

Abstract
The standard of care for cancer patients includes surgical resection, radiation and chemotherapy with cytotoxic chemotherapy drugs usually part of the treatment. However, these drugs are commonly associated with cardiotoxicity, ototoxicity, nephrotoxicity, peripheral neuropathy and myelosuppression. Strategies to deliver cytotoxic chemotherapy drugs while reducing secondary toxicity and increasing tumor dosing would therefore be desirable. This goal can be achieved through the use of controlled release, drug carrier systems. The aim of this review is to provide an overview of clinically used drug carrier systems and recently developed approaches for drug-biomaterial conjugation.
Graphical Abstract
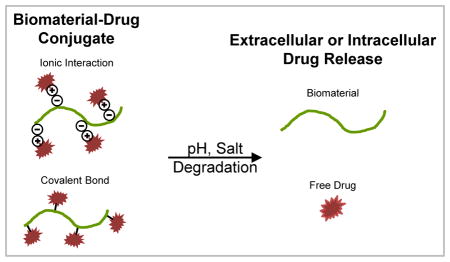
Introduction
Cytotoxic chemotherapy drugs have been the front line for cancer treatments for decades.1 Intravenous administration is the most common route of administration. However, secondary toxicities significantly limit dosing, which impedes both the use and the benefits of the drugs. Side effects include cardiotoxicity, ototoxicity, nephrotoxicity, peripheral neuropathy and myelosuppression.2–6 Therefore, while a cytotoxic drug may be extremely effective in treating the specific cancer, the optimal dose required may not be achievable without undue side effects to the patient. Biomaterial-based drug delivery systems have been utilized to preferentially deliver drugs to cancer sites and thereby limit systemic toxicity. These drug delivery systems are administered systemically as nanoparticle formulations or locally via implantable or injectable materials. The chemical structures of common chemotherapeutic agents investigated in drug release systems are depicted in Figure 1. Drug release from these delivery modes is achieved through various processes, including diffusion, linker degradation or biomaterial degradation. In this review, clinically used drug carrier systems will be described, followed by details on drug carrier systems based on drug-biomaterial interactions through ionic, covalent or cisplatin complexation mechanisms. The clinically used drug carrier system, Gliadel, will be described, however, other polymeric systems utilizing hydrolysis and diffusion for drug release will not be discussed as these have been reviewed extensively elsewhere.7–10
Figure 1. Chemical structure of several chemotherapy agents investigated in sustained release formats.

(A) Anthracyclines - doxorubicin, 1; idarubicin, 2; and mitoxantrone, 3. (B) Topoisomerase II inhibitor - etoposide, 4. (C) Alkylating agents - cisplatin, 5; and carmustine, 6. (D) Antimetabolic agents - methotrexate, 7; and gemcitabine, 8. (E) Mitotic inhibitors - paclitaxel, 9; docetaxel, 10; and vincristine, 11.
Clinical Systems
There is extensive research towards developing improved delivery systems for chemotherapy drugs, however, few of these systems have received regulatory approval from the Food and Drug Administration (FDA). The approved systems have significantly improved patient outcomes compared to conventional techniques. Outside of the United States, drug eluting systems that combine pre-existing cancer treatment strategies (see Direct Injection, Drug Eluting Microspheres below) have been approved for use in Canada and the European Union. The approaches that are clinically approved both in the United States, Canada and Europe are described in the subsequent sections.
Liposomal Formulations: Doxil®, DaunoXome®, Marqibo® Abraxane®, Myocet® and Caleyx®
Liposomes are spherical vesicles composed of polar lipid molecules that self-assemble into lipid bilayers that are structurally analogous to the cell membrane. These vesicles can be used to carry and release hydrophobic and hydrophilic payloads. There are currently three FDA approved liposome formulations, Doxil®, DaunoXome® and Marqibo®, which carry doxorubicin (1, DOX), daunorubicin or vincristine (11, VCR), respectively. Myocet® and Caelyx® are used outside of the United States as a liposome DOX carrier. Liposome formulations have been reviewed extensively elsewhere.11–13 Doxil® and Caleyx® liposomes are formulated with a PEGylated outer lipid layer which reduces opsonization and clearance by the mononuclear phagocyte system.14 These liposomes have increased blood circulation time compared to the free drug, 3–4 days versus 25 hours.15 The other liposome formulations are readily cleared by the mononuclear phagocyte system, resulting in liposome degradation and drug release into the circulation system.15 The benefits of liposomal vesicles for chemotherapeutic drug delivery include decreased systemic toxicity and increased payload to the tumor site via enhanced permeability and retention (EPR).16
Human Serum Albumin Nanoparticles: Abraxane®
A significant limitation with many chemotherapeutic drugs is their low solubility in water. To systemically deliver these drugs, surfactants and organic solvents are often employed to increase solubility. However, these additives can introduce additional side effects, including disruption of biological membranes and hemolysis.17 A hypersensitivity reaction to cremophor EL-solubilized paxlitaxel (9, PTX) is an example; while the exact mechanism of this drug-induced hypersensitivity is unknown. Premedication with steroids can be used to minimize hypersensitivity.18 Exploiting the interaction of hydrophobic molecules with albumin has been used to overcome solubility limitations, thereby eliminating the need for solubilizing agents.19 Abraxane® is non-covalent human serum albumin-bound PTX approved by the FDA for breast, non-small cell lung and pancreatic cancers. Abraxane® is reconstituted with normal saline eliminating the need for premedicating with steroids. Furthermore, this albumin-based nanoparticle system may utilize the albumin receptor (gp60)-mediated pathway for transcytosis through endothelial cells to gain access to tumor cells.20–22
Implantable Polymer Wafers: Gliadel®
A significant challenge to treating brain tumors is overcoming the blood-brain barrier. One way to overcome this challenge is to directly place chemotherapeutic drugs into the tumor space. Gliadel® is an implanted poly(anhydride) wafer containing carmustine (6) for the treatment of glioblastoma. Poly(anhydrides) are a class of polymers suited for slow release of hydrophobic drugs due to their biocompatibility, low initial water uptake and rapid surface-mediated degradation kinetics. Gliadel® wafers release carmustine over 5 days via a combination of diffusion and degradation/diffusion kinetics. In animal studies, the entire polymer wafer was degraded within 6–8 weeks,23 while, in humans, portions of the wafers were observed months after the initial implant.24 This represents the only locally administered, sustained release treatment option for solid tumors approved in the United States.
Direct Injection, Drug Eluting Microspheres: DEBDOX®, DEBIRI® and Hepasphere®
Transarterial chemoembolization (TACE) is a procedure used in hypervascularized tumors, such as primary liver tumors, to block blood supply via filling the supplying arteries with an embolic agent.25 Additionally, chemotherapeutic drugs are directly injected into the artery during the procedure (mitomycin, DOX or cisplatin). A number of embolization options are available, including rapidly hardening liquids (n-butyl-2-cyanoacrylate), ethanol that hardens the endothelium lining the blood vessels, poly(vinyl alcohol) microspheres, gelatin microspheres, degradable starch microspheres or metal coils which induce blood clotting.26–31 These embolic agents, particularly the microsphere formulations, can be used to co-administer and slowly release chemotherapeutic drugs. BTG International has marketed sulfated poly(vinyl alcohol) microspheres (DC Beads®) loaded with DOX (DEBDOX®) or irinotecan (DEBIRI®) for hypervascularized tumors or malignant colorectal cancers metastasized to the liver, respectively.32,33 Merit Medical Systems has marketed sodium acrylate alcohol copolymer microspheres (Hepaspheres®) loaded with DOX for hypervascularized liver tumors.34 These formulations represent a method of loading and sustained release, which exploit the electrostatic interactions between the negatively charged microspheres and positive charged drugs. These products are not approved for use in the United States.
Electrostatic Interactions and Ion Pairing
Utilizing electrostatic interactions to bind drugs to polymers is a promising drug delivery approach. Many chemotherapeutic drugs have charged groups (amines, carboxylic acids and phosphates) making this approach broadly applicable. Electrostatic interactions have been used extensively in the biomaterial literature to crosslink hydrogel networks, for example calcium crosslinked alginate and glycerol phosphate disodium salt-crosslinked chitosan hydrogels.35,36 Furthermore, utilization of electrostatic interaction to form hydrophobic ion pairing is a favorable approach to limit solubility and encapsulate hydrophilic drugs into polymer systems. Examples of cationic drugs containing primary, secondary and tertiary amines as well as common anionic materials used for electrostatic and ion pairing are outlined in Figure 2.
Figure 2. Structure of anionic and cationic molecules used for chemotherapeutic agent delivery systems.

(A) Examples of cationic chemotherapeutic agents: doxorubicin, 1; mitoxantrone, 3; and vincristine, 11. Blue highlights primary amine (square), secondary amine (circle) and tertiary amine (triangle). (B) Examples of carboxylate-containing polymers and polar hydrocarbons: alginate, 12; poly(γ-glutamic acid), 13; and oleic acid, 14. Carboxylate groups highlighted with red boxes. (C) Examples of sulfonate-containing polymers and polar hydrocarbons: dextran sulfate, 15; taurodeoxylate, 16; and tetradecyl sulfate, 17. Sulfonate groups highlighted with green boxes.
One approach to ion pairing is the use of fatty acids as an anion to interact with cationic drugs. When the ion pair forms, the hydrocarbon of the fatty acid reduces the solubility. This, in combination with neutralizing the drug charge, often results in a precipitant that can be further processed into drug delivery vehicles. Zhang et al. used a VCR-oleic acid (14, OA) ion complex to engineer a VCR sustained release system.37 The VCR-OA ion complex was developed into nanoparticles using the warmed emulsion technique. MCF-7 cell exposed to the VCR-OA nanoparticles had increased uptake of VCR when compared to the free drug-treated cells. This correlated to an approximate 15% increase in cellular apoptosis.
