

Abstract
Taking 12-N-p-chlorobenzyl sophoridinol 2 as a lead, a series of novel sophoridinic derivatives with various 3′-substituents at the 11–side chain were synthesized and evaluated for their anticancer activity from sophoridine (1), a natural antitumor medicine. Among them, the sophoridinic ketones 5a–b, alkenes 7a–b and sophoridinic amines 14a–b displayed reasonable antiproliferative activity with IC50 values ranging from 3.8 to 5.4 μmol/L. Especially, compounds 5a and 7b exhibited an equipotency in both adriamycin (AMD)-susceptible and resistant MCF-7 breast carcinoma cells, indicating a different mechanism from AMD. The primary mechanism of action of 5a was to arrest the cell cycle at the G0/G1 phase, consistent with that of parent compound 1. Thus, we consider 12-chlorobenzyl sophoridinic derivatives with a tricyclic scaffold to be a new class of promising antitumor agents with an advantage of inhibiting drug-resistant cancer cells.
KEY WORDS: Sophoridinic derivatives, Synthesis, Structure–activity relationship, Anticancer, Drug resistance
Graphical abstract
Taking compound 2 as the lead, structure–activity relationship (SAR) investigation was continuously developed on the variations of 3'-CH2OH at the 11–side chain from sophoridine, a Chinese natural antitumor medicine. Among them, compound 5a displayed a potent antiproliferative activity with an advantage of inhibiting drug-resistant cancer cells.

1. Introduction
Several quinolizidine alkaloids extracted from Chinese herb Kushen have demonstrated potential antitumor activity1, 2, 3, 4. Sophoridine (1, Fig. 1), the main active ingredient of Kushen extracts, was approved by the Chinese FDA to treat malignant neoplasm in 20055, 6. Its mechanism is to inhibit the DNA topoisomerase I (topo I) activity and induce cell cycle arrest at the G0/G1 phase7, 8, 9. From compound 1, as depicted in Fig. 1, we have successfully identified that 12-N-substitued sophoridinol derivatives were a promising class of anticancer agents with a novel chemical scaffold and ideal druggable parameters10, 11. A representative compound, 12-N-p-chlorobenzyl sophoridinol (2, Fig. 1) displayed a reasonable antiproliferative activity with an IC50 of 9.3 μmol/L, much better than that of the parent 1 (>80 μmol/L)10. Furthermore, it has a couple of advantages such as special scaffold, flexibility structure, high solubility and good safety profiles. In addition, it has an excellent potential for further chemical modifications and optimization10, 11.
Figure 1.

Chemical structures of sophoridine (1) 12-N-p-cholobenzyl sophoridinol (2) and the structural fragments in 2 to be modified.
The previous structure–activity relationship (SAR) revealed that 12-N-p-cholobenzyl substituent was beneficial for the antitumor activity10. Thus, taking compound 2 as a lead, SAR investigation was continuously developed on the variations of 3′-CH2OH at the 11–side chain in an effort to discover more potent antitumor candidates. In the present study, as illustrated in Fig. 1, the 12-N-p-cholobenzyl was retained as a required group for activity, and 3′-CH2OH at 11–attachment was replaced with various substituents and different sophoridinic derivatives were produced. Based on this strategy, a series of sophoridinic ketone, alkene, imine and amine analogs were then constructed and measured. Herein, presented in this study were the synthesis, antitumor assessment, SAR analysis and primary mechanism of action of the representative compounds.
2. Results and discussion
2.1. Synthetic routes
The synthesis of all the newly synthesized compounds was respectively described in Scheme 1, Scheme 2. Each target compound was prepared using commercially available 1 with purity over 98% as the starting material, which was purchased from the Yanchi Dushun Biological and Chemical Co., Ltd. (Shanxi, China).
Scheme 1.

Reagents and conditions: (a) HCl, reflux, 6 h; CH3OH, r.t., 5–6 h, in 2 steps; (b) R1CHO, TEA, 1,2-dichloroethane, reflux, 4 h; STB, reflux, 4 h, in 2 steps; (c) R2MgBr (1.0 equiv.), THF, r.t., 7 h; (d) R2MgBr (excessive), THF, reflux, 7 h; (e) hydrochloride/ether, r.t., 4 h.
Scheme 2.

Reagents and conditions: (a) LiAlH4, THF, r.t.; (b) DMSO, (COCl)2, TEA, DCM, –78 °C, 1 h; (c) HCl/MeOH, r.t., 10 min; (d) NH2OH.HCl, 20% NaOH, r.t., 12 h; (e) methyl benzenesulfonate, THF, r.t.; (f) R2NH2, MeOH, reflux, 4 h; (g) NaBH3CN, MeOH, reflux, 4 h.
