—H stretching bands at 2966.58 cm−1, C—N bands at 1134.28 cm−1 and aliphatic C—H stretching bands at 2563.53 cm−1 (Fig 5a). The spectra of phospholipids showed aliphatic C—H bands from 2863.69 cm−1 to 2922.86 cm−1, C O bands at 17,400 cm−1 and C C stretching bands from 1588.28 cm−1 to 1519.92 cm−1 as shown in Fig 5b. Similar peaks were found in the spectra of F6 formulations. It exhibited aliphatic C—H stretching bands at 1735.14 cm−1, aromatic C C stretching bands at 2384.74 cm−1, alkenyl C C—H stretching bands at 2953.53 cm−1 and C—N bands at 1057.28 cm−1. The major peaks for the pure drug and the formulation were well supported with the theoretical estimation with respect to the functional groups.
Fig. 5.

FT-IR spectra of (A) Terbinafine Hydrochloride, (B) phospholipids and (C) formulation (F6).
Stability studies
Stability studies were carried out for the most satisfactory formulation (F6) at 4 °C and at room temperature 25 °C for 2 months. After two months, the samples were evaluated and DEE was found to be 62.11 ± 2.11% with the vesicle size of 247 ± 1.02 nm. This indicated the stabilizing effect of ethanol. In vitro drug diffusion before and after study was 84.14 ± 1.12% and 82.23 ± 1.10% after 12 h respectively (Fig. 3). It can be explained due to effect of temperature on gel-to-liquid transition of lipid bilayer.
Fig. 3.
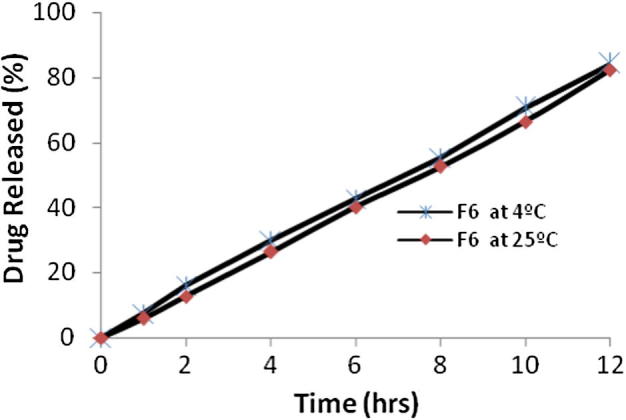
Comparison of in-vitro drug release profiles for the formulation (F6) at 4 °C and 25 °C.
Conclusions
TH loaded ethosomal formulation was successfully prepared by loading phospholipids and ethanol, and ethosomal gel based formulations were prepared with hydrophilic polymer Carbopol 934P. It can serve as a useful vehicle for the delivery of TH through the affected part of the skin for extended period of time. This study also revealed that ethosomal gel (F6∗) resides at targeted site for a relatively longer period of time with a zero order release profile. It signifies the improved patient compliance.
Conflict of Interest
The authors have declared no conflict of interest.
Compliance with Ethics Requirements
This article does not contain any studies with human or animal subjects.
Acknowledgments
The authors are thankful to Orchid Pharmaceuticals Chennai, India, and Sandoz chemicals, Mumbai, India, for generously giving the sample of terbinafine hydrochloride and propylene glycol.
Footnotes
Peer review under responsibility of Cairo University.

References
- 1.Touitou E., Dayan N., Bergelson L., Godin B., Eliaz M. Ethosomes as novel vesicular carriers for enhanced delivery: characterization and skin penetration properties. J Control Release. 1999;65:403–418. doi: 10.1016/s0168-3659(99)00222-9. [DOI] [PubMed] [Google Scholar]
- 2.Bendas E.R., Tadros M.I. Enhanced transdermal delivery of salbutamol sulfate via ethosomes. AAPS Pharmscitech. 2007;8:1–7. doi: 10.1208/pt0804107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Essa A.E., Bonner M.C., Barry B.W. Electroporation and ultradeformable liposomes; human skin barrier repair by phospholipids. J Control Release. 2003;92:163–172. doi: 10.1016/s0168-3659(03)00326-2. [DOI] [PubMed] [Google Scholar]
- 4.Godin B., Touitou E. Mechanism of bacitracin permeation enhancement through the skin and cellular membranes from an ethosomal carrier. J Control Release. 2003;94:365–379. doi: 10.1016/j.jconrel.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 5.Touitou E., Godin B., Dayan N. Intracellular delivery mediated by ethosomal carrier. Biomaterials. 2001;22:3055–3059. doi: 10.1016/s0142-9612(01)00052-7. [DOI] [PubMed] [Google Scholar]
- 6.Elsayed M.A., Abdallah Y.O., Naggar F.V., Khalafallah N.M. Lipids vesicles for skin delivery of drugs: reviewing three decades of research. Int J Pharm. 2006;332:1–16. doi: 10.1016/j.ijpharm.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 7.Verma D.D., Verma S., Blume G., Fahr A. Particle size of liposomes influences dermal delivery of substances into skin. Int J Pharm. 2003;258:141–151. doi: 10.1016/s0378-5173(03)00183-2. [DOI] [PubMed] [Google Scholar]