Ma et al. used ion pairing of DOX and idarubicin (2) with sodium taurodeoxycholate (16) and sodium tetradecyl sulfate (17) to form water insoluble precipitants.38 These precipitants were then formed into solid lipid nanoparticles using a warm microemulsion technique. Varying the ion pair ratio resulted in a bell shaped curve of drug precipitate to total drug. Further experiments were performed with the optimal ion pair ratio. The nanoparticles formed were around 75 nm, within the optimal range for utilizing the EPR effect. Nanoparticles are also able to bypass the P-glycoprotein efflux pumps (one of the main drug resistance mechanisms). Idarubicin nanoparticles were administered intravenously to an orthotopic human colon cancer model (HCT-15 human colon cells implanted in the subcutaneous space) and a leukemia mouse model (intravenously implanted with P388 murine leukemia cells). DOX nanoparticles were administered intravenously to a mouse leukemia model (P388/ADR murine leukemia cells implanted in the intraperitoneal space). The idarubicin nanoparticles behaved similarly to the free drug in terms of mouse survival time. The DOX nanoparticles increased mouse survival time when compared to free drug.
Dextran sulfate (15) has been used by multiple groups for ion pairing with cationic drugs. Dextran sulfate nanoparticles containing DOX can be formed by mixing the drug and polymer.39 Nearly 100% encapsulation efficiency is achieved at a drug/dextran weight ratio of 0.5. In vitro release from these nanoparticles showed an initial rapid release of 14% over the first 24 hours and a total release of 32% through 15 days.
Dextran sulfate has also been used to increase the encapsulation efficiency of the cationic drug mitoxantrone (3, MTO) in lipid nanoparticles formed via a microemulsion-sonication technique.40 Dextran sulfate increased the encapsulation efficiency of MTO by 27% (1:0.5, MTO:dextran charge ratio) and 62% (1:1, MTO:dextran charge ratio) compared the nanoparticles produced without dextran sulfate. Additionally, dextran sulfate extended the MTO release time from 12 hours (92.3% released) to 72 hours (86.9% released) and resulted in a more linear release profile. In vivo, the nanoparticles increased the mean plasma MTO concentration when compared to free MTO administration. Additionally, the nanoparticles increased the drug concentration in the liver, spleen and brain while decreased the drug distribution in the heart, kidney, and lung. In vitro, the nanoparticles decreased the IC50 of MTO in MTO-resistant MCF-7 cells (MCF-7/MX) presumably by bypassing P-glycoprotein efflux pumps.
VCR complexed-dextran sulfate nanoparticles have also been formed using the warmed microemulsion technique.41,42 Release of VCR into phosphate buffer saline was low (<18% over 6 days). Adding calcium to the phosphate buffered saline increased VCR release to nearly 100% over 6 days. In vivo, VCR plasma concentration was higher with nanoparticle administration when compared to free VCR administration. Nanoparticles increased spleen and liver VCR concentration while decreasing heart and kidney VCR concentration. This result is comparable to the previous section pertaining to MTO-containing nanoparticles. However, VCR was difficult to measure in the brain; labeling the nanoparticles with cumarin-6 allowed for quantification of the nanoparticle delivery to the brain.41,42 The nanoparticles increased cumarin-6 concentration in the brain at 60 minutes and 360 minutes post-intravenous injection. Therefore, the VCR-loaded nanoparticles may have enhanced utility in spleen, liver and brain cancers.
Electrostatic complexes may also be used to enhance the drug release profile from poly (ester) -based delivery vehicles that typically have a rapid release of hydrophilic drugs. Ling et al. developed VCR-loaded poly(glutamic acid-co-lactic acid) nanoparticles using dextran sulfate to increase encapsulation efficiency and enhance VCR release kinetics.43 The nanoparticles were formed by self-assembly and nanoprecipitation techniques to form VCR/dextran sulfate-loaded nanoparticles with a PEG shell and lecithin monolayer coating; nanoparticles void of dextran sulfate were used as controls. Dextran sulfate increased the encapsulation efficiency of VCR (93.6% versus 64.7%) and extended the release from 89.4% over 24 hours to 80.4% over 96 hours. Orally delivered VCR/dextran sulfate nanoparticles increased the maximum serum concentration and serum retention time when compared to free VCR administration. VCR/dextran sulfate nanoparticle treatment of MCF7/Adr cells (P-glycoprotein overexpressing cells) increased cellular uptake of VCR when compared to soluble-VCR treated cells.
Poly(γ-glutamic acid) (13, γ-PGA) is another example of a polyanionic polymer. Manocha et al. used electrostatic interaction to bind DOX to γ-PGA.44 Varying the weight ratios of DOX to γ-PGA was evaluated. All weight ratios evaluated resulted in almost 100% complexation efficiency. When the DOX weight ratio was 2:1 and 3:1 (DOX: γ-PGA), the entire complex formed a precipitant. Lower weight ratios gave water-soluble complexes with a colloidal suspension of approximately 80 nm nanoparticles. DOX release from the complex increased with decreasing pH of the release media. Hellman et al. further modified the DOX: γ-PGA complex system with the addition of chitosan. Chitosan is a polycation of repeating disaccharide units.45 A weight ratio of 1:10 (DOX:γ-PGA) was used. This solution complex was added dropwise to a stirred chitosan solution to form ion-complexed particles. The particle diameter increased with increasing concentration of the DOX:γ-PGA added to the chitosan solution (ranging from 169 nm to 546 nm). Varying the chitosan concentration did not significantly change the particle diameter. The DOX:γ-PGA:CS particles were cytotoxic to the squamous cell carcinoma line HN-5a.
Alginate (12) is a polycation of repeating disaccharide units that can be readily formed into gels using ionic and covalent conjugation methods.46 Redox-responsive alginate nanogels can be formed by EDC crosslinking a microemulsion of alginate with cystamine.47,48 This forms a redox-degradable polymer network where, upon degradation, increased drug release. Redox-responsive materials are particularly interesting for cancer therapeutics as increased glutathione, a reducing molecule, has been found in some cancer cells.49 Additionally, high levels of γ-interferon-inducible lysosomal thiol reductase, an enzyme that reduces disulfide bonds, are found in endosomes and lysosomes.50 Maciel et al. used redox-responsive alginate nanogels to bind DOX.51 The reducing agent DTT increased DOX release from the nanogel. Furthermore, the DOX-loaded alginate nanogels increased the cytotoxic effect of DOX in CAL-72 cells (IC50 = 0.9 μM for DOX-loaded alginate nanogels versus IC50 = 2.0 μM for free DOX).
Laponite® (LP) is a synthetic clay composed of silicate nanodisks. LP swells in water forming a clear hydrogel. It is anionic capable of electrostatically bind cationic molecules. Wang et al. evaluated LP nanodisks for DOX delivery (LP/DOX nanodisks).52 The LP/DOX nanodisks exhibited pH-dependent release kinetics. Furthermore, LP/DOX nanodisks increased in vitro DOX cytotoxicity in KB carcinoma cells. The DOX IC50 was 2.45 and 3.02 times lower with LP/DOX nanodisk administration versus free drug after 24 and 48 hours, respectively. Li et al. evaluated the LP/DOX nanodisks in vivo using a mouse subcutaneous carcinoma (KB cells) model.53 LP/DOX nanodisks treatment decreased the relative tumor volume, increased DOX tumor concentration and increased animal survival when compared to free DOX treated mice. Gonçalves et al. coated the LP/DOX nanodisks with alginate (LP/DOX/AG nanohybrids).54 Due to the negative charge of aliginate, coating with alginate decreased the burst release of DOX. The LP/DOX/AG nanohybrids exhibited increased in vitro DOX cytotoxicity as compared to free-DOX.
DOX interchelates into DNA inhibiting topoisomerase II-induced relaxation of DNA supercoils. A two-step mechanism has been proposed where DOX rapidly binds to the DNA groove via electrostatic interaction55,56 followed by slow interchelation of the aromatic DOX core into the GC sequence.55 Dong et al. developed a pH/enzyme responsive delivery system using the DNA/DOX complex.57 The DNA/DOX complex was combined with cationic gelatin (CPX1). This complex can be degraded by gelatinases and DNases. CPX1 was then combined with PEG-hystidine-alginate forming the pH responsive portion of the complex (CPX2). Exposure of CPX1 and CPX2 to tumor tissue homogenate supernatant induced DOX release from both formulations. However, liver tissue homogenate supernatant induced DOX release from CPX1 but little release from CPX2, suggesting that the CPX2 formulation may exhibit enhanced tumor-specific release. The in vivo biodistribution of DOX administered as CPX1 or CPX2 complexes compared to free DOX was evaluated in a mouse heterotopic sarcoma (S180 cells) model. CPX2 enhanced DOX distribution to the tumor while decreasing DOX distribution to the liver. Both CPX1 and CPX2 decreased DOX distribution to the heart, kidney, spleen and lung. All DOX treatment groups decreased tumor volume; CPX2 had the strongest tumor effect followed by CPX1 than free DOX. Additionally, 100% of the CPX2 treated animals survived through week 4 (n=10, 1 death at week 4) when compared to 80% for CPX1 (2 deaths at week 3.5) and 30% for free DOX (4 deaths at week 2.5 and 3 survivals at week 4).
Zhu et al. further modified CPX1 with an outer layer of negatively charged human serum albumin (HDD).58 DOX did not release from HDD after 6 hours in phosphate buffered saline. Exposure of HDD to capthesin B, gelatinase and DNase I induced DOX release. DOX biodistribution was evaluated in a mouse orthotopic hepatoma (Heps cells) model. HDD increased DOX accumulation in the tumor than free DOX administration. Additionally, HDD treatment decreased DOX accumulation in the heart, liver and kidney. DOX cytotoxicity was also reduced with HDD compared to free DOX administration; HDD treated mice had better body weight maintenance, increased survival and decreased tumor weight.