The synthesis of the desired products 12-N-p-cholobenzyl sophoridinic ketones 5a–b and 12-N-p-cholobenzyl sophoridinic alkenes 7a–b was displayed in Scheme 1. The intermediate 3 was obtained through hydrolysis and esterization of 1 in an overall yield of 95%10, 11. Then the condensation of 3 with 4-chlorobenzaldehyde followed by the reduction with sodium triacetoxyborohydride (STB) afforded the key intermediate 4 in 53% yield12. The nucleophilic-addition reaction of 4 with one molar equivalent of p-Cl-PhMgBr and p-F-PhMgBr in dry THF at room temperature provided products 5a–b with yields of 61%–62%13, while the reaction of 4 with excessive amount of Grignard reagents at reflux temperature generated tertiary alcohols 6a–c in 45%–58% yields, which were dehydrated under acidic conditions to give the target alkenes 7a–b with yields of 32%–40%.
As illustrated in Scheme 2, the reduction of 4 in the presence of LiAlH4 generated sophoridinol 8 in 86% yield, which was converted to sophoridinic aldehyde 9 through a Swern oxidation reaction in 56% yield14. The aldol condensation of 9 in HCl/CH3OH afforded hemiacetal 10 at room temperature with the yield of 67%, while the condensation of 9 with hydroxylamine hydrochloride afforded hydroxyl oxime 11 in a 72% yield, which went through a methyl etherification to give the oxime ether 12 in 57% yield. The sophoridinic imines 13a–c were obtained via the condensation of 9 and three amines, which were then reduced by sodium cyanoborohydride (NaBH3CN) to afford 14a–b in good yields. All desired products were purified by flash chromatography using CH2Cl2/CH3OH as the gradient eluent.
2.2. SAR analysis for antitumor activity
All the target compounds were evaluated for their cytotoxic activity in human HepG2 hepatoma cell line using Taxol as a positive control using the MTT assay15. Structures of the 11-sophoridinic derivatives and their anti-proliferative activity were shown in Table 1. The ClogP values were calculated by ChemBioOffice software (Version 12.0).
Table 1.
SAR of the target compounds for their anti-proliferative activities in HepG2 cells.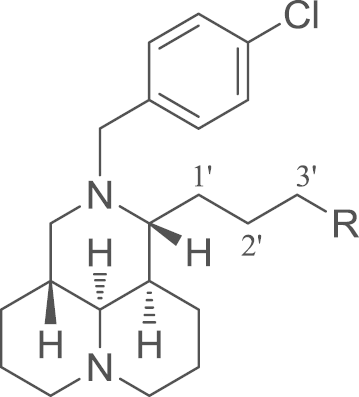
| Compd. | R | IC50 (μmol/L) | ClogP |
|---|---|---|---|
| 2 |  |
9.3±0.4 | 4.5 |
| 5a | 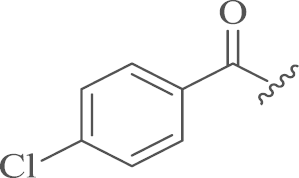 |
4.9±1.2 | 7.1 |
| 5b | 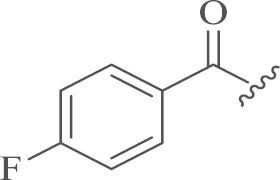 |
4.0±0.9 | 7.0 |
| 6a |  |
16.3±2.8 | 5.9 |
| 7a | 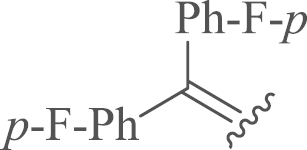 |
4.3±1.3 | 10.0 |
| 7b | 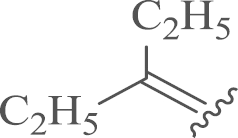 |
3.8±1.0 | 8.0 |
| 10 | 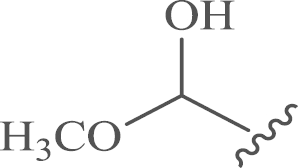 |
17.3±2.1 | 4.5 |
| 11 |  |
11.2±1.2 | 4.6 |
| 12 | 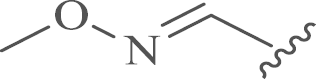 |
>50 | 4.8 |
| 13a | 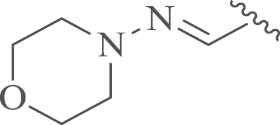 |
11.8±0.7 | 4.9 |
| 14a | 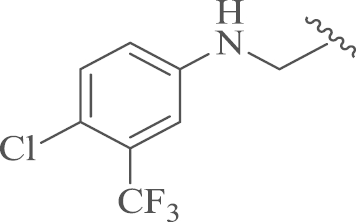 |
4.3±0.1 | 7.6 |
| 14b | 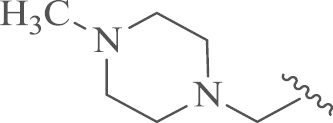 |
5.4±0.2 | 4.5 |
| Taxol | 15.6±2.3 | 4.7 | |
Taking compound 2 as a lead, the SAR investigation was mainly focused on the variation of 3'-CH2OH at the 11–side chain with 12-N-p-cholobenzyl as a required group for antitumor activity. Firstly, in replacing the CH2OH group with ketones, the resultant compounds 5a–b with higher lipophilicity (ClogP>7.0) displayed improved cytotoxic activity with the IC50 values of 4.9 μmol/L and 4.0 μmol/L, respectively. Introduction of two methyl groups on the α-carbon adjacent to the hydroxy group led to a tertiary butyric alcohol 6a, which showed a decreased activity with an IC50 value of 16.3 μmol/L. The replacement of 3′-CH2OH with an alkene group on the 3'-position created more lipophilic 7a–b, which showed significant improvement on the antiproliferative activity with the IC50 values of 4.3 and 3.8 μmol/L, respectively, much better than that of 2 and Taxol. The results suggested that a higher ClogP values might be beneficial for good potency against cancer.
