

Abstract
Type 1 diabetes mellitus (T1DM) as one of the most well-known autoimmune disease, results from the destruction of β-cells in pancreas by autoimmune process. T1DM is fatal without insulin treatment. The expansion of alternative treatment to insulin is a dream to be fulfilled. Currently autoimmunity is considered as main factor in development of T1DM. So manipulation of the immune system can be considered as alternative treatment to insulin. For the past decades, vitamin A has been implicated as an essential dietary micronutrient in regulator of immune function. Despite major advantage in the knowledge of vitamin A biology, patients who present T1DM are at risk for deficiency in vitamin A and carotenoids. Applying such evidences, vitamin A treatment may be the key approach in preventing T1DM.
Keywords: Diabetes, Autoimmune, Pancreas, Insulin, Vitamin A
Core tip: Diet modification and vitamin supplementation is a practical treatment approach for autoimmune diseases. However few broadly studies have been conducted on the use of vitamin A in the treatment of type 1 diabetes. Our objective is to consolidate the current literature to better delineate the vitamin A on immune pathway involved in formation of type 1 diabetes.
INTRODUCTION
Type 1 diabetes mellitus (T1DM) is a well-known autoimmune disease that is characterized by a state of T cell-mediated selective deficiency of absolute or relative insulin-producing β-cells[1-4]. Despite modern medical management, T1DM is still one of the most common chronic childhood diseases[5]. T1DM eventually ends up in several disorders including renal failure, ketoacidosis, heart disease, stroke and visionless[1]. It is estimated that T1DM affects 497100 children under 15 years globally[5]. Currently administration of exogenous insulin has been the core strategy of treatment for patient with T1DM[4,6]. However it is not without complications. So, alternative preventive and treatment approaches to insulin are required. It has been shown that manipulation of the immune system with altering the course of the disease can be consider as alternative preventive and treatments approach to insulin[7]. The effects of vitamin A on immune system have been studied more than any other nutrients[8]. The concept of modulation of immune system by vitamin A dates back to the early twenties and the work of Green et al[9,10] who reported that vitamin A and β-carotene have “anti-infective” properties. Today, dietary vitamin A and its derivatives are recognized as crucial agents for normal immune system function and regulation[11].
Interestingly, recent studies have demonstrated that vitamin A deficiency leads to defects in glucose-stimulated insulin secretion[12]. In addition presence of relatively high levels of cellular retinol binding protein (RBP), cellular retinoic acid (RA) binding proteins, transthyretin (TTR) and RBP in pancreatic rat islets have been documented[13-15]. Despite major advantage in the knowledge of vitamin A biology, patients who present T1DM are at risk for deficiency in vitamin A and carotenoids[16,17]. It has been shown that bioavailability and plasma concentrations of vitamin A, TTR, retinol, and RBP fall in children and adults with T1DM[18-22].
The action of vitamin A to control immune response has led to a growing hypothesis of potential role of vitamin A in T1DM as an autoimmune disease. This review highlights new information regarding to vitamin A and RA in regulation of immune responses in patient with T1DM. So, the purpose of the current study was to thoroughly review the function of vitamin A in the immunology of T1DM.
RESEARCH
We performed a comprehensive literature search of the subject using MEDLINE and PubMed, for “vitamin A”, OR “retinoic acid” OR “retinol” AND “type 1 diabetes mellitus” OR “T1DM”. All papers fulfilling the above criteria were considered. All papers obtained in the search were fully discussed by the authors. References lists of all original published articles were scanned to find additional eligible studies.
VITAMIN A/RA METABOLISM AND SIGNALING
Vitamin A is an essential nutrient that can be acquired from the diet either as preformed vitamin A [primarily as retinyl ester (RE), retinol, and in much smaller amount as RA] or provitamin A carotenoids[23]. This vitamin has been well known for its critical function in embryonic development, vision and the nervous system, as well as in regulation and development of the immune system[24]. Both dietary vitamin A as vegetable and fruit-derived carotenoids and REs from animal sources are converted to retinol within the lumen of the small intestine or the intestinal mucosa and then enzymatically re-esterified with long-chain fatty acids within the enterocyte to form RE[23,25-27]. REs are packaged into chylomicrons together with other dietary lipids and secreted into the lymphatic system[27]. The liver is the primary organ for storage of vitamin A, where the retinol form of vitamin is esterifies by lecithin: Retinol acyl transferase and stored as a RE[28,29]. To meet the tissue vitamin A needs, retinol released into the circulation from liver and bound to its specific transport protein, retinol-binding protein (RBP or RBP4)[30,31].
In the liver and the peripheral stream, vitamin A is mainly in the form of retinol and REs. Although the function of vitamin A is applied in its metabolite form RA[28]. So, its precursors must be converted to RA by a two-step process[32]. First, retinol is hydrolyzed into retinal by ubiquitous alcohol dehydrogenase, and then irreversible hydrolysis reaction allows the formation of RA[33]. Regulation of gene expression by RA, and the discovery of RA receptor and retinoic acid X receptor which are specific receptors for the active metabolites of vitamin A such as all trans and 9-cis-retinoic acids, provided fundamental documents for the understanding retinoids effects on immune function[34-36]. RA is inactivated by CYP26A1, CYP26B1 and CYP26C1[28,29].
PREDIABETES STAGE
Type 1 diabetes is an autoimmune disease, which result from development of islet autoantibodies against proteins in insulin producing beta cells and immune-mediated destruction of insulin producing beta cells in the pancreas[37]. These individuals with antibody positive within many years are at risk of developing T1DM[38-40]. Progressive autoimmune β-cell damage usually precedes the clinical onset of diabetes, and occurs years before any clinical symptom of T1DM[41,42]. This long pre-diabetes phase making T1DM as a predictable disease, and provides an opportunity to prevent individuals with active insulitis from developing clinical disease[37].
So T1DM will be a preventable disease by the intervention targeting the manipulating of immune system. In this context one approach is the trimolecular complex, including a self-reactive CD4 T cell, insulin, and HLA molecule[43].
TYPE 1 DIABETES AND INNATE IMMUNE RESPONSES
The body's first defense system against microorganism invasion is the innate immune system[44]. Unlike the adaptive immunity, the response mounted by the innate immune system is relatively nonspecific, that mediated primarily by macrophages, dendritic cells (DCs), and granulocytes, basically functioning as phagocytes and APCs[45].
The innate immune response depends on the recognition of the microbial-associated molecular patterns (MAMPs), through special cell receptors called pattern recognition receptors (PRRs)[46,47]. PRRs enable innate immune system to sense and recognize specific microbial compounds known as MAMPs[46]. PRRs comprise at least three distinct families: RA-inducible gene-I-like helicases, nucleotide oligomerization domain-like receptors (NLRs), and Toll-like receptors (TLRs)[48].
The TLR family, best known and characterized in mammals, is composed of 13 receptors, which when activated cause activation of the immune system[49]. TLRs are able to recognize extracellular and endocytosed ligands[50]. Activation of TLR starts a cascade of pro-inflammatory reactions that leads to increased expression of specific cytokines, chemokines, and co-stimulatory molecules[51]. In type 1 diabetes, it has been demonstrated that TLRs, as the result of autoreactive processes directed against self antigens, may be priming an unwarranted adaptative immune response[52]. According to a study of Devaraj et al[53] the monocytes attained from type 1 diabetic patients expressed TLR2 and TLR4 more than the control group. Furthermore shown in these patients the TLRs activity as well as the targets of the downstream TLR signaling including nuclear factor-κB (NF-κB), MyD88, and TIR-domain-containing adapter-inducing interferon (IFN)-β were all respectively more expressed. So, TLR2 and TLR4 signaling may have significant role in development of type 1 diabetes[54]. However, TLR3 is not required for onset of autoimmune diabetes, while TLR9-deficient compared to TLR9 heterozygotes mice showed a significantly decreased incidence of diabetes[55].
NLRs and C-type lectin receptors have not been reported to be directly related to autoimmunity. However, they may trigger autoimmune responses or initiate the adaptive immune system by autoimmune mechanisms[56].
Macrophage
The early studies indicated the role of macrophages in the pathogenesis of T1DM[57]. It has been reported that the islet infiltrates of young non-obese diabetic (NOD) mice contain macrophages and if the influx of these cells into the pancreas is inhibited, development of type 1 diabetes is prevented[58]. In addition, according to animal models macrophages produce proinflammatory cytokines such as tumor necrosis factor (TNF)-α and interleukin (IL)-1β which could be pathogenic for B cells[59,60]. Cytotoxic T cells are activated in the presence of macrophages, which subsequently destroy pancreatic β-cells[61]. Overall, the current evidence supports a pathogenic role for macrophages in initiation and development of T1DM.