Silk has been investigated in multiple formats for sustained release of DOX.59–62 Silk is a protein, block-copolymer composed of multiple hydrophobic domains separated by smaller hydrophilic domains. Silk can be formed into multiple material formats, including films, hydrogels and particles using various processing techniques.63 The tunable crystallinity, due to the control of formation of β-sheets via the hydrophobic domains, confers substantial stability and mechanical robustness. Seib et al. showed that by incubating silk films in a DOX solution, DOX adsorbed into silk films60 through hydrophobic and charge interactions. Silk nanoparticles were loaded with DOX in a similar manner.61 Silk hydrogels were formed by sonication induced gelation; DOX was loaded prior to complete gelation.59 All of these DOX-loaded silk material formats exhibited sustained release of DOX through at least 30 days. DOX-loaded silk films and hydrogels were evaluated in an orthotopic mouse breast cancer model (injection of MDA-MB-231 cells transfected with luciferase for in vivo imaging). Films were placed on the tumor; hydrogels were injected into the tumor. DOX-loaded silk films significantly decreased tumor growth, evidenced by decreased luciferase signal per mouse and decreased tumor weight, when compared to intravenous DOX administration and immediately dissolved DOX-loaded silk films (a local delivery control). DOX-loaded silk hydrogels decreased tumor weight when compared to intravenous DOX administration.
Covalent Chemotherapeutic Conjugates
Covalent coupling methods have been explored extensively for delivering chemotherapeutic drugs (Figure 3). Numerous coupling methods can be used, resulting in different degrees of stability. Amide bonds are formed through the reaction of an amine with a carboxylic acid (Figure 3A). These linkages are stabile with minimal hydrolysis under physiological pH conditions. These bonds are hydrolyzed by proteolysis and under extreme pH conditions. Proteolytic enzymes are elevated in the tumor microenvironment making amide bonds a facile approach. In contrast, ester bonds, formed through the reaction of an alcohol with an ester, have a limited pH stability range and readily degrade at pH values deviating from neutral pH. The tumor microenvironment is slightly acidic, which will increase the hydrolysis of ester bonds. Additionally, ester linkages are readily hydrolyzed in the acidic environment of lysosomes. Another example of acid hydrolyzable bonds are hydrozones. Hydrazones are formed through the reaction of a hydrazide with an aldehyde. The exact conjugation desired is dependent on the available reactive groups as well as the desired stability and release mechanism. In this section, recent literature on covalent polymer/drug conjugates will be reviewed.
Figure 3. Structure of degradable linkers commonly used for polymer-drug conjugates.

(A) Linker susceptible to proteolysis: amide. (B) Linkers susceptible to hydrolysis: ester and anhydride. (C) Linker susceptible to hydrolysis under acidic conditions: hydrazone. For the ester, anhydride and amide linkers, R and R1 are either the carrier or the drug. For the hydrazone, R is the carrier and R2 and R3 are the drug.
Amide Linker
Gemcitabine (8, GEM) provides a primary amine group for amide bond formation. Chitkara et al. conjugated GEM to the carboxylic acid group of poly(ethylene glycol)-block-poly(2-methyl-2-carboxyl-propylene carbonate).64 The conjugate contained the polar poly(ethylene glycol) and non-polar GEM conjugate which spontaneously formed 23.6 ± 4 nm micelles (GEM micelles) under aqueous conditions. GEM micelles exhibited sustained release in PBS (pH 5.5). The addition of capthesin B to the PBS increased GEM release. GEM release and stability were evaluated in serum where GEM degrades to the metabolite dFdU. Free GEM degraded in serum to dFdU within 24 hours. With GEM micelles, a larger amount of intact GEM was detected in serum than the metabolite. This indicates that GEM micelles confirm stability to the conjugated GEM. In a mouse pancreatic adenocarcinoma (MIA PaCa-2 cell) xenograft model, GEM micelles significantly decreased tumor growth and weight. GEM micelle treated tumors contained more TUNEL-positive stained cells when compared to control tumors and free GEM treated tumors, indicating that the GEM micelles induced significantly more apoptosis.
Methotrxate (7, MTX) has been conjugated to PEGylated chitosan nanoparticles [(MTX-PEG)-CS-NPs] using EDC/NHS to form the amide linkage.65 MTX served two roles, (1) a folate analog binding to the folate receptor for internalization of the (MTX-PEG)-CS-NPs66,67 and (2) once inside the cells the amide bond degraded, releasing MTX, where MTX then conferred its cytotoxic behavior. MTX release from the (MTX-PEG)-CS-NPs in serum at pH 7.4, 6.5 and 5.0 was about 10% of the total drug loading with a slight trend towards increased release with decreased pH. At pH 5, MTX release significantly increased with the exposure to proteases. This data suggests that the (MTX-PEG)-CS-NPs would be stabile in blood circulation and then release MTX in the acidic, high protease environment of lysosomes. HeLa cells are folate receptor overexpressing cancer cells. (MTX-PEG)-CS-NPs increased the cytotoxicity of MTX (compared to soluble MTX) in HeLa cells but not in healthy fibroblasts which do not overexpress folate receptors. To evaluate the FA receptor targeting activity, (MTX-PEG)-CS-NPs were fluorescently tagged with FITC [FITC-(MTX-PEG)-CS-NPs]. FITC-(MTX-PEG)-CS-NPs were effectively taken up by HeLa cells. When the cells were exposed to high levels of folate, for competitive binding, FITC-(MTX-PEG)-CS-NPs uptake was inhibited.
Ester Linker
Wei et al. developed cholesterol-coupled HA (CHA) nanogels with ester linked cytotoxic drugs 68. The cytotoxic drugs investigated were etoposide (4), salinomycin and curcumin. Release of etoposide and salinomycin from the CHA nanogels was nearly linear at pH 7.4 over 20 days and 30 days, respectively. Curcumin is known to have limited stability under aqueous conditions.69 Therefore, the stability of curcumin released from the CHA nanogels was investigated after 24 hours in PBS. Less than 10% curcumin degradation was observed with the CHA nanogels compared to almost complete degradation of the free drug. CHA-drug nanogels increased cytotoxicity for all of the drugs investigated. In MDA-MB231/F cells (breast carcinoma cells with resistance to floxuridine), the CHA-etoposide IC50 was 3 μM compared to 12.3 μM (free etoposide); the CHA-salinomycin IC50 was 0.9 μM compared to 5.1 μM (free salinomycin). CHA-curcumin was evaluated with the MIA PaCa-2 cell line. The CHA-curcumin IC50 was 9 μM compared to 19 μM (free curcumin). The CHA IC50 was >5 mg/mL. Therefore, all CHA-drug groups exhibited enhanced cytotoxicity when compared to soluble drug groups.
Hydroxyethyl starch (HES) has been studied as a high-molecular weight carrier for MTX.70 HES is used clinically as a plasma volume expander to prevent shock after severe blood loss.71 The carboxylic acid on MTX was coupling to alcohols on HES via an ester bond. Stability of the conjugate was studied in PBS and plasma at pH 7.2. The HES-MTX half-life in plasma and PBS was 65.5 hours and 121.5 hours, respectively, indicating enzymatic degradation of the ester linkage. Tumor treatment was evaluated in NOD/SCID mice subcutaneously inoculated MV-4-11 leukemia cells. The HES-MTX conjugate increased mice survival compared to free MTX treatment. Furthermore, the HES-MTX conjugate reduced the tumor volume compared to free MTX treatment starting at day 8 post-treatment. After day 8, the HES-MTX conjugate treated tumors did not increase in tumor volume, suggesting tumor growth control at least through 22 days.
Namgung et al. reported on a nano-assembly of polymer cyclodextrin (pCD) and polymer paxlitaxel (pPTX).72 Cyclodextrin is known to form inclusion complexes with hydrophobic drugs.73 PTX forms a stable inclusion complex with β-cyclodextrin increasing its water solubility.74 Individual polymers containing both molecules, CD and PTX, were formed by reacting the hydroxyl groups on the drug with maleic anhydride (MAnh) on poly(isobutylenealt-MAnh) [poly(IB-alt-MAnh), molecular weight (Mw) 6 kDa] and poly(methyl vinyl ether-alt-MAnh) [poly(MVE-alt-MAnh), Mw 80 kDa], respectively. This reaction formed an ester linkage between the small molecule and polymer. During dialysis to remove unconjugated small molecules, the unreacted maleic anhydride converted to carboxylic acid resulting in water soluble polymer conjugates. The pPTX/pCD nano-assembly was formed by dissolving pPTX and pCD in a water/ethanol solvent followed by lyophilization; the average nano-assembly radius in water was 54.6±11.6 nm. The pPTX/pCD nano-assembly increased PTX cytotoxicity in three cancer cell lines: breast carcinoma MCF-7, cervical carcinoma HeLa and colorectal adenocarcinoma HCT-8 cells. The increased PTX cytotoxicity was reported to be due to passive intracellularization of the nanoparticles followed by esterase degradation which disintegrates the nano-assembly. To support this, the authors showed that exposing the pPTX/pCD nano-assembly to esterases from porcine liver decreased the diameter to less than 1 nm in 10 minutes. Exposing the nano-assembly to heat-denatured esterase did not facilitate nano-assembly disintegration, only the formation of larger aggregates. Furthermore, exposing the nano-assembly to serum did not change the particle size showing that the nano-assembly could remain stable in blood circulation. The nano-assembly was evaluated in mouse xenograft colorectal adenocarcinoma model (subcutaneous injection of HCT-8 cells). pPTX/pCD nano-assembly decreased the tumor growth when compared to soluble PTX and pPTX treatment. While all PTX treatments induced tumor regression through day 12 post-treatment, pPTX/pCD nano-assembly had minimal tumor regrowth thereafter; soluble PTX and pPTX treatment tumors were larger than the initial tumor volume at the end of the study. The pPTX/pCD nano-assembly was also evaluated in a mouse xenograft breast adenocarcinoma model (subcutaneous injection of HCT-8 cells). All PTX treatments in this model were less effective than in the HCT-8 xenograft mouse model. The MDA-M231 cells are known to be insensitive to PTX-induced apoptosis.75 MDA-M231 cells overexpress IL-4 receptor making this a potential focus for targeted therapeutics.76–78 Further modification of the nano-assembly with an AP-1 peptide which binds to IL-4 receptor79,80 increased the tumor suppression effect of the pPTX/pCD nano-assembly in the MDA-M231 xenograft mouse model.