Secondly, the generated hemiacetal 10, hydroxyl oxime 11, oxime ether 12 and imines 13a–c abolished cytotoxic activity partially or completely with IC50 between 11.8 and >50 μmol/L. The constructed aromatic amine 14a and aliphatic amine 14b showed a significantly improved activity with IC50 values of 4.3 and 5.4 μmol/L, respectively. It seemed that the introduction of a suitable methyleneamine group on the 3′-position might greatly enhance the activity against cancer.
Considering their potent anticancer effects as well as reasonable ClogP values, compounds 5a and 7b were chosen as the representative compounds for further investigation.
2.3. Anti-resistant tumor effect of 5a and 7b
Evaluation of compounds 5a and 7b against drug-resistant cancer cell lines were then carried out. We measured their activity against human wild MCF-7 and adriamycin (AMD)-resistant MCF-7 (MCF-7/AMD) breast carcinoma cells using AMD as a reference control16. As depicted in Fig. 2, AMD was active against wild type MCF-7, and completely inactive in the MCF-7/AMD cells. Meanwhile, compounds 5a and 7b were equipotent or almost equipotent in both MCF-7 cell lines, suggesting a different mode of action from AMD. As compound 5a exhibited an equivalent antiproliferative effect against both MCF-7 cell lines, thus was selected for the next study.
Figure 2.

IC50 (μmol/L) value comparison of 5a and 7b in MCF-7 and MCF-7/AMD breast carcinoma cell lines.
2.4. Mechanism of action of 5a
To verify the possible change of mechanism of action after the structure modifications, flow cytometric analysis in the HepG2 cells was conducted. The HepG2 cells were treated for 24 h without or with 5a at concentrations of 1.25, 2.5 and 5.0 μg/mL, respectively. As shown in Fig. 3, compound 5a arrested the HepG2 cells at the G0/G1 phase as anticipated, indicating a similar mechanism of action to that of its parent compound 111.
Figure 3.

Blockage of cell cycle in HepG2 cells treated without or with 5a at different concentrations for 24 h.
3. Conclusions
A variety of novel sophoridinic derivatives, such as sophoridinic ketones, alkenes, imines and amines, were synthesized and evaluated for their antitumor activity. SAR analysis indicated that the introduction of a suitable methyl amine group on the 3'-position could greatly improve the potency. Compounds 5a and 7b exhibited an equipotent effect in both AMD-susceptible and AMD-resistant breast carcinoma cells, indicating a different mechanism from AMD. The mechanism of action of 5a was to arrest the cell cycle at the G0/G1 phase, consistent with that of 1. The SAR results provided powerful information for further modifications and optimization of this novel scaffold to identify anticancer candidates that might be active in drug-resistant cancer cells.
4. Experimental section
4.1. Chemistry
Melting point (m.p.) was obtained with a CXM-300 melting point apparatus and uncorrected. The 1H NMR spectra were performed on a Varian Inova 400 MHz spectrometer (Varian, San Francisco, CA, USA) and 13C NMR on a Bruker Avance III 400 spectrometer in CD3OD or (CD3)2SO, using Me4Si as a internal standard. ESI high-resolution mass spectra (HR-MS) were recorded on an Autospec UItima-TOF mass spectrometer (Micromass UK Ltd., Manchester, UK). Flash chromatography was performed on a CombiflashRf 200 machine (Teledyne, Nebraska, USA) using silica gel having particle size of 0.038 mm.
4.1.1. General procedures for the synthesis of compound 5
To a solution of 4 (0.50 g, 1.2 mmol) in anhydrous THF (20 mL), 1 mol/L 4-chlorophenylmagnesium bromide or 4-fluorophenylmagnesium bromide (1.2 mL) was slowly added at 0 °C and then stirred at room temperature until TLC analysis showed the completion of the reaction. Then the reaction was quenched by the addition of saturated ammonium chloride solution (5 mL). After the solvent was removed by condensation, dichloromethane (30 mL) was added, and the resultant mixture was washed by water (20 mL) and brine (20 mL), dried over anhydrous sodium sulfate, concentrated, and the residue was purified by flash column chromatography on silica gel with CH2Cl2/CH3OH as the eluents to afford target compounds.