DCs
DCs, a group of diverse intrinsic effectors which have two main actions relevant to T cell immune system controlling, including: Presentation of antigens to T cells and determining the nature of T cell response[62]. In vivo, as compared to healthy controls, DCs were located around the pancreatic islets in type 1 diabetic patients[63]. DCs present in the pancreatic islets of type 1 diabetic patient which suggests that these cells have a direct or indirect role in β-cell destruction[63]. In T1DM, DCs function as important APCs, DCs degrade the T cell response to antigen presentation[63]. Together, these studies support a diabetogenic role for DCs in the initiation steps of this disease.
Natural killer cells
Natural killer (NK) cells can recognize and kill virus-infected cells through a number of different mechanisms; in addition NK cells have a critical role in immune regulation[64]. Data on NK cells in patients with T1DM are inconclusive[1]. Following stimulation by pro-inflammatory cytokines, NK cells generate a large quantity of cytokines such as IFN-γ, TNF-α, and granulocyte macrophage-colony-stimulating factor (GM-CSF)[65]. According to animal models, NK cells can play a consequential role in the development of T1DM; however human studies accomplished in this field are rare. Evidence from animal model and man have shown that NK cells are potentially involved both in progression and protective of type 1 diabetes, thus suggesting a dual role for these cells in type 1 diabetes pathogenesis[66]. NK cells shown differently role in different stages of diseases to the disease pathogenesis[66]. These cells are the main source of IFN-γ, and therefore, they regulate the intensity of the immune attack in diabetes and also the progression of insulitis to diabetes[67].
The NK receptors consist of two main families including NKG2A, for HLA E molecules, and the killer cell immunoglobulin-like receptors (KIR), for the recognition of HLA A, B, and C molecules[68]. Increased frequency of KIR gene haplotypes has been observed in patients with type 1 diabetes[68].
In the blood of diabetic patients and in lymphoid tissues of NOD mice NK cell function has been impaired. Also, in type 1 diabetic patients a slight decrease of NKG2D expression has been observed. However animal studies suggested that NK cells activity was detected only in the early pre-diabetic infiltrates[1]. Most pancreatic NK cells of NOD mice became hypo-responsive during the later stages of diabetes development, as it is detected by lower cytokine secretion and a higher tendency for degranulation as a reaction toward antibodies which are distinct for receptor activation[69].
According to the available evidence, NK cells can exhibit protective functions in β-cell autoimmunity conceivably by the down-regulation of T-cell lymphocytes and by the generation of IFN-γ.
Neutrophils
Neutrophils are a part of the immune system which do not act specifically. These cells have a key role in the host immune system against different bacterial infections during the early host response to infection[1]. Neutrophils express many chemokine receptors, including CXCR1 and CXCR2, which respond to early chemokines released by macrophages. Neutrophils also express chemotactic receptors for complement, lipid mediators, and bacterial products[70,71]. So, neutrophils react to different chemo-attractants including lipid mediators, complement fragments and bacterial products[72,73].
Many studies for the roles of neutrophils in the pathogenesis of diabetic complications have been carried out and ended with many controversies. According to previous studies, neutrophil dysfunction in chemotaxis, phagocytosis, killing bacteria and the release of superoxide in type 1 diabetic patients and animal models are not a cause but an effect of disease[74-77].
VITAMIN A AND INNATE IMMUNE SYSTEM
Macrophage
The available evidence has introduced retinoids as important regulators of monocytic/macrophages function[78-81]. According to RA effect on monocytic/macrophages, it is shown that RA restrains the secretion of cytokines that promote the production of Th1-type cells and also it increases the secretion of cytokines that promote the production of Th2-type cells[82]. Macrophages secrete cytokines like TNF and nitric oxide (NO) under activation conditions[83]. RA affects the secretion of major cytokines generated by macrophages, including TNF-α, IL-1, IL-6, and IL-12[81]. A number of studies have shown that all-trans-RA extremely reduced the mRNA levels of TNF, regulates NO production, and increases IL-1 generation[82].
Kim et al[84] studied the impact of RA on a mouse model macrophage and its oblique effect on T cells. In their study they pretreated the macrophages with RA and precedingly activated them with lipopolysaccharides. In regard to the previous study, it was shown that RA inhibited the production of pro-inflammatory mediators (IL-12 production) by activating macrophages and the macrophages treated with RA when applied as antigen presenting cells (APCs) decreased the T-cell production of IFN-γ and increase the generation of IL-4. Collectively RA signaling seems to set up a Th2-Treg non-inflammatory base[84,85]. Using RA-treated macrophages as APCs in co-cultures, result in IL-12 reduction and also T cell-derived IFN-γ and IL-4 levels down-regulated and up-regulated, significantly[86]. Supplementation with vitamin A at 6500 IU/d for 6 mo in 6 patients with common variable immunedeficiency, who had low serum retinol concentrations, decreased the TNF-α level in comparison to onset levels[87]. The overall results show that supplementing with the preformed vitamin A may decrease the production of particular proinflammatory cytokines [monocyte-derived DCs (MoDCs), TNF-α and IL-6] by macrophages[8] (Figure 1).
Figure 1.
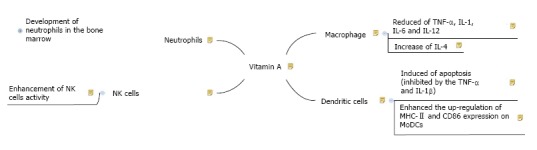
Schematic diagram for the pathways of vitamin A effects on innate immune system. NK: Natural killer; TNF: Tumor necrosis factors; IL: Interleukin; MoDCs: Monocyte-derived dendritic cells.
DCs
DCs, the primitive guardian cells which activate the development of adaptive immunity, can act as APCs and establish immune responses[88]. Therefore, RA’s influence on this kind of cell could have a major role in initiating the adaptive immunity[79]. Apoptosis was induced by retinoids in immature MoDCs, this is inhibited by the secretion of cytokines like TNF-α and IL-1β. Also, retinoids enhanced the up-regulation of MHC-II and CD86 expression on MoDCs[79]. IL-4 and RA act synergistically on some populations of DCs and reduce the production of proinflammatory cytokines[89,90]. Furthermore RA could regulate the immunosuppressive properties of human tolerogenic DC and also mediate the transformation of B cells into B-regs[91] (Figure 1).
NK cells
NK cells, part of the innate immune system, are critical in the first line of defense against tumors and viral infections[92]. These cells play an immune-regulatory role in antibody production and cell-mediated immunity through their production of various cytokines[93]. Previous investigations reported that vitamin A deficiency has a fundamental effect on NK cell lytic activity in young rodents[94]. Deficiency of vitamin A reduces the activity of NK cells and the ability of spleen cells to produce IFN after mitogen stimulation[95,96]. According to in vitro and in vivo studies, using physiologic or high concentrations of retinoids result in an enhancement of NK cell activity[97-99]. However, the mechanism for this stimulation is not fully clears[99]. A U-shaped relationship between vitamin A and NK cells has suggested which both low and high doses of vitamin A may have deleterious effects on NK cell hematopoiesis, differentiation or function[100]. In addition there is an interesting progressive relationship between the degree of vitamin A deficiency and the observed immune-suppression[101] (Figure 1).
Neutrophils
The importance of the neutrophils is recognized in animals with neutropenia or a deficiency of any key neutrophils enzymes[102,103]. In these animals, mild infections can be life threatening[102,103]. The neutrophils differentiates requires the oxidized form of retinol, RA[104,105]. The development of neutrophils in the bone marrow is controlled by the genes that are modulated by RA receptors, and RA in cultures accelerates maturation of neutrophils[106,107]. According to previous studies, treatment with RA or vitamin A could restore the level of neutrophils and the capacity of superoxide-production in calves and rats significantly[108,109]. Vitamin A deficient rats had significantly higher numbers of hyper-segmented neutrophils (67%) relative to those in the control rats[103]. However the data on the relationship between vitamin A and neutrophils function in humans are sparse and inconclusive[8] (Figure 1).
TYPE 1 DIABETES AND ADAPTIVE IMMUNE RESPONSES
The adaptive immune system, which is the body’s second defense system against pathogens, functions by its antigen-specific structure which distinguishes foreign molecules by their antigens which is mediated by the interaction between T cells and APCs. It is generates long-term response by using immunological memory. T cells and T cell receptor are the essential section in the adaptative immune system[1].