PTX modified with a hydrolyzable succinate group (dicarboxylic acid forming a diester linker) and conjugated to low molecular weight chitosan (LMWC-PTX) has been evaluated.81,82 Three different degrees of PTX modification were evaluated: LMWC-PTX L, 4.8 wt%, LMWC-PTX M, 7.9 wt%, and LMWC-PTX H, 11.1 wt%. The LMWC-PTX spontaneously formed nanoparticles under aqueous conditions. Increasing the conjugated weight percentage increased the nanoparticle size (21.6 ± 12.7 nm, 67.6 ± 42.0 nm and 135.6 ± 33.1 nm). In murine breast cancer 4T1 cells, LMWC-PTX L and LMWC-PTX M conjugates decreased the IC50 of PTX (soluble PTX: 12.4 ± 27.9 ng/mL, LMWC-PTX L: 0.26 ± 0.57 ng/mL, LMWC-PTX M: 2.71 ± 3.3 ng/mL and LMWC-PTX H: 14.11 ± 27.27 ng/mL). In vivo, all LMWC-PTX conjugates exhibited higher serum concentrations through 24 hours than soluble PTX, which was cleared in 8 hours. The antitumor efficacy of LMWC-PTX H nanoparticles was evaluated in a murine 4T1 breast cancer model. LMWC-PTX H significantly decreased the tumor volume through 23 days as compared to soluble PTX administration.
Hydrazone Linkage
Kaminskas et al. developed a DOX conjugated generation five PEGylated poly(lysine) dendrimer (D-DOX) through a hydrazone bond.83–85 The D-DOX was compared to DOX-loaded liposomes (L-DOX), a clinical DOX formulation, and solution DOX administration in a mouse carcinoma model (subcutaneous injection of Walker 256 cells) and a mouse xenograft breast cancer model (subcutaneous injection of matrigel containing MDA-MB231 cells).83,84 All DOX treatment groups exhibited a similar effect on tumor volume growth (in both the carcinoma and breast cancer tumor models). In the mouse carcinoma model, the D-DOX treated mice had comparable body mass changes to PBS treated mice. L-DOX and solution DOX treated mice had comparably reduced body mass.83 Three days after administration, the DOX concentration in the tumor was higher in the D-DOX and L-DOX treated mice compared to free DOX-treated mice. The DOX concentration in the spleen, liver and heart was lower in D-DOX than L-DOX treated mice; D-DOX and L-DOX treated mice had lower DOX concentration in each organ investigated than soluble DOX-treated mice. Furthermore, mice treated with L-DOX rapidly developed palmar plantar erythematosis (PPE), or hand-foot syndrome; whereas D-DOX treated mice took a longer time and higher DOX dose before developing PPE. In the mouse breast cancer model, the maximum tolerable dose (MTD) was 10, 5 and 4.5 mg/kg for DOX solution, L-DOX and D-DOX treated mice, respectively.84 When mice were dosed with each respective MTD, comparable tumor growth suppression and body mass changes were observed. Taken together, these studies show that D-DOX treatment facilitated similar tumor suppression as L-DOX with the benefit of reduced systemic toxicity.
El-Dakdouki et al. developed DOX conjugated hyaluronic acid containing supermagnetic iron oxide nanoparticles (DOX/HA-SPIONs) through a hydrazone bond.86,87 The iron oxide nanoparticles are a contrast agent for magnetic resonance imaging, making the DOX/HA-SPIONs traceable in vivo. DOX release from DOX/HA-SPIONs was shown to be pH dependent, with increased release at pH 4.5 and pH 4.0 compared to pH 7.4.87 The DOX/HA-SPIONs had lower DOX release at pH 7.4 than reported for other DOX-conjugated nanoparticles88,89 which may be attributed to the different linker used90 or the buffering effect of HA. In both SKOV-3 and NCI/ADR-RES cells (both human ovarian cancer-derived cell lines), DOX/HA-SPIONs increased the cytotoxicity of DOX compared to solution DOX treatment.87 Fluorescence imaging of the cells showed increased DOX internalization in the DOX/HA-SPIONs treated cells. The SKOV-3 cells were transfected with luciferase and injected into the intraperitoneal cavity of mice.86 Mice were treated with DOX/HA-SPIONs, solution DOX or phosphate buffered saline (PBS). PBS-treated mice had extensive bioluminescence throughout the intraperitoneal cavity indicative of extensive tumor growth. Solution DOX-treatment decrease bioluminescence as compared to PBS control. However, DOX/HA-SPION treatment had the lowest bioluminescence. Furthermore, DOX/HA-SPION treatment increased mouse survival.
Jäger et al. reported on core-shell polymer nanoparticles with encapsulated DOX and encapsulated docetaxel (10, DTXL).91 DOX was encapsulated via a hydrazone linker to poly(N-(2-hydroxypropyl)methacrylamide)-cholesterol (PHPMA-chol-DOX). A nanoprecipitate and self-assembly protocol was used to form nanoparticles composed of a (poly(butylene succinate-co-butylene dilinoleate)) and docetaxel core coated with PHPMA-chol-DOX (NPDTXL-DOX). Core nanoparticles (NP0), core-shell nanoparticles (NP1), core-shell nanoparticles containing DTXL (NPDTXL) and core-shell nanoparticles containing DOX (NPDOX) were all evaluated as controls. DOX release increased at pH 5.0 as compared to pH 7.0; as expected, DTXL release was not pH. The anticancer activity of the nanoparticles was evaluated in a mouse syngeneic lymphoma model (EL-4 lymphoma cells). All drug-loaded nanoparticle groups had lower tumor volumes than PBS controls. When comparing soluble drug to individual drug-loaded nanoparticles, there was no difference between soluble drug and nanoparticle treated tumor volumes (DOX vs NPDOX and DTXL vs NPDTXL). NPDTXL-DOX treatment significantly decreased tumor volume with almost no tumor regrowth. Additionally, NPDTXL-DOX treatment increased mouse survival time compared to all other treatment groups.
Self-assembled DOX conjugated PEGylated-poly(glutamic acid)2 has been characterized for their cytotoxicity in vitro.92 The DOX conjugated PEGylated-poly(glutamic acid)2 self assembles into a core containing DOX with a PEGylated outer shell (DOX-conjugated micelles) after dissolution in DMSO and dialyzing in water. DOX release increased under acidic conditions compared to neutral pH confirming the retained pH sensitivity of the hydrazone linker inside of the self-assembled micelles. The DOX-conjugated micelles were internalized by HeLa cells and exerted a cytotoxic response. However, the intracellular fluorescence of DOX was decreased in to DOX-conjugated micelle treatment group versus free-DOX treatment group.
Platinum-Based Complexes
Cisplatin (5) is a commonly used chemotherapeutic drug. It contains platinum complexed with two ammonia molecules and two chloride ions through coordinate bonds. Cisplatin interferes with cell division by crosslinking DNA, which in turn activates apoptosis.93 In order for this to occur, cisplatin must first undergo ligand exchange where a chloride ion is replaced by water, thereby conferring its active form. Cisplatin aquation occurs in low chloride environments (i.e. water) and can be induced using silver nitrate.94 Aquated cisplatin is also able to complex with multiple chemical groups, including hydroxyls, amines, carboxylates and amides making this a potential mechanism for forming polymer/cisplatin complexes (Figure 4). Subsequent exposure to chloride rich environments reverses the complex and aquation (Figure 4A), thereby reverting back to cisplatin. Platinum based polymer-drug conjugates has been extensively reviewed elsewhere.95 In this review, recent polymer-based delivery systems will be discussed.
Figure 4. Structures of cisplatin conjugates.

(A) Cisplatin is reversibly converted to the active molecule via aquation in low chloride environments. (B) Aquated cisplatin reaction with carboxylate groups forming a monocarboxylato complex, 18a; and dicarboxylato complex, 18b. (C) Monocarboxylato cisplatin complex reaction with the oxygen of an amide group forming an O → Pt coordinate bond, 19a; and nitrogen of an amide group forming an N → Pt coordinate bond, 19b.
γ-PGA-based polymer systems have been investigated for complexation and release of cisplatin (monocarboxylato, 18a, and dicarboxylato, 18b, complexes, Figure 4B).96 This polymer-drug complex exhibited an initial burst release followed by a slow sustained release profile. When compared to soluble drug, the polymer-drug conjugate exhibited reduced cytotoxicity in vitro. In vivo, administration of the polymer-drug conjugate eliminated the body weight reduction exhibited by soluble drug–treated mice. Furthermore, the polymer-drug conjugate increased survive rate and decrease tumor growth of BALB/cA nude mice bearing human breast tumor Bcap-37 cells as compared to free-drug administration and PBS controls.96 In another study, the γ-PGA-cisplatin conjugate was compared to non-lethal soluble drug administration of cisplatin, carboplatin and oxaliplatin.97 Carboplatin and oxaliplatin are platinum derivatives with reduced systemic cytotoxicity compared to cisplatin. In this study, the γ-PGA-cisplatin conjugate exhibited comparable tumor volume control while eliminating the body mass loss observed by soluble cisplatin administration. Comparable body mass change was observed with the γ-PGA-cisplatin conjugate, PBS controls and soluble carboplatin and oxaliplatin treatment. Taken together, these studies show that the γ-PGA-cisplatin conjugate exhibited significant therapeutic potential as compared to free-cisplatin administration and the two cisplatin derivatives, carboplatin and oxaliplatin.