- 8.Cosco D., Celia C., Cilurzo F., Trapasso E., Paolino D. Colloidal carriers for the enhanced delivery through the skin. Expert Opin Drug Deliv. 2008;5:737–755. doi: 10.1517/17425247.5.7.737. [DOI] [PubMed] [Google Scholar]
- 9.Touitou E., Dayan N., Bergelson L., Godin B., Eliaz M. Ethosomes—novel vesicular carriers for enhanced delivery: characterization and skin penetration properties. J Control Release. 2000;65:403–418. doi: 10.1016/s0168-3659(99)00222-9. [DOI] [PubMed] [Google Scholar]
- 10.Sinha V.R., Bansal K., Kaushik R., Kumria R., Trehan A. Poly-epsilon-caprolactone microspheres and nanospheres: an overview. Int J Pharm. 2004;278:1–23. doi: 10.1016/j.ijpharm.2004.01.044. [DOI] [PubMed] [Google Scholar]
- 11.Pardeike J., Hommoss A., Müller R.H. Lipid nanoparticles (SLN, NLC) in cosmetic and pharmaceutical dermal products. Int J Pharm. 2009;366:170–184. doi: 10.1016/j.ijpharm.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 12.Bendas E.R., Tadros M.I. Enhanced transdermal delivery of salbutamol sulfate via ethosomes. AAPS Pharmscitech. 2007;8:E107. doi: 10.1208/pt0804107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhalaria M.K., Naik S., Misra A.N. Ethosomes: a novel delivery system for anti-fungal drugs in the treatment of topical fungal disease. Ind J Exp Biol. 2009;(47):368–375. [PubMed] [Google Scholar]
- 14.Bouwstra J.A., Honeywell-Nguyen P.L. Skin structure and mode of action of vesicles. Adv Drug Deliv Rev. 2002;54(Suppl. 1):S41–55. doi: 10.1016/s0169-409x(02)00114-x. [DOI] [PubMed] [Google Scholar]
- 15.Newland J.G., Rahman A.S.M. Update on terbinafine with a focus on dermatophytoses. Clin Cosmet Investig Dermatol. 2009;2(2):49–63. doi: 10.2147/ccid.s3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sakine T.T., Ozgen O. Novel topical formulations of Terbinafine-HCl for treatment of onychomycosis. Eur J Pharm Sci. 2013;48(6):628–636. doi: 10.1016/j.ejps.2012.12.014. [DOI] [PubMed] [Google Scholar]
- 17.Godin B., Touitou E., et al. A new approach for treatment of deep skin infections by an ethosomal antibiotic preparation, an in vivo study. J Antimicrob Chemother. 2005;55:989–994. doi: 10.1093/jac/dki125. [DOI] [PubMed] [Google Scholar]
- 18.Rakesh P.P., Hardik H.P., Ashok H.B. Formulation and evaluation of carbopol gel containing liposomes of ketoconazole. Int J Drug Deliv Technol. 2009;1(2):42–45. [Google Scholar]
- 19.Patel K.K. A validated RP-HPLC method for determination of terbinafine hydrochloride in pharmaceutical solid dosage form. Int J of Pharm Technol. 2012;4(3):4663–4669. [Google Scholar]
- 20.Iizhar S.A., Mansor A., Arief M. Formulation and characterization of mucoadhesive buccal films of trimetazidine di hydrochloride. Lat Am J Pharm. 2015;34(8):1585–1593. [Google Scholar]
- 21.Vaibhav D., Dinesh M.J., Jain N.K., Tathagata D., Manoj N., Saraf D. Dermal and transdermal delivery of an anti-psoriatic agent via ethanolic liposomes. J Control Release. 2007;123:148–154. doi: 10.1016/j.jconrel.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 22.Subheet J., Ashok K.T., Bharti S., Jain N.K. Formulation and evaluation of ethosomes for transdermal delivery of lamivudine. AAPS Pharmscitech. 2007;8(4):E1–E9. doi: 10.1208/pt0804111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dayan Nava, Touitou E. Carriers for skin delivery of trihexyphenidyl HCl: ethosomes vs liposomes. Biomaterials. 2000;21:1879–1885. doi: 10.1016/s0142-9612(00)00063-6. [DOI] [PubMed] [Google Scholar]
- 24.Sandhu P., Bilandi A., Kumar S., Rathore D., Bhardwaj S. Additives in topical dosage forms. IJPCBS. 2012;2(1):78–96. [Google Scholar]
- 25.Pathan I.B., Nandure H., Syed S.M. Transdermal delivery of ethosomes as a novel vesicular carrier for paroxetine hydrochloride: in vitro evaluation and in vivo study. Marmara Pharm J. 2016;20:1–6. [Google Scholar]
- 26.Vaibhav D., Dinesh M., Manoj N., Vikas J., Jain N.K. Enhanced transdermal delivery of an anti-HIV agent via ethanolic liposomes. Nanomed Nanotechnol Biol Med. 2010;6(2):590–596. doi: 10.1016/j.nano.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 27.Tiwari S., Mistry P., Patel V. SLNs based on co-processed lipids for topical delivery of terbinafine hydrochloride. J Pharm Drug Dev. 2014;2(14):1–8. [Google Scholar]