It is currently accepted that T cells play an important role in type 1 diabetes pathogenesis[110]. These cells are the most important players in the autoimmune attack of β-cells[110]. Anti-islet T cells, including both: CD4 and CD8 T cells have been observed in patients with T1DM[111]. It has been shown that transfer of anti-islet specific CD4 or CD8 T cells can lead to diabetes, as follows; insulitis and diabetes can be adoptively transferred by T cells from diabetic mice into non-diabetic mice, whereas B cells are not needed[111-113]. In fact insulitis seen in T1DM is induced by diabetogenic T cells that then recruit heterogeneous mixture of cells[114].
CD8 T cells directly attack β-cells by MHC type 1 expressed on pancreatic β-cells, therefor in the absence of beta-2 microglobulin which reduces MHC type 1 or in the status of beta-restricted MHC type 1 deficiency, it is adequate to stop diabetes development and to avoid β-cell demolition in NOD mice[115,116].
CD4 T cells are activated by β-cell APCs, and they mainly provide cytokines such as IL-21 to help both B cells and CD8 T cells, which is required for the development of T1DM in NOD mice. CD4 T cells secrete IFN-γ, stimulating macrophages to release other cytokines, such as IL-1β, TNF-α, and free radicals, which are toxic to β-cells[44].
Lymphocytes can kill β-cells directly through a cytotoxic process or by the secretion of proinflammatory cytokines, such as IL-1β, IFN-γ; they also release free radicals, which destructs the pancreatic β-cells. Cytokines induce the production of inducible NO synthase which results in NO production and NO synthesis influences the β-cell death[117]. Free radicals can induce, in turn, apoptosis and necrosis of β-cells[118].
In the disease progression phase, both T and B lymphocytes can be activated against self antigens in an islet lesion and trigger an immunological response that leads to the destruction of pancreatic β-cells[119,120] (Figure 2).
Figure 2.
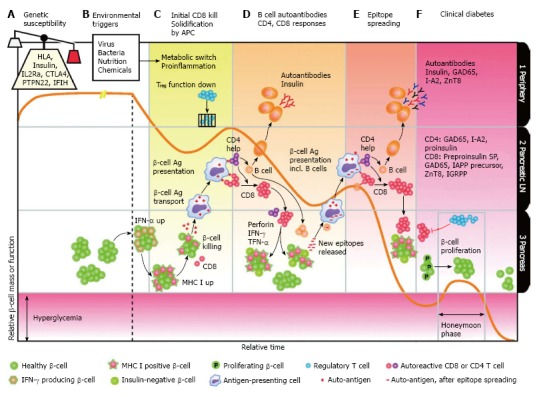
Immunology of type 1 diabetes[115]. TNF: Tumor necrosis factors; IL: Interleukin; MoDCs: Monocyte-derived dendritic cells; APCs: Antigen presenting cells; IFN: Interferon; HLA: Human leukocyte antigen.
VITAMIN A AND ADAPTIVE IMMUNE RESPONSES
A study by Iwata et al[121] for the first time described the role of RA in the biology of T cells. Recently evidenced T-cell immune-competence can be affected by vitamin A deficiency[8].
Transforming growth factor (TGF)-β which is a suppressor of Th1 and Th2 differentiation and the inducer of transforming T-cells to Tregs or to Th17, mediates RA in the process of formation, differentiation and inhibition or activation of Th1, Th2, Th17 and Treg lymphocytes[122]. The main impact of RA in lymphocyte differentiation by TGF-β is the transformation of Th17[123,124]. RA has a two way affected on Th17 which on one hand it promotes its differentiation and on the other hand it downregulates it[125]. Th17 appears to be the example of customized immunity for special types of pathogens, but the abnormal Th17 responses could be involved in an exceeding number of autoimmune dysfunctions[126]. In some types of irregular immune responses, the defective form of RA via a genetic or an environmental mechanism intermediates the complex regulation of Th17[127].
A growing number of evidence indicates that vitamin A is involved in the modulation of IL-10 production. IL-10, secreted by Th2-helper T cells, restricts the production of pro-inflammatory Th1-type cytokines, such as IFN-γ and IL-2, in both T and NK cells. This is a major mechanism in reducing the inflammatory responses to some defects[128].
A recent review demonstrated that in the status of vitamin A deficiency, Th1-cells mediate immune responses and when vitamin A is supplemented it induces Th2 immune responses[129]. Results from a number of studies that investigated the effect of vitamin A on infections that lead to one of the two immune responses, Th1 or a Th2 response, intimate that the immunological functions of vitamin A are specific for every pathogene and may involve other parts of the immune system other than Th1 or a Th2. Further studies are needed to examine the mechanism of vitamin A supplementation on differential of Th1/Th2 responses comparing to the baseline vitamin A status in humans and the specific pathogens that cause disorders[8].
Overall, there is no exact evidence to state for a direct role of vitamin A supplementation on cytokine secretion or lymphocyte production. One principle reason for the wide range of results in studies is the specific immune response toward each pathogen which may affect the impact of vitamin A on T-cell function. Also, vitamin A may have momentary effects on intermediary factors of T-cell-dependent immunity which may not have been noticed in some population studies. Few randomized clinical trials have been accomplished on the topic of vitamin A supplementation on the proliferation or activation of B lymphocytes[8].
REVIEWS ON T1DM AND VITAMIN A
In the status of lack of balance between different subtypes of T-cells, autoimmune diseases occur[130]. For instance, IFN-γ-producing CD4 or CD8T effectors (Teff) cells activation and expansion and/or reduction of the number or function of CD4T regulatory (Treg) cells, can result in autoimmune diseases[130-134]. Present evidence showed that both CD4+ and CD8+ Teff cells are related in the initiation and further development of type 1 diabetes[135-137]. CD4+ and CD8+ Teff cells which are typically reactive with antigens on the β-cells in the pancreas can cause type 1 diabetes, an autoimmune disease[138]. Recent studies introduced IL-17-producing CD4 Th17 cells as a new generation of Teff cells that trigger potent inflammatory responses resulting in autoimmune disorders[139]. These results suggest that the establishment of effective in vivo immune-tolerance can be considered as practical strategy to treat autoimmune diseases such as type 1diabetes. However this approach requires simultaneous targeting of more than one T-cell population subset. Therefore, immune tolerance induced by clinically relevant agents or methods affecting various T-cell subtypes could demonstrate an effective way for treating human autoimmune disorders[140].
Vitamin A and its derivatives are potent immune tolerance agents by its ability to transform Th1 to Th2 lymphocytes[138,140]. Vitamin A regulates the adaptive and innate immune responses by different mechanisms for example, its high-level can diminish development of Th1 and promote development of Th2 responses[141]. Vitamin A supplementation results in a decrease in serum pro-inflammatory cytokines, such as TNF-α and IFN-γ, and an increase in the immunosuppressive cytokine IL-10[87,142]. Such immune modulation by vitamin A could decrease the development risk of autoimmune diseases[143]. Present evidence reported high level of dietary vitamin A may have major effects on down regulating inflammatory immune cells and reducing the damage caused by oxidation in the islets that contribute to dysfunction of β cells. An animal study conducted by Zunino et al[138] showed that intervention with a diet rich in vitamin A inhibited the development of type I diabetes in mice by reducing or delaying of the infiltration of immune cells in to the islets.
Furthermore vitamin A plays a role in the release of insulin and glucagon hormones[12], and therefore has profound effects throughout the body in the regulation of glucose homeostasis[12,13]. Vitamin A in active form has an important role in the secretion and release of insulin in the langerhanse islets cells. The presence of RA binding proteins in pancreatic islet cells could probably explain the significance of vitamin A for optimal islet function[12]. Furthermore RA may be involved in regulation of the hormones released by the islet cells[12]. Deficient islet cells were defective in hormone release when exposed to graded levels of glucose[12,13]. Vitamin A deficiency results in change in pancreatic tissue quality, which could conceivably increase digestion with the collagenase[144].
In an animal study, NOD mice were divided into 3 groups and treated with 250 IU of vitamin A per gram of their daily food or treated with 1% freeze-dried grape powder in their diet or a control diet for 7 mo. After 7 mo, in the control group 71% of the mice had a blood sugar level more than 13.9 mmol/L (full-blown T1DM) whereas only 25% of the mice in the vitamin A group and 33% of the grape powder group reached the above blood sugar level. Furthermore TNF-α, an inflammatory marker in T1DM patients, in the vitamin A and grape powder groups was respectively lower compared to the control group[145]. These results suggested that polyphenols or vitamin A in the diet protect beta-cell islets against autoimmune inflammatory attacks and have the potency to decrease the formation of autoimmune diseases such as type 1 diabetes[146].