Stable nanoparticles formulated from a cisplatin-prodrug conjugated via a pH responsive hydrazone linker have been developed.98 Cisplatin was conjugated to two PEG-poly(lactic acid) (PEG-PLA) polymers generating Bi(PEG-PLA-linker)-cisplatin. This conjugate formed a hydrophobic cisplatin core decorated with a PEGylated nanoparticle surface. The polymer-prodrug nanoparticles exhibit controllable drug loading, stable pH-triggered release and cytotoxicity against ovarian cancer cells.
Hyperbranched polymers have been explored for conjugating cisplatin.99 Hyperbranched polyglycerols (PG2) and polyesters (HP40) are commercially available and both have terminal hydroxyl groups. The terminal hydroxyl groups are readily modified with carboxylic acid groups using succinic anhydride. Loading of carboxylic acid-modified PG2 and HP40 with cisplatin resulted in 25% w/w and 50% w/w cisplatin loading, respectively. The two hyperbranched systems exhibited different release kinetics. PG2 release cisplatin in saline continuously through 7 days, whereas little release was observed in water. However, cisplatin release from HP40 was similar in water and saline suggesting a weaker binding of cisplatin to HP40 compared to PG20. This work shows an interesting comparison of similar polymer systems with vastly different release kinetics, suggesting a potential for tuning release profiles based on the class of polymer systems used.
Hyaluronic acid is a naturally occurring polysaccharide that contains carboxylic acid groups and binds to the cell surface receptor CD44. CD44 is found in numerous malignant cells, including many ovarian cancers. Li et al. showed that cisplatin complexed-hyaluronic acid could form stable nanoparticles (Hyplat).100 Hyplat uptake was observed by CD44-positive cells, but not CD44-negative cells. Furthermore, Hyplat induced a 2–3 fold increase in cellular platinum levels when compared to free drug. When administered into the intraperitoneal cavity of mice, nanoparticle-loaded cisplatin was present for a longer time compared to free drug (8 hours versus 2 hours). In a mouse CD44+ xenograft tumor model, Hyplat treatment induced a higher tumor cisplatin concentration than free drug. Additionally, Hyplat treatment increased survival time to free-drug administration. In a mouse CD44+ xenograft tumor model, Hyplat and free-cisplatin treated mice had similar cisplatin tumor levels.
A novel polymer-cisplatin system has been developed taking into account interesting structure-activity relationship.101 In this work, poly(isobutylene-alt-maleic anhydride) was hydrolyzed to form poly(isobutylene-alt-maleic acid) (PIMA). Each monomeric unit could interact with one cisplatin molecule; the complete reaction of the monomeric units with cisplatin resulted in gelation. However, decreasing the molar ratio of cisplatin to monomeric unit allowed for the tunable formation of nanoparticles of different average diameter. Nanoparticles could be formatted with a diameter of 80–140 nm by using a 15:1 Pt:polymer ratio within the ideal range to utilized the EPR effect. However, PIMA-cisplatin nanoparticles induced similar in vitro cytotoxicity as PIMA alone, but was less cytotoxic than free cisplatin. PIMA was modified with glucosamine (PIMA-GA) via amide bond formation between one carboxylate per monomeric unit and the amine-group of glucosamine. Varying the pH during cisplatin binding induced different conjugation to the amide group: at pH less than seven, an amide O→Pt coordinate bond formed (19a, Figure 4C), at pH greater than seven, an amide N→Pt coordinate bond formed (19b, Figure 4C) as determined by NMR. Both conjugation chemistries resulted in nanoparticle formation at the 15:1 ratio. The PIMA-GA O→Pt nanoparticles induced greater in vitro cisplatin release and cellular cytotoxicity as compared to PIMA-GA N→Pt nanoparticles, presumably due to a weaker interaction between the O→Pt coordinate bond versus the N→ Pt coordinate bond. Mice bearing either a breast cancer or ovarian cancer model treated with PIMA-GA O→Pt nanoparticles exhibited comparable body mass changes to vehicle treatment. Furthermore, PIMA-GA O→Pt nanoparticles treatment reduced cisplatin accumulation in the kidney, spleen and liver, eliminated cisplatin-induce apoptosis in the kidney and decreasing tumor growth. This represents a unique way to alter the cisplatin-polymer conjugation resulting in enhance in vitro and in vivo responses.
Sengupta et al. developed self-assembled cisplatin nanoparticles (SACNs) using a lipid-film hydration self-assembly method102 with cholesterol-succinic acid-platinum (II) molecule, phosphatidylcholine and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000].103 Cisplatin was chelated to the succinic acid-modified cholesterol forming a monocarboxylato and an O→Pt coordinate bond. SACNs increased cisplatin cytotoxicity in the Lewis lung carcinoma (LLC) and 4T1 breast cancer cell lines. SACNs were also tested in the cisplatin-resistance hepatocellular carcinoma cell line 7404-CP20. SACNs decreased the cisplatin IC50 from 42.84 μM (free drug administration) to 3.02 μM. In an in vivo mouse breast cancer model, SACN treated mice had better maintained body weight and increased animal survival. SACNs decreased kidney and spleen cisplatin accumulation and increased tumor cisplatin accumulation. Furthermore, immunostaining for KIM-1, a kidney injury marker, showed no KIM-1 in SACNs-treated mouse kidney. However, substantial KIM-1 staining was observed in the free-cisplatin treatment group.
Tauro et al. evaluated matrix metalloproteinase- (MMP-) triggerable release of cisplatin from PEGDA hydrogels of low (574 Da) and high (400 Da) PEG chain molecular weight.104 First, PEGDA was modified with the MMP-cleavable peptide CDLGG using Michael addition of the acrylate and free thiol groups. Hydrogels were formed via free-radical polymerization using ammonium persulfate and N,N,N,N-tetramethylethylenediamine. Cisplatin was reacted to the peptide C-terminus. Nonspecific release of cisplatin from the hydrogel was observed. PEG(4000)DA-cisplatin hydrogels exhibited increased cisplatin release when exposed to MMP-2 and MMP-9. However, PEG(574)DA-cisplatin hydrogels exhibited similar cisplatin release with and without MMP-2 and MMP-9 exposure, likely due to inhibited MMP diffusion into the PEG(574)DA-cisplatin hydrogel. PEG(4000)DA-cisplatin hydrogels induced similar cytotoxicity to soluble cisplatin with MMP2 and MMP9 addition to the cell culture media.
Conclusions
In the pursuit of more effective cancer treatment options, emerging sustained release systems using conjugation/interaction techniques are increasing in interest and beginning to emerge clinically. Successful delivery systems will support sustained release over days to weeks, thereby reducing or eliminating frequent drug administration. These systems allow for increase efficacy of the administered drug, reducing the dose necessary to facilitate an equivalent response, as well as reducing systemic side effects. When choosing the appropriate delivery material, it is important to consider potential advantageous drug/material interactions that can favor drug binding and release, including inherent electrostatic interactions and degradable covalent linkers. Furthermore, engineered interactions can be incorporated into the sustained release system via co-encapsulation with polymers known to interact with the drug or encapsulating hydrophobic ion-pairs. Systemically delivered approaches, including tumor targeting ligands on the surface of the sustained release system (i.e. folate, methotrexate, and cancer-targeting antibodies) will further increase the tumor targeting effect and the efficacy of the chemotherapy drug. Emerging combined drug conjugation and tumor targeting schemes offer unique advantages that should lead to better clinical outcomes.