4.1.1.1. 12-N-p-Chlorobenzyl sophoridinic-4′,4-chlorophenyl ketone (5a)
The title compound was prepared from 4 and 4-chlorophenylmagnesium bromide using the same manner as described above. Yield: 62%; white solid; m.p. 83–84 °C; 1H NMR (500 MHz, DMSO-d6) δ 7.97 (d, J=8.1 Hz, 2H), 7.60 (d, J=8.5 Hz, 2H), 7.38–7.29 (m, 4H), 3.61 (d, J=14.1 Hz, 1H), 3.54–3.42 (m, 2H), 3.41–3.32 (m, 4H), 3.20–3.08 (m, 1H), 3.06–2.93 (m, 2H), 2.65–2.57 (m, 1H), 2.40–2.25 (m, 2H), 2.25–2.14 (m, 2H), 2.13–2.03 (m, 1H), 1.94–1.74 (m, 3H), 1.73–1.53 (m, 3H), 1.52–1.35 (m, 3H), 1.28–1.14 (m, 1H); 13C NMR (126 MHz, DMSO-d6) δ 198.8, 138.6, 137.9, 135.1, 131.1, 129.8 (2), 129.75 (2), 128.7 (2), 128.1 (2), 62.2, 58.5, 56.8, 51.6, 49.9, 44.1, 38.0, 35.0, 27.3, 26.0, 22.4, 22.2, 21.6, 21.2, 17.6; HR-MS: Calcd. for C28H34ON2Cl2 [M+H]+, 485.2121, Found, 485.2123.
4.1.1.2. 12-N-p-Chlorobenzyl sophoridinic-4′,4-fluorophenyl ketone (5b)
The title compound was prepared from 4 and 4-fluorophenyl magnesium bromide using the same manner as described above. Yield: 61%, m.p.: 87–89 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.98 (d, J=8.5 Hz, 2H), 7.60 (d, J=8.5 Hz, 2H), 7.43–7.20 (m, 4H), 3.74–3.43 (m, 2H), 3.41–3.32 (m, 4H), 3.20–2.87 (m, 5 H), 2.73–2.55 (m, 1H), 2.41–2.26 (m, 2H), 2.27–2.09 (m, 2H), 1.99–1.74 (m, 3 H), 1.74–1.55 (m, 3H), 1.55–1.33 (m, 3H), 1.27–1.11 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 199.3, 139.2, 138.4, 135.7, 131.7, 130.3 (2), 130.3 (2), 129.3 (2), 128.6 (2), 62.8, 59.0, 57.3, 52.2, 50.5, 44.7, 38.5, 35.5, 27.9, 26.5, 23.0, 22.7, 22.2, 21.7, 18.1; HR-MS: Calcd. for C28H34ON2FCl [M+H]+, 469.2416, Found, 469.2416.
4.1.2. General procedures for the synthesis of compounds 6 and 7
4.1.2.1. -4′,4′ dimethyl sophoridinol (6)
To a solution of 4 (0.50 g, 1.2 mmol, 1.0 equiv.) in anhydrous THF (20 mL), solution of methylmagnesium bromide (4.0 equiv.) in THF was slowly added at 0 °C and then stirred at room temperature until TLC analysis showed completion of the reaction. Then the reaction was quenched by the addition of saturated ammonium chloride solution (5 mL). After the solvent was removed by condensation, dichloromethane (30 mL) was added, and the resultant mixture was washed by water (20 mL) and brine (20 mL), dried over anhydrous sodium sulfate, concentrated, and the residue was purified by flash column chromatography on silica gel with CH2Cl2/CH3OH as the eluents to afford target compound 6. Yield: 45%, m.p.: 91–93 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.28–7.76 (m, 2 H), 7.66–7.42 (m, 2 H), 4.28–4.05 (m, 1 H), 3.89–3.61 (m, 1 H), 3.37–3.02 (m, 5 H), 3.00–2.74 (m, 3 H), 2.76–2.54 (m, 2 H), 2.06–1.79 (m, 5 H), 1.74–1.58 (m, 5 H), 1.60–1.27 (m, 4 H), 1.00–0.76 (m, 6 H); 13C NMR (101 MHz, DMSO-d6) δ 134.7, 133.0, 132.9 (2), 129.3 (2), 80.6, 60.9, 55.6, 52.0, 51.9, 44.1, 39.4, 34.0, 33.4, 26.0, 25.2, 22.7, 21.7, 21.1, 17.6, 13.0, 9.0 (2); HR-MS: Calcd. for C24H37ON2Cl [M+H]+, 405.2667, Found, 405.2677.