In addition it has been showed that all-trans retinoic acid (ATRA), a potent derivative of vitamin A treatment restricted both CD4+ and CD8+ IFN-γ producing cells without affecting CD4+ IL-17-producing cells. ATRA treatment also affects the function and activation status of CD8+ T-cells[140]. Macrophages generate less TNF-α which in return reduces the production of chemokines which promote the recruitment of immune cells in to the islets including IP-10, RANTES, and MIP-1b, and also intracellular adhesion molecule-1[147,148]. According to histological studies, non diabetic animal treated with ATRA did not have insulitis, indicating that ATRA may have also inhibited T-cells trafficking to and infiltration in to islets, thus preventing diabetes. Furthermore, recently in vitro and in vivo trials indicated that ATRA treatment may result in upregulation of Foxp3+ Treg cells and reduction of Th1 and Th17 cell differentiation[125,141,149]. Protective effects of ATRA are impairing in the status of inadequacy of donors Foxp3+ CD4+ Treg cells[140]. Overall, these evidences support the idea that vitamin A and its derivatives exerted it’s autoimmune-protective effect, at least in part, by inhibiting both CD4+ and CD8+ IFN-γ-producing Teff cells with no effects on IL-17-producing Teff cells, and inducing the production of Treg cells. However despite the fact that ATRA treatment inhibited the in vitro differentiation of Th17 cells did not alter the Th17 cell population[125,141,149,150]. Although Th17 cells are important players in pathogenesis of some autoimmune diseases including experimental autoimmune encephalitis and autoimmune arthritis, its function in type 1 diabetes is not yet discovered[151-153]. Expression of granzyme B was suppressed by ATRA. In addition ATRA efficiently inhibits infiltration of T-cells into islets, and precluded the progression of insulitis and diabetes. A study conducted by Van et al[140] showed that defect less islets or pre-insulitis were detected in ATRA-treated mice, even after 17 wk of the cell transfer while the control group developed severe destructive insulitis at 2 wk after cell transfer with CD4 CD25.
CONCLUSION
This review reported that both vitamin A and ATRA effectively induced immune tolerance that inhibited islet inflammation and progression to diabetes. In this review, as fully mentioned previously we showed that ATRA treatment had a dual effect, the inhibition of Teff cells and inducing Treg cell proliferation in therapeutic of type 1 diabetes. Nonetheless, the protective effect of ATRA is inhibited when CD4CD25 T-cells, thus a majority of Foxp3 Treg, are drawn down in donor splenocytes. In prediabetic NOD mice with initiated insulitis, ATRA treatment can inhibit the development of T1DM. Nevertheless, the mechanisms demonstrating the role of vitamin A or ATRA treatment in inducing immune tolerance and prevention of autoimmune diseases is not yet clear[138,142,154-156]. So, to further validate and establish the potential of using ATRA for therapy, further studies are needed to evaluate its relative contribution in modulating type 1 diabetes and to show the mechanisms by which vitamin A and ATRA may inhibit the development of autoimmune disorders. Overall, it seems that the use of vitamin A and ATRA via induction of immune tolerance provides an effective method in inhibiting type 1 diabetes.
ACKNOWLEDGMENTS
Our research group would like to thank all subjects who took part in the current study.
Footnotes
Conflict-of-interest statement: No potential conflicts of interest. No financial support.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: July 30, 2015
First decision: September 16, 2015
Article in press: April 11, 2016
P- Reviewer: Gomes A, Liu SH S- Editor: Kong JX L- Editor: A E- Editor: Lu YJ
References
- 1.Szablewski L. Role of immune system in type 1 diabetes mellitus pathogenesis. Int Immunopharmacol. 2014;22:182–191. doi: 10.1016/j.intimp.2014.06.033. [DOI] [PubMed] [Google Scholar]
- 2.Atkinson MA, Eisenbarth GS. Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet. 2001;358:221–229. doi: 10.1016/S0140-6736(01)05415-0. [DOI] [PubMed] [Google Scholar]
- 3.Patterson CC, Dahlquist GG, Gyürüs E, Green A, Soltész G. Incidence trends for childhood type 1 diabetes in Europe during 1989-2003 and predicted new cases 2005-20: a multicentre prospective registration study. Lancet. 2009;373:2027–2033. doi: 10.1016/S0140-6736(09)60568-7. [DOI] [PubMed] [Google Scholar]
- 4.Laing SP, Swerdlow AJ, Slater SD, Botha JL, Burden AC, Waugh NR, Smith AW, Hill RD, Bingley PJ, Patterson CC, et al. The British Diabetic Association Cohort Study, II: cause-specific mortality in patients with insulin-treated diabetes mellitus. Diabet Med. 1999;16:466–471. doi: 10.1046/j.1464-5491.1999.00076.x. [DOI] [PubMed] [Google Scholar]
- 5.Federation ID. Idf diabetes atlas. Brussels: International Diabetes Federation; 2013. [Google Scholar]
- 6.Hassan GA, Sliem HA, Ellethy AT, Salama Mel-S. Role of immune system modulation in prevention of type 1 diabetes mellitus. Indian J Endocrinol Metab. 2012;16:904–909. doi: 10.4103/2230-8210.102989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bougnères PF, Landais P, Boisson C, Carel JC, Frament N, Boitard C, Chaussain JL, Bach JF. Limited duration of remission of insulin dependency in children with recent overt type I diabetes treated with low-dose cyclosporin. Diabetes. 1990;39:1264–1272. doi: 10.2337/diab.39.10.1264. [DOI] [PubMed] [Google Scholar]
- 8.Villamor E, Fawzi WW. Effects of vitamin a supplementation on immune responses and correlation with clinical outcomes. Clin Microbiol Rev. 2005;18:446–464. doi: 10.1128/CMR.18.3.446-464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green HN, Mellanby E. Vitamin A as an anti-infective agent. Br Med J. 1928;2:691–696. doi: 10.1136/bmj.2.3537.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green H, Mellanby E. Carotene and vitamin A: The anti-infective action of carotene. Brit J Exper Path. 1930;11:81–89. [Google Scholar]
- 11.Ross AC. Vitamin A and retinoic acid in T cell-related immunity. Am J Clin Nutr. 2012;96:1166S–1172S. doi: 10.3945/ajcn.112.034637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berdanier C. Vitamin A needs in diabetes mellitus. Sight and Life Newsletter. 2003;1:3–15. [Google Scholar]
- 13.Berdanier CD, Everts HB, Hermoyian C, Mathews CE. Role of vitamin A in mitochondrial gene expression. Diabetes Res Clin Pract. 2001;54 Suppl 2:S11–S27. doi: 10.1016/s0168-8227(01)00331-x. [DOI] [PubMed] [Google Scholar]
- 14.Chertow BS, Blaner WS, Baranetsky NG, Sivitz WI, Cordle MB, Thompson D, Meda P. Effects of vitamin A deficiency and repletion on rat insulin secretion in vivo and in vitro from isolated islets. J Clin Invest. 1987;79:163–169. doi: 10.1172/JCI112778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Driscoll HK, Adkins CD, Chertow TE, Cordle MB, Matthews KA, Chertow BS. Vitamin A stimulation of insulin secretion: effects on transglutaminase mRNA and activity using rat islets and insulin-secreting cells. Pancreas. 1997;15:69–77. [PubMed] [Google Scholar]
- 16.Olmedilla B, Granado F, Gil-Martinez E, Blanco I, Rojas-Hidalgo E. Reference values for retinol, tocopherol, and main carotenoids in serum of control and insulin-dependent diabetic Spanish subjects. Clin Chem. 1997;43:1066–1071. [PubMed] [Google Scholar]
- 17.Granado F, Olmedilla B, Botella F, Simal A, Blanco I. Retinol and alpha-tocopherol in serum of type 1 diabetic patients with intensive insulin therapy: a long term follow-up study. Nutrition. 2003;19:128–132. doi: 10.1016/s0899-9007(02)00908-5. [DOI] [PubMed] [Google Scholar]