References
- 1.DeVita VT, Chu E. A history of cancer chemotherapy. Cancer Res. 2008;68:8643–8653. doi: 10.1158/0008-5472.CAN-07-6611. [DOI] [PubMed] [Google Scholar]
- 2.Chatterjee K, Zhang J, Honbo N, Karliner JS. Doxorubicin Cardiomyopathy. Cardiology. 2010;115:155–162. doi: 10.1159/000265166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanigan MH, Devarajan P. Cisplatin nephrotoxicity: molecular mechanisms. Cancer Ther. 2003;1:47–61. [PMC free article] [PubMed] [Google Scholar]
- 4.Boyette-Davis JA, Cata JP, Driver LC, Novy DM, Bruel BM, Mooring DL, Wendelschafer-Crabb G, Kennedy WR, Dougherty PM. Persistent chemoneuropathy in patients receiving the plant alkaloids paclitaxel and vincristine. Cancer Chemother Pharmacol. 2013;71:619–626. doi: 10.1007/s00280-012-2047-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruggiero A, Trombatore G, Triarico S, Arena R, Ferrara P, Scalzone M, Pierri F, Riccardi R. Platinum compounds in children with cancer: toxicity and clinical management. Anticancer Drugs. 2013;24:1007–1019. doi: 10.1097/CAD.0b013e3283650bda. [DOI] [PubMed] [Google Scholar]
- 6.Gale R. Antineoplastic chemotherapy myelosuppression: mechanisms and new approaches. Exp Hematol. 1984;13:3–7. [PubMed] [Google Scholar]
- 7.Sadat Tabatabaei Mirakabad F, Nejati-Koshki K, Akbarzadeh A, Yamchi MR, Milani M, Zarghami N, Zeighamian V, Rahimzadeh A, Alimohammadi S, Hanifehpour Y, et al. PLGA-based nanoparticles as cancer drug delivery systems. Asian Pac J Cancer Prev. 2014;15:517–535. doi: 10.7314/apjcp.2014.15.2.517. [DOI] [PubMed] [Google Scholar]
- 8.Cherng JY, Hou TY, Shih MF, Talsma H, Hennink WE. Polyurethane-based drug delivery systems. Int J Pharm. 2013;450:145–162. doi: 10.1016/j.ijpharm.2013.04.063. [DOI] [PubMed] [Google Scholar]
- 9.Uhrich KE, Cannizzaro SM, Langer RS, Shakesheff KM. Polymeric systems for controlled drug release. Chem Rev. 1999;99:3181–3198. doi: 10.1021/cr940351u. [DOI] [PubMed] [Google Scholar]
- 10.Sampath P, Brem H. Implantable slow-release chemotherapeutic polymers for the treatment of malignant brain tumors. Cancer control: journal of the Moffitt Cancer Center. 1998;5:130–137. doi: 10.1177/107327489800500204. [DOI] [PubMed] [Google Scholar]
- 11.Janknegt R. Liposomal formulations of cytotoxic drugs. Support Care Cancer. 1996;4:298–304. doi: 10.1007/BF01358884. [DOI] [PubMed] [Google Scholar]
- 12.Huynh NT, Passirani C, Saulnier P, Benoit JP. Lipid nanocapsules: a new platform for nanomedicine. Int J Pharm. 2009;379:201–209. doi: 10.1016/j.ijpharm.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 13.Silverman JA, Deitcher SR. Marqibo® (vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother Pharmacol. 2013;71:555–564. doi: 10.1007/s00280-012-2042-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan X, Scherphof GL, Kamps JA. Liposome opsonization. J Liposome Res. 2005;15:109–139. doi: 10.1081/lpr-64971. [DOI] [PubMed] [Google Scholar]
- 15.Dawidczyk CM, Kim C, Park JH, Russell LM, Lee KH, Pomper MG, Searson PC. State-of-the-art in design rules for drug delivery platforms: lessons learned from FDA-approved nanomedicines. J Controlled Release. 2014;187:133–144. doi: 10.1016/j.jconrel.2014.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41:189–207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 17.Strickley RG. Solubilizing excipients in oral and injectable formulations. Pharm Res. 2004;21:201–230. doi: 10.1023/b:pham.0000016235.32639.23. [DOI] [PubMed] [Google Scholar]
- 18.Kintzel PE. Prophylaxis for paclitaxel hypersensitivity reactions. Ann Pharmacother. 2001;35:1114–1117. doi: 10.1345/aph.10287. [DOI] [PubMed] [Google Scholar]
- 19.Fasano M, Curry S, Terreno E, Galliano M, Fanali G, Narciso P, Notari S, Ascenzi P. The extraordinary ligand binding properties of human serum albumin. IUBMB Life. 2005;57:787–796. doi: 10.1080/15216540500404093. [DOI] [PubMed] [Google Scholar]
- 20.Miele E, Spinelli GP, Miele E, Tomao F, Tomao S. Albumin-bound formulation of paclitaxel (Abraxane(®) ABI-007) in the treatment of breast cancer. Int J Nanomedicine. 2009;4:99–105. doi: 10.2147/ijn.s3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.John TA, Vogel SM, Tiruppathi C, Malik AB, Minshall RD. Quantitative analysis of albumin uptake and transport in the rat microvessel endothelial monolayer. Am J Physiol Lung Cell Mol Physiol. 2003;284:L187–196. doi: 10.1152/ajplung.00152.2002. [DOI] [PubMed] [Google Scholar]
- 22.Vogel SM, Minshall RD, Pilipovic M, Tiruppathi C, Malik AB. Albumin uptake and transcytosis in endothelial cells in vivo induced by albumin-binding protein. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1512–1522. doi: 10.1152/ajplung.2001.281.6.L1512. [DOI] [PubMed] [Google Scholar]
- 23.Dang W, Daviau T, Ying P, Zhao Y, Nowotnik D, Clow CS, Tyler B, Brem H. Effects of GLIADEL® wafer initial molecular weight on the erosion of wafer and release of BCNU. J Controlled Release. 1996;42:83–92. [Google Scholar]
- 24.Brem H, Mahaley MS, Jr, Vick NA, Black KL, Schold SC, Jr, Burger PC, Friedman AH, Ciric IS, Eller TW, Cozzens JW, et al. Interstitial chemotherapy with drug polymer implants for the treatment of recurrent gliomas. J Neurosurg. 1991;74:441–446. doi: 10.3171/jns.1991.74.3.0441. [DOI] [PubMed] [Google Scholar]
- 25.Van Ha TG. Transarterial chemoembolization for hepatocellular carcinoma. Semin Intervent Radiol. 2009;26:270–275. doi: 10.1055/s-0029-1225672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berghammer P, Pfeffel F, Winkelbauer F, Wiltschke C, Schenk T, Lammer J, Müller C, Zielinski C. Arterial hepatic embolization of unresectable hepatocellular carcinoma using a cyanoacrylate/lipiodol mixture. Cardiovasc Intervent Radiol. 1998;21:214–218. doi: 10.1007/s002709900247. [DOI] [PubMed] [Google Scholar]
- 27.Kirchhoff T, Chavan A, Galanski M. Transarterial chemoembolization and percutaneous ethanol injection therapy in patients with hepatocellular carcinoma. Eur J Gastroenterol Hepatol. 1998;10:907–910. doi: 10.1097/00042737-199811000-00001. [DOI] [PubMed] [Google Scholar]
- 28.Tadavarthy SM, MOLLER JH, AMPLATZ K. Polyvinyl alcohol (Ivalon)—a new embolic material. Am J Roentgenol. 1975;125:609–616. doi: 10.2214/ajr.125.3.609. [DOI] [PubMed] [Google Scholar]
- 29.Albert M, Kiefer MV, Sun W, Haller D, Fraker DL, Tuite CM, Stavropoulos SW, Mondschein JI, Soulen MC. Chemoembolization of colorectal liver metastases with cisplatin, doxorubicin, mitomycin C, ethiodol, and polyvinyl alcohol. Cancer. 2011;117:343–352. doi: 10.1002/cncr.25387. [DOI] [PubMed] [Google Scholar]
- 30.Kirikoshi H, Saito S, Yoneda M, Fujita K, Mawatari H, Uchiyama T, Higurashi T, Goto A, Takahashi H, Abe Y. Outcome of transarterial chemoembolization monotherapy, and in combination with percutaneous ethanol injection, or radiofrequency ablation therapy for hepatocellular carcinoma. Hepatol Res. 2009;39:553–562. doi: 10.1111/j.1872-034X.2009.00490.x. [DOI] [PubMed] [Google Scholar]
- 31.Laurent A. Microspheres and nonspherical particles for embolization. Tech Vasc Interv Radiol. 2007;10:248–256. doi: 10.1053/j.tvir.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 32.Lammer J, Malagari K, Vogl T, Pilleul F, Denys A, Watkinson A, Pitton M, Sergent G, Pfammatter T, Terraz S. Prospective randomized study of doxorubicin-eluting-bead embolization in the treatment of hepatocellular carcinoma: results of the PRECISION V study. Cardiovasc Intervent Radiol. 2010;33:41–52. doi: 10.1007/s00270-009-9711-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lencioni R, de Baere T, Burrel M, Caridi JG, Lammer J, Malagari K, Martin RC, O’Grady E, Real MI, Vogl TJ. Transcatheter treatment of hepatocellular carcinoma with doxorubicin-loaded DC Bead (DEBDOX): technical recommendations. Cardiovasc Intervent Radiol. 2012;35:980–985. doi: 10.1007/s00270-011-0287-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malagari K, Pomoni M, Moschouris H, Kelekis A, Charokopakis A, Bouma E, Spyridopoulos T, Chatziioannou A, Sotirchos V, Karampelas T. Chemoembolization of hepatocellular carcinoma with hepasphere 30–60 μm. Safety and efficacy study. Cardiovasc Intervent Radiol. 2014;37:165–175. doi: 10.1007/s00270-013-0777-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tønnesen HH, Karlsen J. Alginate in Drug Delivery Systems. Drug Dev Ind Pharm. 2002;28:621–630. doi: 10.1081/ddc-120003853. [DOI] [PubMed] [Google Scholar]