4.1.2.2. 12-N-p-Chlorobenzyl-4′,4′-di-4-fluorophenyl sophoridinene (7a)
Compound 7a was obtained from 4-fluorophenylmagnesium bromide and 4 stirring at refluxing temperature following the similar procedure with 6 and acidification by HCl/diethyl ether as a white solid. Yield: 32%, m.p.: 64–66 °C; 1H NMR (600 MHz, CD3OD) δ 7.67–7.57 (m, 2H), 7.55–7.48 (m, 1H), 7.45–7.25 (m, 3H), 7.20–7.03 (m, 6H), 4.59–4.39 (m, 1H), 4.41–4.16 (m, 1H), 3.97–3.79 (m, 1H), 3.66–3.49 (m, 1H), 3.49–3.33 (m, 3H), 3.21–3.02 (m, 2H), 3.00–2.77 (m, 2H), 2.35–2.23 (m, 2H), 2.14–1.92 (m, 5H), 1.92–1.80 (m, 2H), 1.79–1.56 (m, 1H), 1.53–1.36 (m, 2H), 1.39–1.21 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 160.2 (2), 134.5, 129.3 (2), 128.7(2), 128.6 (4), 126.7 (2), 125.5, 115.4, 115.1, 114.9 (4), 54.7, 53.0, 51.5, 50.8, 48.6, 44.1, 34.5, 26.5, 26.0, 24.9, 21.7, 20.8, 17.4, 15.1; HR-MS: Calcd. for C34H37N2F2Cl [M+H]+, 547.2686, Found, 547.2686.
4.1.2.3. -4′,4′-diethyl sophoridinene bihydrochloride (7b)
Compound 7a was obtained from ethylmagnesium bromide and 4 stirring at refluxing temperature following the similar procedures with 6 and acidification by HCl/diethyl ether as a white solid. Yield: 40%, m.p.: 77–79 °C; 1H NMR (500 MHz, DMSO-d6) δ 11.67 (s, 1H), 11.20 (s, 1H), 8.29–7.79 (m, 2H), 7.66–7.19 (m, 2H), 4.62–4.47 (m, 4H), 4.46–4.33 (m, 1H), 4.31–4.09 (m, 1H), 3.96–3.67 (m, 1H), 3.35–3.23 (m, 2H), 3.23–3.02 (m, 3H), 3.01–2.79 (m, 1H), 2.77–2.54 (m, 1H), 2.51–2.43 (m, 1H), 2.02–1.77 (m, 5H), 1.78–1.61 (m, 5H), 1.61–1.29 (m, 4H), 0.94–0.84 (m, 4H), 0.84–0.67 (m, 2H); 13C NMR (126 MHz, DMSO-d6) δ 143.6, 140.4 (2), 134.1 (2), 132.4, 128.8, 117.3, 80.1, 60.3, 55.0, 54.3, 51.5, 44.0 (2), 38.8, 32.4, 25.4, 24.6, 22.1, 21.1, 17.0, 12.9, 12.4, 8.5 (2); HR-MS: Calcd. for C26H39N2Cl·2HCl [M–2HCl+H]+, 415.2874, Found, 415.2871.
4.1.3. 12-N-p-Chlorobenzyl sophoridinic hemiacetal (10)
A dry, 250-mL, three-necked, round-bottomed flask equipped with a magnetic stirrer and a dropping funnel was charged with 100 mL of dry dichloromethane under an atmosphere of nitrogen. After cooling to −78 °C, oxalyl chloride (1.0 mL, 12 mmol ) and dry dimethyl sulfoxide (1.5 mL, 20 mmol) were added dropwise to the stirred solution. After 5 min, 8 (3.7 g, 10 mmol) in 20 mL of dichloromethane was added with stirring. The mixture was stirred for an additional 0.5 h at −78 °C, and freshly distilled triethylamine (5.6 mL, 40 mmol) was added. The mixture was then allowed to warm to room temperature over 0.5 h, whereupon 50 mL of water is added. The phases are separated, and the aqueous phase is extracted three times with 50 mL of diethyl ether or dichloromethane. The combined organic phases were washed successively with water (50 mL) and brine (50 mL). The organic layer was dried over anhydrous magnesium sulfate, and the solvent was removed under reduced pressure, and the residue was purified by flash column chromatography on silica gel with CH2Cl2/CH3OH as the eluents to afford compound 9 as a yellow viscous solid, to which 1 mol/L hydrochloric acid-methanol solution (10 mL) was added and stirred at room temperature for 10 min, removal of the solvent generated target compound 10 as a white solid. Yield: 67%, m.p.: 64–66 °C; 1H NMR (500 MHz, CD3OD) δ 7.38–7.28 (m, 4 H), 4.91 (s, 3H), 3.68–3.46 (m, 2H), 3.20–3.05 (m, 1H), 3.00–2.74 (m, 2H), 2.65–2.46 (m, 1H), 2.39–2.28 (m, 1H), 2.25–1.97 (m, 4H), 1.94–1.69 (m, 4H), 1.72–1.48 (m, 6H), 1.47–0.94 (m, 6H); 13C NMR (126 MHz, CD3OD) δ 140.2, 133.4, 131.0 (2), 129.2 (2), 99.6, 65.0, 64.9, 60.2, 58.9, 55.1, 52.6, 46.2, 39.1, 30.2, 28.4, 26.4, 26.0, 23.8, 23.7, 23.5, 20.0; HR-MS: Calcd. for C23H35O2N2Cl [M+H]+, 407.2459, Found, 407.2459.