- 18.Kobbah AM, Hellsing K, Tuvemo T. Early changes of some serum proteins and metals in diabetic children. Acta Paediatr Scand. 1988;77:734–740. doi: 10.1111/j.1651-2227.1988.tb10739.x. [DOI] [PubMed] [Google Scholar]
- 19.Basu TK, Tze WJ, Leichter J. Serum vitamin A and retinol-binding protein in patients with insulin-dependent diabetes mellitus. Am J Clin Nutr. 1989;50:329–331. doi: 10.1093/ajcn/50.2.329. [DOI] [PubMed] [Google Scholar]
- 20.Nakamura A, Osonoi T, Terauchi Y. Relationship between urinary sodium excretion and pioglitazone-induced edema. J Diabetes Investig. 2010;1:208–211. doi: 10.1111/j.2040-1124.2010.00046.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jain SK, McVie R, Duett J, Herbst JJ. The effect of glycemic control on plasma prealbumin levels in type-1 diabetic children. Horm Metab Res. 1993;25:102–104. doi: 10.1055/s-2007-1002052. [DOI] [PubMed] [Google Scholar]
- 22.Granado F, Olmedilla B, Gil-Martínez E, Blanco I, Millan I, Rojas-Hidalgo E. Carotenoids, retinol and tocopherols in patients with insulin-dependent diabetes mellitus and their immediate relatives. Clin Sci (Lond) 1998;94:189–195. doi: 10.1042/cs0940189. [DOI] [PubMed] [Google Scholar]
- 23.Gottesman ME, Quadro L, Blaner WS. Studies of vitamin A metabolism in mouse model systems. Bioessays. 2001;23:409–419. doi: 10.1002/bies.1059. [DOI] [PubMed] [Google Scholar]
- 24.Niederreither K, Dollé P. Retinoic acid in development: towards an integrated view. Nat Rev Genet. 2008;9:541–553. doi: 10.1038/nrg2340. [DOI] [PubMed] [Google Scholar]
- 25.Blomhoff R, Blomhoff HK. Overview of retinoid metabolism and function. J Neurobiol. 2006;66:606–630. doi: 10.1002/neu.20242. [DOI] [PubMed] [Google Scholar]
- 26.Napoli JL. Physiological insights into all-trans-retinoic acid biosynthesis. Biochim Biophys Acta. 2012;1821:152–167. doi: 10.1016/j.bbalip.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vogel S, Gamble M, Blaner W. Biosynthesis, absorption, metabolism and transport of retinoids. In: Retinoids , Section A, edotors , editors. Berlin Heidelberg: Springer; 1999. pp. 31–95. [Google Scholar]
- 28.O’Byrne SM, Blaner WS. Retinol and retinyl esters: biochemistry and physiology. J Lipid Res. 2013;54:1731–1743. doi: 10.1194/jlr.R037648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vieira AV, Schneider WJ, Vieira PM. Retinoids: transport, metabolism, and mechanisms of action. J Endocrinol. 1995;146:201–207. doi: 10.1677/joe.0.1460201. [DOI] [PubMed] [Google Scholar]
- 30.Quadro L, Hamberger L, Colantuoni V, Gottesman ME, Blaner WS. Understanding the physiological role of retinol-binding protein in vitamin A metabolism using transgenic and knockout mouse models. Mol Aspects Med. 2003;24:421–430. doi: 10.1016/s0098-2997(03)00038-4. [DOI] [PubMed] [Google Scholar]
- 31.Sporn M, Roberts A, Goodman D. The retinoids: Biology, chemistry and medicine. New York: Raven Press; 1994. p. 351. [Google Scholar]
- 32.Duester G. Families of retinoid dehydrogenases regulating vitamin A function: production of visual pigment and retinoic acid. Eur J Biochem. 2000;267:4315–4324. doi: 10.1046/j.1432-1327.2000.01497.x. [DOI] [PubMed] [Google Scholar]
- 33.Dowling JE, Wald G. The biological function of vitamin A acid. Proc Natl Acad Sci USA. 1960;46:587–608. doi: 10.1073/pnas.46.5.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giguere V, Ong ES, Segui P, Evans RM. Identification of a receptor for the morphogen retinoic acid. Nature. 1987;330:624–629. doi: 10.1038/330624a0. [DOI] [PubMed] [Google Scholar]
- 35.Mangelsdorf DJ, Ong ES, Dyck JA, Evans RM. Nuclear receptor that identifies a novel retinoic acid response pathway. Nature. 1990;345:224–229. doi: 10.1038/345224a0. [DOI] [PubMed] [Google Scholar]
- 36.Pillay K, Coutsoudis A, Agadzi-Naqvi AK, Kuhn L, Coovadia HM, Janoff EN. Secretory leukocyte protease inhibitor in vaginal fluids and perinatal human immunodeficiency virus type 1 transmission. J Infect Dis. 2001;183:653–656. doi: 10.1086/318535. [DOI] [PubMed] [Google Scholar]
- 37.Simmons KM, Michels AW. Type 1 diabetes: A predictable disease. World J Diabetes. 2015;6:380–390. doi: 10.4239/wjd.v6.i3.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harrison LC, Honeyman MC, DeAizpurua HJ, Schmidli RS, Colman PG, Tait BD, Cram DS. Inverse relation between humoral and cellular immunity to glutamic acid decarboxylase in subjects at risk of insulin-dependent diabetes. Lancet. 1993;341:1365–1369. doi: 10.1016/0140-6736(93)90940-i. [DOI] [PubMed] [Google Scholar]
- 39.Kaufman DL, Clare-Salzler M, Tian J, Forsthuber T, Ting GS, Robinson P, Atkinson MA, Sercarz EE, Tobin AJ, Lehmann PV. Spontaneous loss of T-cell tolerance to glutamic acid decarboxylase in murine insulin-dependent diabetes. Nature. 1993;366:69–72. doi: 10.1038/366069a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Orban T, Sosenko JM, Cuthbertson D, Krischer JP, Skyler JS, Jackson R, Yu L, Palmer JP, Schatz D, Eisenbarth G. Pancreatic islet autoantibodies as predictors of type 1 diabetes in the Diabetes Prevention Trial-Type 1. Diabetes Care. 2009;32:2269–2274. doi: 10.2337/dc09-0934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Exton JH. Mechanisms of hormonal regulation of hepatic glucose metabolism. Diabetes Metab Rev. 1987;3:163–183. doi: 10.1002/dmr.5610030108. [DOI] [PubMed] [Google Scholar]
- 42.Haller MJ, Atkinson MA, Schatz D. Type 1 diabetes mellitus: etiology, presentation, and management. Pediatr Clin North Am. 2005;52:1553–1578. doi: 10.1016/j.pcl.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 43.Michels AW. Targeting the trimolecular complex. Clin Immunol. 2013;149:339–344. doi: 10.1016/j.clim.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pietropaolo M, Surhigh JM, Nelson PW, Eisenbarth GS. Primer: immunity and autoimmunity. Diabetes. 2008;57:2872–2882. doi: 10.2337/db07-1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 46.Janeway CA, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 47.Elia PP, Tolentino YF, Bernardazzi C, de Souza HS. The role of innate immunity receptors in the pathogenesis of inflammatory bowel disease. Mediators Inflamm. 2015;2015:936193. doi: 10.1155/2015/936193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wells JM, Rossi O, Meijerink M, van Baarlen P. Epithelial crosstalk at the microbiota-mucosal interface. Proc Natl Acad Sci USA. 2011;108 Suppl 1:4607–4614. doi: 10.1073/pnas.1000092107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 50.Hornef MW, Normark BH, Vandewalle A, Normark S. Intracellular recognition of lipopolysaccharide by toll-like receptor 4 in intestinal epithelial cells. J Exp Med. 2003;198:1225–1235. doi: 10.1084/jem.20022194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takeda K, Akira S. Toll-like receptors. Curr Protoc Immunol. 2015;109:14.12.1–14.12.10. doi: 10.1002/0471142735.im1412s109. [DOI] [PubMed] [Google Scholar]
- 52.Martin DA, Elkon KB. Autoantibodies make a U-turn: the toll hypothesis for autoantibody specificity. J Exp Med. 2005;202:1465–1469. doi: 10.1084/jem.20052228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Devaraj S, Dasu MR, Rockwood J, Winter W, Griffen SC, Jialal I. Increased toll-like receptor (TLR) 2 and TLR4 expression in monocytes from patients with type 1 diabetes: further evidence of a proinflammatory state. J Clin Endocrinol Metab. 2008;93:578–583. doi: 10.1210/jc.2007-2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pino SC, Kruger AJ, Bortell R. The role of innate immune pathways in type 1 diabetes pathogenesis. Curr Opin Endocrinol Diabetes Obes. 2010;17:126–130. doi: 10.1097/MED.0b013e3283372819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wong FS, Hu C, Zhang L, Du W, Alexopoulou L, Flavell RA, Wen L. The role of Toll-like receptors 3 and 9 in the development of autoimmune diabetes in NOD mice. Ann N Y Acad Sci. 2008;1150:146–148. doi: 10.1196/annals.1447.039. [DOI] [PubMed] [Google Scholar]