- 36.Kim GO, Kim N, Kim DY, Kwon JS, Min BH. An electrostatically crosslinked chitosan hydrogel as a drug carrier. Molecules. 2012;17:13704–13711. doi: 10.3390/molecules171213704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang T, Zheng Y, Peng Q, Cao X, Gong T, Zhang Z. A novel submicron emulsion system loaded with vincristine–oleic acid ion-pair complex with improved anticancer effect: in vitro and in vivo studies. Int J Nanomedicine. 2013;8:1185–1196. doi: 10.2147/IJN.S41775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma P, Dong X, Swadley CL, Gupte A, Leggas M, Ledebur HC, Mumper RJ. Development of idarubicin and doxorubicin solid lipid nanoparticles to overcome Pgp–mediated multiple drug resistance in leukemia. J Biomed Nanotechnol. 2009;5:151–161. doi: 10.1166/jbn.2009.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yousefpour P, Atyabi F, Farahani EV, Sakhtianchi R, Dinarvand R. Polyanionic carbohydrate doxorubicin-dextran nanocomplex as a delivery system for anticancer drugs: in vitro analysis and evaluations. Int J Nanomedicine. 2011;6:1487–1496. doi: 10.2147/IJN.S18535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang P, Ling G, Pan X, Sun J, Zhang T, Pu X, Yin S, He Z. Novel nanostructured lipid-dextran sulfate hybrid carriers overcome tumor multidrug resistance of mitoxantrone hydrochloride. Nanomed Nanotechnol Biol Med. 2012;8:185–193. doi: 10.1016/j.nano.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 41.Aboutaleb E, Atyabi F, Khoshayand MR, Vatanara AR, Ostad SN, Kobarfard F, Dinarvand R. Improved brain delivery of vincristine using dextran sulfate complex solid lipid nanoparticles: optimization and in vivo evaluation. J Biomed Mater Res A. 2014;102:2125–2136. doi: 10.1002/jbm.a.34890. [DOI] [PubMed] [Google Scholar]
- 42.Aboutaleb E, Dinarvand R. Vincristine-dextran complex loaded solid lipid nanoparticles for drug delivery to the brain. Proceedings of World Academy of Science, Engineering and Technology. 2012;6:611–615. [Google Scholar]
- 43.Ling G, Zhang P, Zhang W, Sun J, Meng X, Qin Y, Deng Y, He Z. Development of novel self-assembled DS-PLGA hybrid nanoparticles for improving oral bioavailability of vincristine sulfate by P-gp inhibition. J Controlled Release. 2010;148:241–248. doi: 10.1016/j.jconrel.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 44.Manocha B, Margaritis A. Controlled release of doxorubicin from doxorubicin/γ-polyglutamic acid ionic complex. J Nanomater. 2010;2010 doi: 10.1155/2010/780171. [DOI] [Google Scholar]
- 45.Hellmers F, Ferguson P, Koropatnick J, Krull R, Margaritis A. Characterization and in vitro cytotoxicity of doxorubicin-loaded γ-polyglutamic acid-chitosan composite nanoparticles. Biochem Eng J. 2013;75:72–78. [Google Scholar]
- 46.Augst AD, Kong HJ, Mooney DJ. Alginate Hydrogels as Biomaterials. Macromol Biosci. 2006;6:623–633. doi: 10.1002/mabi.200600069. [DOI] [PubMed] [Google Scholar]
- 47.Khdair A, Chen D, Patil Y, Ma L, Dou QP, Shekhar MP, Panyam J. Nanoparticle-mediated combination chemotherapy and photodynamic therapy overcomes tumor drug resistance. J Controlled Release. 2010;141:137–144. doi: 10.1016/j.jconrel.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khdair A, Handa H, Mao G, Panyam J. Nanoparticle-mediated combination chemotherapy and photodynamic therapy overcomes tumor drug resistance in vitro. Eur J Pharm Biopharm. 2009;71:214–222. doi: 10.1016/j.ejpb.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 49.Morstyn G, Russo A, Carney DN, Karawya E, Wilson SH, Mitchell JB. Heterogeneity in the radiation survival curves and biochemical properties of human lung cancer cell lines. J Natl Cancer Inst. 1984;73:801–807. [PubMed] [Google Scholar]
- 50.Arunachalam B, Phan UT, Geuze HJ, Cresswell P. Enzymatic reduction of disulfide bonds in lysosomes: characterization of a gamma-interferon-inducible lysosomal thiol reductase (GILT) Proc Natl Acad Sci USA. 2000;97:745–750. doi: 10.1073/pnas.97.2.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maciel D, Figueira P, Xiao S, Hu D, Shi X, Rodrigues J, Tomás H, Li Y. Redox-responsive alginate nanogels with enhanced anticancer cytotoxicity. Biomacromolecules. 2013;14:3140–3146. doi: 10.1021/bm400768m. [DOI] [PubMed] [Google Scholar]
- 52.Wang S, Wu Y, Guo R, Huang Y, Wen S, Shen M, Wang J, Shi X. Laponite nanodisks as an efficient platform for Doxorubicin delivery to cancer cells. Langmuir. 2013;29:5030–5036. doi: 10.1021/la4001363. [DOI] [PubMed] [Google Scholar]
- 53.Li K, Wang S, Wen S, Tang Y, Li J, Shi X, Zhao Q. Enhanced in vivo antitumor efficacy of doxorubicin encapsulated within laponite nanodisks. ACS Appl Mater Interfaces. 2014;6:12328–12334. doi: 10.1021/am502094a. [DOI] [PubMed] [Google Scholar]
- 54.Goncalves M, Figueira P, Maciel D, Rodrigues J, Qu X, Liu C, Tomas H, Li Y. pH-sensitive Laponite(®)/doxorubicin/alginate nanohybrids with improved anticancer efficacy. Acta Biomater. 2014;10:300–307. doi: 10.1016/j.actbio.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 55.Pérez-Arnaiz C, Busto N, Leal JM, García B. New Insights into the Mechanism of the DNA/Doxorubicin Interaction. J Phys Chem B. 2014;118:1288–1295. doi: 10.1021/jp411429g. [DOI] [PubMed] [Google Scholar]
- 56.Rizzo V, Sacchi N, Menozzi M. Kinetic studies of anthracycline-DNA interaction by fluorescence stopped flow confirm a complex association mechanism. Biochemistry (Mosc) 1989;28:274–282. doi: 10.1021/bi00427a038. [DOI] [PubMed] [Google Scholar]
- 57.Dong L, Xia S, Wu K, Huang Z, Chen H, Chen J, Zhang J. A pH/enzyme-responsive tumor-specific delivery system for doxorubicin. Biomaterials. 2010;31:6309–6316. doi: 10.1016/j.biomaterials.2010.04.049. [DOI] [PubMed] [Google Scholar]
- 58.Zhu Q, Jia L, Gao Z, Wang C, Jiang H, Zhang J, Dong L. A tumor environment responsive doxorubicin-loaded nanoparticle for targeted cancer therapy. Mol Pharm. 2014;11:3269–3278. doi: 10.1021/mp4007776. [DOI] [PubMed] [Google Scholar]
- 59.Seib FP, Pritchard EM, Kaplan DL. Self-assembling doxorubicin silk hydrogels for the focal treatment of primary breast cancer. Adv Funct Mater. 2013;23:58–65. doi: 10.1002/adfm.201201238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Seib FP, Kaplan DL. Doxorubicin-loaded silk films: Drug-silk interactions and in vivo performance in human orthotopic breast cancer. Biomaterials. 2012;33:8442–8450. doi: 10.1016/j.biomaterials.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seib FP, Jones GT, Rnjak-Kovacina J, Lin Y, Kaplan DL. pH-dependent anticancer drug release from silk nanoparticles. Adv Healthc Mater. 2013;2:1606–1611. doi: 10.1002/adhm.201300034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chiu B, Coburn J, Pilichowska M, Holcroft C, Seib FP, Charest A, Kaplan DL. Surgery combined with controlled-release doxorubicin silk films as a treatment strategy in an orthotopic neuroblastoma mouse model. Br J Cancer. 2014;111:708–715. doi: 10.1038/bjc.2014.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rockwood DN, Preda RC, Yucel T, Wang X, Lovett ML, Kaplan DL. Materials fabrication from Bombyx mori silk fibroin. Nat Protoc. 2011;6:1612–1631. doi: 10.1038/nprot.2011.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chitkara D, Mittal A, Behrman SW, Kumar N, Mahato RI. Self-assembling, amphiphilic polymer–gemcitabine conjugate shows enhanced antitumor efficacy against human pancreatic adenocarcinoma. Bioconj Chem. 2013;24:1161–1173. doi: 10.1021/bc400032x. [DOI] [PubMed] [Google Scholar]
- 65.Luo F, Li Y, Jia M, Cui F, Wu H, Yu F, Lin J, Yang X, Hou Z, Zhang Q. Validation of a Janus role of methotrexate-based PEGylated chitosan nanoparticles in vitro. Nanoscale Res Lett. 2014;9:1–13. doi: 10.1186/1556-276X-9-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Deutsch JC, Elwood PC, Portillo RM, Macey MG, Kolhouse JF. Role of the membrane-associated folate binding protein (folate receptor) in methotrexate transport by human KB cells. Arch Biochem Biophys. 1989;274:327–337. doi: 10.1016/0003-9861(89)90446-3. [DOI] [PubMed] [Google Scholar]
- 67.Kelemen LE. The role of folate receptor α in cancer development, progression and treatment: Cause, consequence or innocent bystander? Int J Cancer. 2006;119:243–250. doi: 10.1002/ijc.21712. [DOI] [PubMed] [Google Scholar]
- 68.Wei X, Senanayake TH, Warren G, Vinogradov SV. Hyaluronic acid-based nanogel–drug conjugates with enhanced anticancer activity designed for the targeting of CD44-positive and drug-resistant tumors. Bioconj Chem. 2013;24:658–668. doi: 10.1021/bc300632w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Manju S, Sreenivasan K. Conjugation of curcumin onto hyaluronic acid enhances its aqueous solubility and stability. J Colloid Interface Sci. 2011;359:318–325. doi: 10.1016/j.jcis.2011.03.071. [DOI] [PubMed] [Google Scholar]
- 70.Goszczyński T, Filip-Psurska B, Kempińska K, Wietrzyk J, Boratyński J. Hydroxyethyl starch as an effective methotrexate carrier in anticancer therapy. Pharmacol Res Per. 2014;2:e00047. doi: 10.1002/prp2.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Myburgh JA, Finfer S, Bellomo R, Billot L, Cass A, Gattas D, Glass P, Lipman J, Liu B, McArthur C, et al. Hydroxyethyl starch or saline for fluid resuscitation in intensive care. N Engl J Med. 2012;367:1901–1911. doi: 10.1056/NEJMoa1209759. [DOI] [PubMed] [Google Scholar]