4.1.4. 12-N-p-lorobenzyl sophoridinic hydroxyl oxime bihydrochloride (11)
To a mixture of hydroxylamine hydrochloride (0.25 g, 3.6 mmol) in 20% sodium hydroxide solution (70 mL), 9 (1.1 g, 3.0 mmol) was added and the mixture was stirred at room temperature until TLC analysis showed the completion of the reaction. The insoluble solid was filtered off and the solvent was removed by condensation, and the residue was purified by flash column chromatography on silica gel with CH2Cl2/CH3OH as the eluents to obtain the free form of 11 as colorless oil, which was then acidified by 3 mol/L HCl/ether (10 mL) to afford 11 as a white solid. Yield: 72%, m.p.: 91–92 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.81 (s, 1H), 10.44 (s, 1H), 7.53–7.14 (m, 4H), 3.64–3.48 (m, 2H), 3.42–3.33 (m, 3H), 3.22–3.05 (m, 2H), 3.01–2.89 (m, 1H), 2.66–2.57 (m, 1H), 2.41–1.97 (m, 7H), 1.93–1.71 (m, 3H), 1.68–1.33 (m, 6H), 1.32–1.14 (m, 2H); 13C NMR (126 MHz, DMSO-d6) δ 149.7, 139.3, 131.7, 130.3 (2), 128.6 (2), 63.2, 58.9, 57.3, 50.3, 44.6, 35.5, 29.6, 27.9, 26.5, 25.1, 24.4, 23.0, 22.8, 22.3, 18.1; HR-MS: Calcd. for C22H32ON3Cl·2HCl [M–2HCl+H]+, 390.2306, Found, 390.2305.
4.1.5. 12-N-p-Chlorobenzyl sophoridinic methyl ether (12)
To a solution of free 11 (1.95 g, 5.0 mmol) in anhydrous THF (50 mL) under nitrogen, methyl benzenesulfonate (2.64 g, 20.0 mmol) was added. The mixture was stirred at room temperature until TLC analysis showed the completion of the reaction. The reaction was quenched by methanol (5 mL), and condensed, the residue was purified by flash column chromatography on silica gel with CH2Cl2/CH3OH as the eluents to obtain the free form of 12 as a yellow solid. Yield: 57%, m.p.: 83–85 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.49–7.14 (m, 5H), 3.52 (s, 3H), 3.42–3.23 (m, 3H), 3.23–3.02 (m, 2H), 3.03–2.91 (m, 1H), 2.76–2.56 (m, 1H), 2.43–1.89 (m, 7H), 1.91–1.65 (m, 4H), 1.71–1.36 (m, 6H), 1.34–0.99 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 150.6, 149.7, 139.3, 131.7, 130.3 (2), 128.6 (2), 63.2, 59.0, 57.3, 52.2, 50.3, 44.6, 35.6, 29.6, 27.9, 26.5, 24.4, 23.0, 22.8, 22.3, 18.1; HR-MS: Calcd. for C23H34ON3Cl [M+H]+, 404.2463, Found, 404.2482.
4.1.6. 12-N-p-Chlorobenzyl-4′-N-morpholin-sophoridinic imine (13)
To a mixture of 9 (1.1 g, 3.0 mmol) in anhydrous methanol (50 mL), 4-amine-morpholin (0.43 mL, 4.5 mmol) was added and the mixture was refluxed for 4 h before cooled to room temperature. The solvent was condensed and dichloromethane (50 mL) was added, and the mixture was washed successively with water (30 mL) and saturated ammonium chloride solution (30 mL). The mixture was evaporated and the residue was purified by flash column chromatography on silica gel with CH2Cl2/CH3OH as the eluents to obtain the target compound 13 as a white solid. Yield: 74%, m.p.: 59–61 °C; 1H NMR (500 MHz, DMSO-d6) δ 7.38–7.30 (m, 4H), 6.93 (t, J=5.2 Hz, 1H), 3.73–3.65 (m, 4H), 3.61–3.44 (m, 2H), 3.01–2.89 (m, 1H), 2.88–2.75 (m, 6H), 2.71–2.60 (m, 1H), 2.50–2.35 (m, 2H), 2.25–1.87 (m, 6H), 1.82–1.62 (m, 3H), 1.58–1.37 (m, 5H), 1.31–1.21 (m, 2H), 1.17–1.10 (m, 1H), 1.01 (dd, J=20.5, 11.7 Hz, 1H); 13C NMR (126 MHz, DMSO-d6) δ 140.4, 139.7, 131.4, 130.2 (2), 128.5 (2), 66.1 (2), 63.9, 58.5, 57.7, 54.4, 52.5 (2), 52.4, 51.6, 45.4, 38.0, 32.9, 29.8, 26.8, 26.1, 25.5, 22.6, 19.2; HR-MS: Calcd. for C26H39ON4Cl [M+H]+, 459.2885, Found, 459.2885.