- 56.Beyan H, Buckley LR, Yousaf N, Londei M, Leslie RD. A role for innate immunity in type 1 diabetes? Diabetes Metab Res Rev. 2003;19:89–100. doi: 10.1002/dmrr.341. [DOI] [PubMed] [Google Scholar]
- 57.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 58.Hutchings PR, Cooke A. The transfer of autoimmune diabetes in NOD mice can be inhibited or accelerated by distinct cell populations present in normal splenocytes taken from young males. J Autoimmun. 1990;3:175–185. doi: 10.1016/0896-8411(90)90139-j. [DOI] [PubMed] [Google Scholar]
- 59.Arnush M, Scarim AL, Heitmeier MR, Kelly CB, Corbett JA. Potential role of resident islet macrophage activation in the initiation of autoimmune diabetes. J Immunol. 1998;160:2684–2691. [PubMed] [Google Scholar]
- 60.Dahlén E, Dawe K, Ohlsson L, Hedlund G. Dendritic cells and macrophages are the first and major producers of TNF-alpha in pancreatic islets in the nonobese diabetic mouse. J Immunol. 1998;160:3585–3593. [PubMed] [Google Scholar]
- 61.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 62.Tisch R, Wang B. Role of plasmacytoid dendritic cells in type 1 diabetes: friend or foe? Diabetes. 2009;58:12–13. doi: 10.2337/db08-1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Summers KL, Behme MT, Mahon JL, Singh B. Characterization of dendritic cells in humans with type 1 diabetes. Ann N Y Acad Sci. 2003;1005:226–229. doi: 10.1196/annals.1288.032. [DOI] [PubMed] [Google Scholar]
- 64.Jost S, Altfeld M. Control of human viral infections by natural killer cells. Annu Rev Immunol. 2013;31:163–194. doi: 10.1146/annurev-immunol-032712-100001. [DOI] [PubMed] [Google Scholar]
- 65.Fauriat C, Long EO, Ljunggren HG, Bryceson YT. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood. 2010;115:2167–2176. doi: 10.1182/blood-2009-08-238469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dotta F, Fondelli C, Falorni A. Can NK cells be a therapeutic target in human type 1 diabetes? Eur J Immunol. 2008;38:2961–2963. doi: 10.1002/eji.200838851. [DOI] [PubMed] [Google Scholar]
- 67.Rodacki M, Svoren B, Butty V, Besse W, Laffel L, Benoist C, Mathis D. Altered natural killer cells in type 1 diabetic patients. Diabetes. 2007;56:177–185. doi: 10.2337/db06-0493. [DOI] [PubMed] [Google Scholar]
- 68.Rodacki M, Milech A, de Oliveira JE. NK cells and type 1 diabetes. Clin Dev Immunol. 2006;13:101–107. doi: 10.1080/17402520600877182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shi FD, Ljunggren HG, La Cava A, Van Kaer L. Organ-specific features of natural killer cells. Nat Rev Immunol. 2011;11:658–671. doi: 10.1038/nri3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ariel A, Fredman G, Sun YP, Kantarci A, Van Dyke TE, Luster AD, Serhan CN. Apoptotic neutrophils and T cells sequester chemokines during immune response resolution through modulation of CCR5 expression. Nat Immunol. 2006;7:1209–1216. doi: 10.1038/ni1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Proebstl D, Voisin MB, Woodfin A, Whiteford J, D’Acquisto F, Jones GE, Rowe D, Nourshargh S. Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J Exp Med. 2012;209:1219–1234. doi: 10.1084/jem.20111622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, Kubes P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 73.Sokol CL, Luster AD. The chemokine system in innate immunity. Cold Spring Harb Perspect Biol. 2015;7:pii: a016303. doi: 10.1101/cshperspect.a016303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nabi AH, Islam LN, Rahman MM, Biswas KB. Polymorphonuclear neutrophil dysfunctions in streptozotocin-induced type 1 diabetic rats. J Biochem Mol Biol. 2005;38:661–667. doi: 10.5483/bmbrep.2005.38.6.661. [DOI] [PubMed] [Google Scholar]
- 75.Shetty N, Thomas B, Ramesh A. Comparison of neutrophil functions in diabetic and healthy subjects with chronic generalized periodontitis. J Indian Soc Periodontol. 2008;12:41–44. doi: 10.4103/0972-124X.44089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hanses F, Park S, Rich J, Lee JC. Reduced neutrophil apoptosis in diabetic mice during staphylococcal infection leads to prolonged Tnfα production and reduced neutrophil clearance. PLoS One. 2011;6:e23633. doi: 10.1371/journal.pone.0023633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alba-Loureiro TC, Hirabara SM, Mendonça JR, Curi R, Pithon-Curi TC. Diabetes causes marked changes in function and metabolism of rat neutrophils. J Endocrinol. 2006;188:295–303. doi: 10.1677/joe.1.06438. [DOI] [PubMed] [Google Scholar]
- 78.Breitman TR, Selonick SE, Collins SJ. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc Natl Acad Sci USA. 1980;77:2936–2940. doi: 10.1073/pnas.77.5.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Geissmann F, Revy P, Brousse N, Lepelletier Y, Folli C, Durandy A, Chambon P, Dy M. Retinoids regulate survival and antigen presentation by immature dendritic cells. J Exp Med. 2003;198:623–634. doi: 10.1084/jem.20030390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jiang YJ, Xu TR, Lu B, Mymin D, Kroeger EA, Dembinski T, Yang X, Hatch GM, Choy PC. Cyclooxygenase expression is elevated in retinoic acid-differentiated U937 cells. Biochim Biophys Acta. 2003;1633:51–60. doi: 10.1016/s1388-1981(03)00072-6. [DOI] [PubMed] [Google Scholar]
- 81.Mohty M, Morbelli S, Isnardon D, Sainty D, Arnoulet C, Gaugler B, Olive D. All-trans retinoic acid skews monocyte differentiation into interleukin-12-secreting dendritic-like cells. Br J Haematol. 2003;122:829–836. doi: 10.1046/j.1365-2141.2003.04489.x. [DOI] [PubMed] [Google Scholar]
- 82.Mehta K, McQueen T, Tucker S, Pandita R, Aggarwal BB. Inhibition by all-trans-retinoic acid of tumor necrosis factor and nitric oxide production by peritoneal macrophages. J Leukoc Biol. 1994;55:336–342. doi: 10.1002/jlb.55.3.336. [DOI] [PubMed] [Google Scholar]
- 83.Sherry B, Cerami A. Cachectin/tumor necrosis factor exerts endocrine, paracrine, and autocrine control of inflammatory responses. J Cell Biol. 1988;107:1269–1277. doi: 10.1083/jcb.107.4.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim BH, Kang KS, Lee YS. Effect of retinoids on LPS-induced COX-2 expression and COX-2 associated PGE(2) release from mouse peritoneal macrophages and TNF-alpha release from rat peripheral blood mononuclear cells. Toxicol Lett. 2004;150:191–201. doi: 10.1016/j.toxlet.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 85.Na SY, Kang BY, Chung SW, Han SJ, Ma X, Trinchieri G, Im SY, Lee JW, Kim TS. Retinoids inhibit interleukin-12 production in macrophages through physical associations of retinoid X receptor and NFkappaB. J Biol Chem. 1999;274:7674–7680. doi: 10.1074/jbc.274.12.7674. [DOI] [PubMed] [Google Scholar]
- 86.Kang SG, Lim HW, Andrisani OM, Broxmeyer HE, Kim CH. Vitamin A metabolites induce gut-homing FoxP3+ regulatory T cells. J Immunol. 2007;179:3724–3733. doi: 10.4049/jimmunol.179.6.3724. [DOI] [PubMed] [Google Scholar]
- 87.Aukrust P, Müller F, Ueland T, Svardal AM, Berge RK, Frøland SS. Decreased vitamin A levels in common variable immunodeficiency: vitamin A supplementation in vivo enhances immunoglobulin production and downregulates inflammatory responses. Eur J Clin Invest. 2000;30:252–259. doi: 10.1046/j.1365-2362.2000.00619.x. [DOI] [PubMed] [Google Scholar]