- 72.Namgung R, Lee YM, Kim J, Jang Y, Lee BH, Kim IS, Sokkar P, Rhee YM, Hoffman AS, Kim WJ. Poly-cyclodextrin and poly-paclitaxel nano-assembly for anticancer therapy. Nat Commun. 2014;5:3702. doi: 10.1038/ncomms4702. [DOI] [PubMed] [Google Scholar]
- 73.Tiwari G, Tiwari R, Rai AK. Cyclodextrins in delivery systems: Applications. J Pharm Bioallied Sci. 2010;2:72–79. doi: 10.4103/0975-7406.67003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cserháti T, Holló J. Interaction of taxol and other anticancer drugs with hydroxypropyl-β-cyclodextrin. Int J Pharm. 1994;108:69–75. doi: 10.1016/0731-7085(95)01263-k. [DOI] [PubMed] [Google Scholar]
- 75.Flores ML, Castilla C, Avila R, Ruiz-Borrego M, Saez C, Japon MA. Paclitaxel sensitivity of breast cancer cells requires efficient mitotic arrest and disruption of Bcl-xL/Bak interaction. Breast Cancer Res Treat. 2012;133:917–928. doi: 10.1007/s10549-011-1864-9. [DOI] [PubMed] [Google Scholar]
- 76.Obiri NI, Siegel JP, Varricchio F, Puri RK. Expression of high-affinity IL-4 receptors on human melanoma, ovarian and breast carcinoma cells. Clin Exp Immunol. 1994;95:148–155. doi: 10.1111/j.1365-2249.1994.tb06029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Toi M, Bicknell R, Harris AL. Inhibition of colon and breast carcinoma cell growth by interleukin-4. Cancer Res. 1992;52:275–279. [PubMed] [Google Scholar]
- 78.Gooch JL, Lee AV, Yee D. Interleukin 4 inhibits growth and induces apoptosis in human breast cancer cells. Cancer Res. 1998;58:4199–4205. [PubMed] [Google Scholar]
- 79.Hong HY, Lee HY, Kwak W, Yoo J, Na MH, So IS, Kwon TH, Park HS, Huh S, Oh GT, et al. Phage display selection of peptides that home to atherosclerotic plaques: IL-4 receptor as a candidate target in atherosclerosis. J Cell Mol Med. 2008;12:2003–2014. doi: 10.1111/j.1582-4934.2008.00189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu XL, Kim JH, Koo H, Bae SM, Shin H, Kim MS, Lee BH, Park RW, Kim IS, Choi K, et al. Tumor-targeting peptide conjugated pH-responsive micelles as a potential drug carrier for cancer therapy. Bioconj Chem. 2010;21:208–213. doi: 10.1021/bc9005283. [DOI] [PubMed] [Google Scholar]
- 81.Kim H, Lee E, Lee IH, Lee J, Kim J, Kim S, Lee Y, Kim D, Choi M, Kim YC. Preparation and therapeutic evaluation of paclitaxel-conjugated low-molecular-weight chitosan nanoparticles. Macromol Res. 2014;22:805–808. [Google Scholar]
- 82.Lee E, Lee J, Lee IH, Yu M, Kim H, Chae SY, Jon S. Conjugated chitosan as a novel platform for oral delivery of paclitaxel. J Med Chem. 2008;51:6442–6449. doi: 10.1021/jm800767c. [DOI] [PubMed] [Google Scholar]
- 83.Kaminskas LM, McLeod VM, Kelly BD, Sberna G, Boyd BJ, Williamson M, Owen DJ, Porter CJ. A comparison of changes to doxorubicin pharmacokinetics, antitumor activity, and toxicity mediated by PEGylated dendrimer and PEGylated liposome drug delivery systems. Nanomed Nanotechnol Biol Med. 2012;8:103–111. doi: 10.1016/j.nano.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 84.Kaminskas LM, McLeod VM, Kelly BD, Cullinane C, Sberna G, Williamson M, Boyd BJ, Owen DJ, Porter CJ. Doxorubicin-conjugated PEGylated dendrimers show similar tumoricidal activity but lower systemic toxicity when compared to PEGylated liposome and solution formulations in mouse and rat tumor models. Mol Pharm. 2012;9:422–432. doi: 10.1021/mp200522d. [DOI] [PubMed] [Google Scholar]
- 85.Kaminskas LM, Kelly BD, McLeod VM, Sberna G, Owen DJ, Boyd BJ, Porter CJ. Characterisation and tumour targeting of PEGylated polylysine dendrimers bearing doxorubicin via a pH labile linker. J Controlled Release. 2011;152:241–248. doi: 10.1016/j.jconrel.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 86.El-Dakdouki MH, Xia J, Zhu DC, Kavunja H, Grieshaber J, O’Reilly S, McCormick JJ, Huang X. Assessing the in vivo efficacy of doxorubicin loaded hyaluronan nanoparticles. ACS Appl Mater Interfaces. 2014;6:697–705. doi: 10.1021/am404946v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.El-Dakdouki MH, Zhu DC, El-Boubbou K, Kamat M, Chen J, Li W, Huang X. Development of multifunctional hyaluronan-coated nanoparticles for imaging and drug delivery to cancer cells. Biomacromolecules. 2012;13:1144–1151. doi: 10.1021/bm300046h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Upadhyay KK, Bhatt AN, Mishra AK, Dwarakanath BS, Jain S, Schatz C, Le Meins JF, Farooque A, Chandraiah G, Jain AK, et al. The intracellular drug delivery and anti tumor activity of doxorubicin loaded poly(γ-benzyl l-glutamate)-b-hyaluronan polymersomes. Biomaterials. 2010;31:2882–2892. doi: 10.1016/j.biomaterials.2009.12.043. [DOI] [PubMed] [Google Scholar]
- 89.Wang F, Wang YC, Dou S, Xiong MH, Sun TM, Wang J. Doxorubicin-tethered responsive gold nanoparticles facilitate intracellular drug delivery for overcoming multidrug resistance in cancer cells. ACS Nano. 2011;5:3679–3692. doi: 10.1021/nn200007z. [DOI] [PubMed] [Google Scholar]
- 90.Lee CC, Cramer AT, Szoka FC, Fréchet JMJ. An intramolecular cyclization reaction is responsible for the in vivo inefficacy and apparent pH insensitive hydrolysis kinetics of hydrazone carboxylate derivatives of doxorubicin. Bioconj Chem. 2006;17:1364–1368. doi: 10.1021/bc060117y. [DOI] [PubMed] [Google Scholar]
- 91.Jäger E, Jäger A, Chytil P, Etrych T, Říhová B, Giacomelli FC, Štěpánek P, Ulbrich K. Combination chemotherapy using core-shell nanoparticles through the self-assembly of HPMA-based copolymers and degradable polyester. J Controlled Release. 2013;165:153–161. doi: 10.1016/j.jconrel.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 92.Sui B, Xu H, Jin J, Gou J, Liu J, Tang X, Zhang Y, Xu J, Zhang H, Jin X. Self-assembled micelles composed of doxorubicin conjugated Y-shaped PEG-poly(glutamic acid)2 copolymers via hydrazone linkers. Molecules. 2014;19:11915–11932. doi: 10.3390/molecules190811915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dasari S, Bernard Tchounwou P. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–378. doi: 10.1016/j.ejphar.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Macka M, Borák J, Kiss F. Separation of some platinum(II) complexes by ionic strength gradient on a solvent-generated ion-exchange sorbent. J Chromatogr. 1991;586:291–295. doi: 10.1016/0021-9673(91)85135-3. [DOI] [PubMed] [Google Scholar]
- 95.Haxton KJ, Burt HM. Polymeric drug delivery of platinum-based anticancer agents. J Pharm Sci. 2009;98:2299–2316. doi: 10.1002/jps.21611. [DOI] [PubMed] [Google Scholar]
- 96.Ye H, Jin L, Hu R, Yi Z, Li J, Wu Y, Xi X, Wu Z. Poly(gamma, L-glutamic acid)-cisplatin conjugate effectively inhibits human breast tumor xenografted in nude mice. Biomaterials. 2006;27:5958–5965. doi: 10.1016/j.biomaterials.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 97.Feng Z, Lai Y, Ye H, Huang J, Xi XG, Wu Z. Poly (gamma, L-glutamic acid)-cisplatin bioconjugate exhibits potent antitumor activity with low toxicity: a comparative study with clinically used platinum derivatives. Cancer Sci. 2010;101:2476–2482. doi: 10.1111/j.1349-7006.2010.01708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Aryal S, Hu CMJ, Zhang L. Polymer cisplatin conjugate nanoparticles for acid-responsive drug delivery. Acs Nano. 2009;4:251–258. doi: 10.1021/nn9014032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Haxton KJ, Burt HM. Hyperbranched polymers for controlled release of cisplatin. Dalton Trans. 2008:5872–5875. doi: 10.1039/b809949a. [DOI] [PubMed] [Google Scholar]
- 100.Li SD, Howell SB. CD44-targeted microparticles for delivery of cisplatin to peritoneal metastases. Mol Pharm. 2009;7:280–290. doi: 10.1021/mp900242f. [DOI] [PubMed] [Google Scholar]
- 101.Paraskar AS, Soni S, Chin KT, Chaudhuri P, Muto KW, Berkowitz J, Handlogten MW, Alves NJ, Bilgicer B, Dinulescu DM. Harnessing structure-activity relationship to engineer a cisplatin nanoparticle for enhanced antitumor efficacy. Proc Natl Acad Sci USA. 2010;107:12435–12440. doi: 10.1073/pnas.1007026107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sengupta S, Eavarone D, Capila I, Zhao G, Watson N, Kiziltepe T, Sasisekharan R. Temporal targeting of tumour cells and neovasculature with a nanoscale delivery system. Nature. 2005;436:568–572. doi: 10.1038/nature03794. [DOI] [PubMed] [Google Scholar]
- 103.Sengupta P, Basu S, Soni S, Pandey A, Roy B, Oh MS, Chin KT, Paraskar AS, Sarangi S, Connor Y. Cholesterol-tethered platinum II-based supramolecular nanoparticle increases antitumor efficacy and reduces nephrotoxicity. Proc Natl Acad Sci USA. 2012;109:11294–11299. doi: 10.1073/pnas.1203129109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tauro JR, Gemeinhart RA. Matrix metalloprotease triggered delivery of cancer chemotherapeutics from hydrogel matrixes. Bioconj Chem. 2005;16:1133–1139. doi: 10.1021/bc0501303. [DOI] [PubMed] [Google Scholar]