4.1.7. General procedures for the synthesis of 14
4.1.7.1. 12-N-p-Chlorobenzyl-4′-(4-chloro-3-trifluoromethylphenyl)sophoridinic amine trihydrochloride (14a)
To a mixture of 9 (1.1 g, 3.0 mmol) in anhydrous methanol (50 mL), 4-chloro-3-trifluoromethylphenylamine (0.8 g, 4.5 mmol) was added and the mixture was refluxed for 4 h, then sodium cyanoborohydride (0.9 g, 4.5 mmol) was added portion wise, and the mixture was refluxed for another 4 h. The mixture was cooled to room temperature and evaporated, then dichloromethane (50 mL) was added, and the mixture was washed successively with water (30 mL) and saturated ammonium chloride solution (30 mL). The mixture was evaporated and the residue was purified by flash column chromatography on silica gel with CH2Cl2/CH3OH as the eluents, then acidified by 3 mol/L HCl/ether (10 mL) and filtrated to obtain the target compound 14a as a white solid. Yield: 68%, m.p.: 71–73 °C; 1H NMR (500 MHz, DMSO-d6) δ 11.55 (s, 1H), 11.12 (s, 1H), 10.93 (s, 1H), 8.12–7.77 (m, 2H), 7.01 (d, J=8.7 Hz, 3H), 6.91–6.74 (m, 2H), 4.52–4.34 (m, 1H), 4.34–4.12 (m, 1H), 3.88–3.69 (m, 1H), 3.65–3.25 (m, 4H), 3.24–2.76 (m, 3H), 2.76–2.55 (m, 3H), 2.43–1.98 (m, 2H), 1.99–1.29 (m, 8H), 1.29–1.14 (m, 2H), 1.03–0.79 (m, 1H); 13C NMR (126 MHz, DMSO-d6) δ 153.9, 146.3, 139.7, 131.5, 130.4, 130.2, 128.9 (2), 128.5 (2), 90.3, 64.1, 60.6, 58.6, 57.7, 56.0, 54.2, 51.5, 45.4, 43.6, 37.8, 30.2, 29.5, 26.9, 25.5, 25.4, 23.1, 23.1, 19.1; HR-MS: Calcd. for C29H36N3F3Cl2·3HCl [M–3HCl+H]+, 554.2311, Found, 554.2313.
4.1.7.2. 12-N-p-Chlorobenzyl-4′-(N-methypiperazine)sophoridinic amine tetrahydrochloride (14b)
Compound 14b was obtained from N-methypiperazine and 9 following the similar procedure with 14a. Yield: 65%, m.p.: 73–75 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.25 (s, 1H), 12.15 (s, 1H), 11.61 (s, 1H), 11.16 (s, 1H), 7.99 (d, J=7.9 Hz, 2H), 7.57 (d, J=7.9 Hz, 2H), 5.07 (s, 3H), 4.50–4.16 (m, 1H), 3.92–3.68 (m, 3H), 3.66–3.35 (m, 5H), 3.34–2.75 (m, 11H), 2.73–2.53 (m, 2H), 2.21–1.58 (m, 9H), 1.60–1.23 (m, 3H), 1.16–0.99 (m, 1H), 1.01–0.80 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 134.7, 133.1, 129.5 (2), 129.2 (2), 65.3, 60.9, 55.5, 55.0, 52.1 (2), 51.9 (2), 49.9, 48.5, 44.6, 42.5, 26.1, 25.1, 23.0, 22.8, 22.3, 21.9, 21.7, 17.6, 15.6; HR-MS: Calcd. for C27H43N4Cl·4HCl [M–4HCl+H]+, 459.3249, Found, 459.3249.
4.2. Biological evaluation
4.2.1. MTT assay
The human tumor cell lines, HepG2 and MCF-7, were obtained from American Type Culture Collection. Cells were routinely cultured in the MEM-EBSS medium (Hyclone, UT) with 10% FBS (Gibco, USA) and 1% penicillinestreptomycin and incubated at 37 °C with 5% CO2. Cells were counted and plated at a density of 4000 cells per well in 96-well plates. After 24 h, cells were treated with different compounds at different concentrations for 48 h. The effect on cell growth inhibition was determined by the MTT assay as previous described. The growth inhibition rate (%) was calculated at each concentration and IC50 value was calculated with Sigmaplot software. Results were obtained from triplicate determinations and shown as mean±SD.
4.2.2. Cell cycle distribution assay
HepG2 cells were treated with 5a at different concentrations for 24 h and then cells were harvested. Cells were fixed with 70% ethanol and stored at –20 °C overnight. The fixed cells were incubated with 200 μg/mL RNase at 37 °C for 30 min and then stained with 50 μg/mL propidium iodide in the dark for 30 min. Cell cycle distribution was then analyzed by flow cytometry using FACS analysis (BD FACSCalibur, USA).