- 88.Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 89.Klebanoff CA, Spencer SP, Torabi-Parizi P, Grainger JR, Roychoudhuri R, Ji Y, Sukumar M, Muranski P, Scott CD, Hall JA, et al. Retinoic acid controls the homeostasis of pre-cDC-derived splenic and intestinal dendritic cells. J Exp Med. 2013;210:1961–1976. doi: 10.1084/jem.20122508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhu B, Buttrick T, Bassil R, Zhu C, Olah M, Wu C, Xiao S, Orent W, Elyaman W, Khoury SJ. IL-4 and retinoic acid synergistically induce regulatory dendritic cells expressing Aldh1a2. J Immunol. 2013;191:3139–3151. doi: 10.4049/jimmunol.1300329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Di Caro V, Phillips B, Engman C, Harnaha J, Trucco M, Giannoukakis N. Retinoic acid-producing, ex-vivo-generated human tolerogenic dendritic cells induce the proliferation of immunosuppressive B lymphocytes. Clin Exp Immunol. 2013;174:302–317. doi: 10.1111/cei.12177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhao Z, Murasko DM, Ross AC. The role of vitamin A in natural killer cell cytotoxicity, number and activation in the rat. Nat Immun. 1994;13:29–41. [PubMed] [Google Scholar]
- 93.Kos FJ. Regulation of adaptive immunity by natural killer cells. Immunol Res. 1998;17:303–312. doi: 10.1007/BF02786453. [DOI] [PubMed] [Google Scholar]
- 94.Ross A, Hammerling U. Retinoids and the immune system. In: Sporn M, Roberts A, Goodman D, editors. The retinoids: Biology, chemistry and medicine. 2nd ed. New York: Raven Press; 1994. pp. 597–630. [Google Scholar]
- 95.Trinchieri G. Biology of natural killer cells. Adv Immunol. 1989;47:187–376. doi: 10.1016/S0065-2776(08)60664-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Michael A, Hackett JJ, Bennett M, Kumar V, Yuan D. Regulation of B lymphocytes by natural killer cells. Role of IFN-gamma. J Immunol. 1989;142:1095–1101. [PubMed] [Google Scholar]
- 97.Alter BP, Potter NU, Li FP. Classification and aetiology of the aplastic anaemias. Clin Haematol. 1978;7:431–465. [PubMed] [Google Scholar]
- 98.Micksche M, Colot M, Uchida A, Kokoschka EM, Lugar TA, Dittrich C, Moser K, Rainer H, Lanzhofer R, Kolb R, et al. Immunomodulation in cancer patients by synthetic biological response modifiers. Cancer Treat Symp. 1985;1:27–35. [Google Scholar]
- 99.Santoni A, Cerruti Sola S, Giovarelli M, Martinetto P, Vietti D, Forni G. Modulation of natural killer activity in mice by prolonged administration of various doses of dietary retinoids. Nat Immun Cell Growth Regul. 1986;5:259–266. [PubMed] [Google Scholar]
- 100.Fortes C, Forastiere F, Agabiti N, Fano V, Pacifici R, Virgili F, Piras G, Guidi L, Bartoloni C, Tricerri A, et al. The effect of zinc and vitamin A supplementation on immune response in an older population. J Am Geriatr Soc. 1998;46:19–26. doi: 10.1111/j.1532-5415.1998.tb01008.x. [DOI] [PubMed] [Google Scholar]
- 101.Bowman TA, Goonewardene IM, Pasatiempo AM, Ross AC, Taylor CE. Vitamin A deficiency decreases natural killer cell activity and interferon production in rats. J Nutr. 1990;120:1264–1273. doi: 10.1093/jn/120.10.1264. [DOI] [PubMed] [Google Scholar]
- 102.Bogomolski-Yahalom V, Matzner Y. Disorders of neutrophil function. Blood Rev. 1995;9:183–190. doi: 10.1016/0268-960x(95)90024-1. [DOI] [PubMed] [Google Scholar]
- 103.Twining SS, Schulte DP, Wilson PM, Fish BL, Moulder JE. Vitamin A deficiency alters rat neutrophil function. J Nutr. 1997;127:558–565. doi: 10.1093/jn/127.4.558. [DOI] [PubMed] [Google Scholar]
- 104.Robertson KA, Emami B, Mueller L, Collins SJ. Multiple members of the retinoic acid receptor family are capable of mediating the granulocytic differentiation of HL-60 cells. Mol Cell Biol. 1992;12:3743–3749. doi: 10.1128/mcb.12.9.3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tsai S, Collins SJ. A dominant negative retinoic acid receptor blocks neutrophil differentiation at the promyelocyte stage. Proc Natl Acad Sci USA. 1993;90:7153–7157. doi: 10.1073/pnas.90.15.7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Maun NA, Gaines P, Khanna-Gupta A, Zibello T, Enriquez L, Goldberg L, Berliner N. G-CSF signaling can differentiate promyelocytes expressing a defective retinoic acid receptor: evidence for divergent pathways regulating neutrophil differentiation. Blood. 2004;103:1693–1701. doi: 10.1182/blood-2002-10-3247. [DOI] [PubMed] [Google Scholar]
- 107.Ribeiro OG, Maria DA, Adriouch S, Pechberty S, Cabrera WH, Morisset J, Ibañez OM, Seman M. Convergent alteration of granulopoiesis, chemotactic activity, and neutrophil apoptosis during mouse selection for high acute inflammatory response. J Leukoc Biol. 2003;74:497–506. doi: 10.1189/jlb.0103039. [DOI] [PubMed] [Google Scholar]
- 108.Zhao Z, Ross AC. Retinoic acid repletion restores the number of leukocytes and their subsets and stimulates natural cytotoxicity in vitamin A-deficient rats. J Nutr. 1995;125:2064–2073. doi: 10.1093/jn/125.8.2064. [DOI] [PubMed] [Google Scholar]
- 109.Higuchi H, Nagahata H. Effects of vitamins A and E on superoxide production and intracellular signaling of neutrophils in Holstein calves. Can J Vet Res. 2000;64:69–75. [PMC free article] [PubMed] [Google Scholar]
- 110.Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. 2010;464:1293–1300. doi: 10.1038/nature08933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Christianson SW, Shultz LD, Leiter EH. Adoptive transfer of diabetes into immunodeficient NOD-scid/scid mice. Relative contributions of CD4+ and CD8+ T-cells from diabetic versus prediabetic NOD.NON-Thy-1a donors. Diabetes. 1993;42:44–55. doi: 10.2337/diab.42.1.44. [DOI] [PubMed] [Google Scholar]
- 112.Bendelac A, Boitard C, Bedossa P, Bazin H, Bach JF, Carnaud C. Adoptive T cell transfer of autoimmune nonobese diabetic mouse diabetes does not require recruitment of host B lymphocytes. J Immunol. 1988;141:2625–2628. [PubMed] [Google Scholar]
- 113.Peterson JD, Pike B, McDuffie M, Haskins K. Islet-specific T cell clones transfer diabetes to nonobese diabetic (NOD) F1 mice. J Immunol. 1994;153:2800–2806. [PubMed] [Google Scholar]
- 114.Lennon GP, Bettini M, Burton AR, Vincent E, Arnold PY, Santamaria P, Vignali DA. T cell islet accumulation in type 1 diabetes is a tightly regulated, cell-autonomous event. Immunity. 2009;31:643–653. doi: 10.1016/j.immuni.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.van Belle TL, Coppieters KT, von Herrath MG. Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev. 2011;91:79–118. doi: 10.1152/physrev.00003.2010. [DOI] [PubMed] [Google Scholar]
- 116.Hamilton-Williams EE, Palmer SE, Charlton B, Slattery RM. Beta cell MHC class I is a late requirement for diabetes. Proc Natl Acad Sci USA. 2003;100:6688–6693. doi: 10.1073/pnas.1131954100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cardozo AK, Ortis F, Storling J, Feng YM, Rasschaert J, Tonnesen M, Van Eylen F, Mandrup-Poulsen T, Herchuelz A, Eizirik DL. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells. Diabetes. 2005;54:452–461. doi: 10.2337/diabetes.54.2.452. [DOI] [PubMed] [Google Scholar]
- 118.Cnop M, Welsh N, Jonas JC, Jörns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005;54 Suppl 2:S97–107. doi: 10.2337/diabetes.54.suppl_2.s97. [DOI] [PubMed] [Google Scholar]
- 119.Conrad B, Weidmann E, Trucco G, Rudert WA, Behboo R, Ricordi C, Rodriquez-Rilo H, Finegold D, Trucco M. Evidence for superantigen involvement in insulin-dependent diabetes mellitus aetiology. Nature. 1994;371:351–355. doi: 10.1038/371351a0. [DOI] [PubMed] [Google Scholar]
- 120.Bottazzo GF, Dean BM, McNally JM, MacKay EH, Swift PG, Gamble DR. In situ characterization of autoimmune phenomena and expression of HLA molecules in the pancreas in diabetic insulitis. N Engl J Med. 1985;313:353–360. doi: 10.1056/NEJM198508083130604. [DOI] [PubMed] [Google Scholar]