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81473248) and Tianjin Medical University General Hospital Funding (ZYYFY2015030).
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
References
- 1.Kyogoku K., Hatayama K., Komatsu M. Constituents of Chinese crude drug "Kushen" (the root of Sophora flavescens Ait.). Isolation of five new flavonoids and formononetin. Chem Pharm Bull. 1973;21:2733–2738. doi: 10.1248/cpb.21.2733. [DOI] [PubMed] [Google Scholar]
- 2.Liang L., Wang X.Y., Zhang X.H., Ji B., Yan H.C., Deng H.Z. Sophoridine exerts an anti-colorectal carcinoma effect through apoptosis induction in vitro and in vivo. Life Sci. 2012;91:1295–1303. doi: 10.1016/j.lfs.2012.09.021. [DOI] [PubMed] [Google Scholar]
- 3.Liu B., Shi R.B. Constituents in the alkaloid fraction of Kushen decoction. China J Chin Mater Med. 2006;31:557–560. [PubMed] [Google Scholar]
- 4.Zhang H., Gu J. Progress of experimental study on treatment of psoriasis by Chinese medicinal monomer and single or compound recipe in Chinese materia medica. Chin J Integr Med. 2007;13:312–316. doi: 10.1007/s11655-007-0312-5. [DOI] [PubMed] [Google Scholar]
- 5.Yan J.G., Yang Y.Q., Wang Y.J., Kan J. Study on effect of sophoridine against bone cancer pain and its mechanism. China J Chin Mater Med. 2013;38:4134–4137. [PubMed] [Google Scholar]
- 6.Yong J.P., Wu X.Y., Lu C.Z. Anticancer advances of matrine and its derivatives. Curr Pharm Des. 2015;21:3673–3680. doi: 10.2174/1381612821666150122123748. [DOI] [PubMed] [Google Scholar]
- 7.Wang W.X., Sun Z.H., Chen H.M., Xu B.N., Wang F.Y. Role and mechanism of sophoridine on proliferation inhibition in human glioma U87MG cell line. Int J Clin Exp Med. 2015;8:464–471. [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Q.R., Li C.H., Fu X.Q., Liu Y.W., Tang J., Fan Q. Effects of sophoridine on the growth and expressions of p53 and vascular endothelial growth factor of transplanted solid tumor SW480 in nude mice. J South Med Univ. 2010;30:1593–1596. [PubMed] [Google Scholar]
- 9.Li X.M., Wu Y.G., Chen S.L., Pan D.X., Wu J.N., Yu Y.H. Antitumor action of sophoridine. Acta Pharmacol Sin. 1987;8:153–158. [PubMed] [Google Scholar]
- 10.Bi C.W., Zhang C.X., Li Y.H., Tang S., Deng H.B., Zhao W.L. Novel N-substituted sophoridinol derivatives as anticancer agents. Eur J Med Chem. 2014;81:95–105. doi: 10.1016/j.ejmech.2014.04.069. [DOI] [PubMed] [Google Scholar]
- 11.Bi C.W., Zhang C.X., Li Y.H., Tang S., Wang S.G., Shao R.G. Synthesis and biological evaluation of sophoridinol derivatives as a novel family of potential anticancer agents. ACS Med Chem Lett. 2014;5:1225–1229. doi: 10.1021/ml500289h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chun B.K., Du J.F., Zhang H.R., Chang W., Ross B.S., Jiang Y. Synthesis of stable isotope labeled analogs of the anti-hepatitis C virus nucleotide prodrugs PSI-7977 and PSI-352938. Nucleosides Nucleotides Nucleic Acids. 2011;30:886–896. doi: 10.1080/15257770.2011.614308. [DOI] [PubMed] [Google Scholar]
- 13.Bhansali P., Hanigan C.L., Perera L., Casero R.A., Jr., Tillekeratne L.M.V. Synthesis and biological evaluation of largazole analogues with modified surface recognition cap groups. Eur J Med Chem. 2014;86:528–541. doi: 10.1016/j.ejmech.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dietz J., Martin S.F. Novel entry to the tricyclic core of stemofoline and didehydrostemofoline. Tetrahedron Lett. 2011;52:2048–2050. doi: 10.1016/j.tetlet.2010.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang H., Xin M.H., Xie X.X., Mao S., Zuo S.J., Lu S.M. Synthesis and antitumor activity evaluation of PI3K inhibitors containing 3-substituted quinazolin-4(3H)-one moiety. Bioorg Med Chem. 2015;23:7765–7776. doi: 10.1016/j.bmc.2015.11.027. [DOI] [PubMed] [Google Scholar]
- 16.Kumar R., Kaur M., Bahia M.S., Silakari O. Synthesis, cytotoxic study and docking based multidrug resistance modulator potential analysis of 2-(9-oxoacridin-10(9H)-yl)-N-phenyl acetamides. Eur J Med Chem. 2014;80:83–91. doi: 10.1016/j.ejmech.2014.04.030. [DOI] [PubMed] [Google Scholar]