- 121.Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity. 2004;21:527–538. doi: 10.1016/j.immuni.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 122.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 123.Korn T, Bettelli E, Gao W, Awasthi A, Jäger A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 125.Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, Cheroutre H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 126.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 127.Fragoso YD, Stoney PN, McCaffery PJ. The evidence for a beneficial role of vitamin A in multiple sclerosis. CNS Drugs. 2014;28:291–299. doi: 10.1007/s40263-014-0148-4. [DOI] [PubMed] [Google Scholar]
- 128.Leal JY, Castejón HV, Romero T, Ortega P, Gómez G, Amaya D, Estévez J. [Serum values of cytokines in children with vitamin A deficiency disorders] Invest Clin. 2004;45:243–256. [PubMed] [Google Scholar]
- 129.Stephensen CB. Vitamin A, infection, and immune function. Annu Rev Nutr. 2001;21:167–192. doi: 10.1146/annurev.nutr.21.1.167. [DOI] [PubMed] [Google Scholar]
- 130.Asano M, Toda M, Sakaguchi N, Sakaguchi S. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J Exp Med. 1996;184:387–396. doi: 10.1084/jem.184.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Amrani A, Verdaguer J, Thiessen S, Bou S, Santamaria P. IL-1alpha, IL-1beta, and IFN-gamma mark beta cells for Fas-dependent destruction by diabetogenic CD4(+) T lymphocytes. J Clin Invest. 2000;105:459–468. doi: 10.1172/JCI8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Savinov AY, Wong FS, Chervonsky AV. IFN-gamma affects homing of diabetogenic T cells. J Immunol. 2001;167:6637–6643. doi: 10.4049/jimmunol.167.11.6637. [DOI] [PubMed] [Google Scholar]
- 133.Wang B, André I, Gonzalez A, Katz JD, Aguet M, Benoist C, Mathis D. Interferon-gamma impacts at multiple points during the progression of autoimmune diabetes. Proc Natl Acad Sci USA. 1997;94:13844–13849. doi: 10.1073/pnas.94.25.13844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Suarez-Pinzon W, Rajotte RV, Mosmann TR, Rabinovitch A. Both CD4+ and CD8+ T-cells in syngeneic islet grafts in NOD mice produce interferon-gamma during beta-cell destruction. Diabetes. 1996;45:1350–1357. doi: 10.2337/diab.45.10.1350. [DOI] [PubMed] [Google Scholar]
- 135.Haskins K, McDuffie M. Acceleration of diabetes in young NOD mice with a CD4+ islet-specific T cell clone. Science. 1990;249:1433–1436. doi: 10.1126/science.2205920. [DOI] [PubMed] [Google Scholar]
- 136.Wicker LS, Leiter EH, Todd JA, Renjilian RJ, Peterson E, Fischer PA, Podolin PL, Zijlstra M, Peterson LB. β2-microglobulin-deficient NOD mice do not develop insulitis or diabetes. Diabetes. 1994;43:500–504. doi: 10.2337/diab.43.3.500. [DOI] [PubMed] [Google Scholar]
- 137.Santamaria P. Effector lymphocytes in islet cell autoimmunity. Rev Endocr Metab Disord. 2003;4:271–280. doi: 10.1023/a:1025156413404. [DOI] [PubMed] [Google Scholar]
- 138.Zunino SJ, Storms DH, Stephensen CB. Diets rich in polyphenols and vitamin A inhibit the development of type I autoimmune diabetes in nonobese diabetic mice. J Nutr. 2007;137:1216–1221. doi: 10.1093/jn/137.5.1216. [DOI] [PubMed] [Google Scholar]
- 139.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 140.Van YH, Lee WH, Ortiz S, Lee MH, Qin HJ, Liu CP. All-trans retinoic acid inhibits type 1 diabetes by T regulatory (Treg)-dependent suppression of interferon-gamma-producing T-cells without affecting Th17 cells. Diabetes. 2009;58:146–155. doi: 10.2337/db08-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Schambach F, Schupp M, Lazar MA, Reiner SL. Activation of retinoic acid receptor-alpha favours regulatory T cell induction at the expense of IL-17-secreting T helper cell differentiation. Eur J Immunol. 2007;37:2396–2399. doi: 10.1002/eji.200737621. [DOI] [PubMed] [Google Scholar]
- 142.Kinoshita K, Yoo BS, Nozaki Y, Sugiyama M, Ikoma S, Ohno M, Funauchi M, Kanamaru A. Retinoic acid reduces autoimmune renal injury and increases survival in NZB/W F1 mice. J Immunol. 2003;170:5793–5798. doi: 10.4049/jimmunol.170.11.5793. [DOI] [PubMed] [Google Scholar]
- 143.Xu J, Storer PD, Chavis JA, Racke MK, Drew PD. Agonists for the peroxisome proliferator-activated receptor-alpha and the retinoid X receptor inhibit inflammatory responses of microglia. J Neurosci Res. 2005;81:403–411. doi: 10.1002/jnr.20518. [DOI] [PubMed] [Google Scholar]
- 144.Shidfar F, Aghasi M, Vafa M, Heydari I, Hosseini S, Shidfar S. Effects of combination of zinc and vitamin A supplementation on serum fasting blood sugar, insulin, apoprotein B and apoprotein A-I in patients with type I diabetes. Int J Food Sci Nutr. 2010;61:182–191. doi: 10.3109/09637480903334171. [DOI] [PubMed] [Google Scholar]
- 145.Zorena K, Myśliwska J, Myśliwiec M, Balcerska A, Lipowski P, Raczyńska K. Interleukin-12 and tumour necrosis factor-alpha equilibrium is a prerequisite for clinical course free from late complications in children with type 1 diabetes mellitus. Scand J Immunol. 2008;67:204–208. doi: 10.1111/j.1365-3083.2007.02054.x. [DOI] [PubMed] [Google Scholar]
- 146.Arnold G. Mouse Study Shows Hope for Vitamin A and Type 1 Diabetes. Available from: http://pitchingdoc.com/fileupload/NOW%20Foods%20Articles/Diabetes/Mouse%20Study%20Shows%20Hope%20for%20Vitamin%20A%20In%20Type%201%20Diabetes.pdf.
- 147.Arimilli S, Ferlin W, Solvason N, Deshpande S, Howard M, Mocci S. Chemokines in autoimmune diseases. Immunol Rev. 2000;177:43–51. doi: 10.1034/j.1600-065x.2000.17716.x. [DOI] [PubMed] [Google Scholar]
- 148.Martin S, van den Engel NK, Vinke A, Heidenthal E, Schulte B, Kolb H. Dominant role of intercellular adhesion molecule-1 in the pathogenesis of autoimmune diabetes in non-obese diabetic mice. J Autoimmun. 2001;17:109–117. doi: 10.1006/jaut.2001.0526. [DOI] [PubMed] [Google Scholar]
- 149.Elias KM, Laurence A, Davidson TS, Stephens G, Kanno Y, Shevach EM, O’Shea JJ. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood. 2008;111:1013–1020. doi: 10.1182/blood-2007-06-096438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, Belkaid Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204:1775–1785. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, Bamba T, Fujiyama Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Batten M, Li J, Yi S, Kljavin NM, Danilenko DM, Lucas S, Lee J, de Sauvage FJ, Ghilardi N. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat Immunol. 2006;7:929–936. doi: 10.1038/ni1375. [DOI] [PubMed] [Google Scholar]
- 153.Koenders MI, Lubberts E, Oppers-Walgreen B, van den Bersselaar L, Helsen MM, Di Padova FE, Boots AM, Gram H, Joosten LA, van den Berg WB. Blocking of interleukin-17 during reactivation of experimental arthritis prevents joint inflammation and bone erosion by decreasing RANKL and interleukin-1. Am J Pathol. 2005;167:141–149. doi: 10.1016/S0002-9440(10)62961-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Miyagawa N, Homma T, Kagechika H, Shudo K, Nagai H. Effect of synthetic retinoid, TAC-101, on experimental autoimmune disease. Pharmacology. 2003;67:21–31. doi: 10.1159/000066783. [DOI] [PubMed] [Google Scholar]
- 155.Osanai M, Nishikiori N, Murata M, Chiba H, Kojima T, Sawada N. Cellular retinoic acid bioavailability determines epithelial integrity: Role of retinoic acid receptor alpha agonists in colitis. Mol Pharmacol. 2007;71:250–258. doi: 10.1124/mol.106.029579. [DOI] [PubMed] [Google Scholar]
- 156.Driscoll HK, Chertow BS, Jelic TM, Baltaro RJ, Chandor SB, Walker EM, Dadgari JM, Pofahl AB. Vitamin A status affects the development of diabetes and insulitis in BB rats. Metabolism. 1996;45:248–253. doi: 10.1016/s0026-0495(96)90062-1. [DOI] [PubMed] [Google Scholar]