


Abstract
Maintenance of proper chromatin states and genomic stability is vital for normal development and health across a range of organisms. Here, we report on the role of KLLN in maintenance of pericentric H3K9 trimethylation (H3K9me3) and genomic stability. Germline hypermethylation of KLLN, a gene uncovered well after the human genome project, has been linked to Cowden cancer-predisposition syndrome (CS) in PTEN wild-type cases. KLLN first identified as a p53-dependent tumor suppressor gene, was believed to bind randomly to DNA and cause S-phase arrest. Using chromatin immunoprecipitation-based sequencing (ChIP-seq), we demonstrated that KLLN binds to DNA regions enriched with H3K9me3. KLLN overexpression correlated with increased H3K9 methyltransferase activity and increased global H3K9me3, while knockdown of KLLN had an opposite effect. We also found KLLN to localize to pericentric regions, with loss of KLLN resulting in dysregulation of pericentric heterochromatin, with consequent chromosomal instability manifested by increased micronuclei formation and numerical chromosomal aberrations. Interestingly, we show that KLLN interacts with DBC1, with consequent abrogation of DBC1 inhibition of SUV39H1, a H3K9 methyltransferase, suggesting the mode of KLLN regulating H3K9me3. These results suggest a critical role for KLLN as a potential regulator of pericentric heterochromatin formation, genomic stability and gene expression.
INTRODUCTION
Perturbations of chromatin organization resulting in genomic instability are a major driving force for inappropriate development and carcinogenesis. Tumor suppressor genes are known to play a major role in the maintenance of epigenetic marks involved in chromatin organization. Germline mutations in one such tumor suppressor gene, PTEN, is responsible for approximately 25% of classically presenting Cowden syndrome (CS), an autosomal dominant cancer predisposition disorder (1) and paradigm of heritable neoplasia. Individuals with CS are at increased risks for malignancies including those of the breast, thyroid, endometrium and kidney (2). Germline hypermethylation of the KLLN promoter has been observed in up to 35% of PTEN mutation negative CS cases (3) and is associated with three-fold increased prevalence of breast cancer and two-fold increased prevalence for renal cell carcinoma compared to PTEN-mutation positive CS (4,5). In the somatic setting, decreased KLLN expression in breast carcinomas compared to adjacent normal tissue is associated with increasing tumor grade and metastases (6). Additionally, 21% of breast carcinomas in The Cancer Genome Atlas (TCGA) project were found to have somatic KLLN deletions (5). These results suggest KLLN mutations and epimutations have roles in both cancer susceptibility and sporadic carcinogenesis.
First reported in 2008 as a tumor suppressor gene, KLLN is both necessary and sufficient for p53-mediated apoptosis in colon cancer cell lines (7). KLLN gene localizes to 10q23 and shares a bidirectional promoter and transcription start site with PTEN (4,7). There are known p53-binding sites on the promoters of both these genes and both are regulated by p53 (7,8). Overexpression of KLLN in breast and prostate cancer cell lines leads to cell death while knockdown of KLLN leads to increased cellular proliferation, clonogenic formation and migration (6,7,9). Therefore, altering KLLN function results in fundamental changes in cell growth indicative of KLLN's role as a tumor suppressor.
KLLN was believed to randomly bind DNA using a distinct DNA binding domain (amino acids 8–50) (7) and was believed to be key for eliciting S and G2 phase checkpoint control in response to genotoxic stress and stalled replication forks (5,7). Naturally occurring germline KLLN variants lead to G2 checkpoint dysfunction (5). Yet, we have been unable to pinpoint G2/S-relevant specific signaling pathways affected by KLLN disruption. KLLN potentially also functions as a transcription factor since it binds the promoters of genes such as TP53, TP73, CHK1 and androgen receptor (AR) gene; and in vitro, KLLN overexpression leads to altered expression of these genes (6,9). Given these seemingly disparate observations, we utilized ChIP-seq to identify global targets of KLLN binding and a combined cytogenetics cum functional approach to determine consequences of KLLN's DNA binding in leading dysregulation of pericentric heterochromatin and genomic instability.
MATERIALS AND METHODS
Cell culture
MCF7 breast cancer cells (PTEN, KLLN and TP53 wild-type) were cultured in DMEM media supplemented with 10% FBS (Life Technologies, Grand Island, NY, USA). ZR-75-30 breast cancer cells (PTEN, KLLN and TP53 wild-type) were cultured in RPMI-1640 media supplemented with 10% FBS (Life Technologies). MCF10A breast epithelial cells were cultured in MEBM media (Lonza, Walkersville, MD, USA) supplemented with components of the MGEM bulletkit (Lonza) and cholera toxin (100 ng/ml) [Sigma Aldrich, St. Louis, MO, USA]. Lymphoblastoid cell lines (LCL or LBL) (reposited at the Genomic Medicine Biorepository, Lerner Research Institute) were cultured in RPMI-1640 media supplemented with 10% FBS. Cell lines were cultured at 37°C and 5% CO2 and passaged using Trypsin-EDTA. All cell lines were purchased from ATCC (Manassas, VA, USA) after 2010 and authenticity was documented by standard STRS analysis per ATCC routine. All cell lines were used during passage 3–15 and routinely tested for mycoplasma.
Overexpression of KLLN by plasmid transfection and siRNA-mediated silencing of KLLN expression
For transfection of either plasmid or siRNA, cells were seeded at 40–50% in appropriate dishes and allowed to attach overnight. For overexpression of KLLN, cells were transfected with 3x FLAG-tagged KLLN in a pCMV vector (Life Technologies) using lipofectamine LTX (Life Technologies) according to the manufacturers protocol. An empty pCMV vector was used as a control. For KLLN knockdown, cells were transfected with KLLN siRNA smartpool using DharmaFECT 1 or Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's instructions. A scrambled siRNA pool (Thermo Fisher Scientific) was used as a control. Cells were collected for analysis 48 h after transfection. QRT-PCR and western blotting was used to confirm overexpression or knockdown of KLLN expression.
RNA collection, reverse transcription and quantitative PCR
RNA was collected using the RNA-easy kit (Qiagen, Valencia, CA, USA) and DNase treatment was done with a subsequent TURBO DNase (Life Technologies) step. Reverse transcription was done using Superscript III (Life Technologies) following the manufacturer's protocol. cDNA was quantified using SYBR Green PCR Master Mix (Life Technologies) on the 7500 Real Time PCR System (Applied Biosystems) using primers specific for KLLN and GAPDH. Data were analyzed using the standard 2−ΔΔCT method.
Western blotting
Harvested cells were lysed using Mammalian Protein Extraction Reagent (Pierce, Rockford, IL, USA) supplemented with protease and phosphatase inhibitors (Sigma Aldrich, St. Louis, MO, USA). Protein concentrations were quantified using the BCA Protein Assay (Pierce) according to manufacturers instructions. Around 20–40 µg of protein mixed with a 6x loading dye containing β-mercaptoethanol was boiled for 10 min before it was run on a 4–15% gradient gel. Protein was transferred to a nitrocellulose membrane using the Trans-Blot Turbo (Bio Rad, Hercules, CA, USA) and membranes blocked for 30 min – 1 h in 3% BSA. Membranes were probed overnight with anti-FLAG M2 at 1:2000 dilution (Sigma-Aldrich). Anti-ACTIN antibody at 1:10 000 dilution (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used as a loading control.
To assess global H3K9 trimethylation by Western blotting, nuclear proteins were collected from cells transfected with KLLN plasmid or siRNA using the NE-PER Nuclear and Cytoplasmic Extraction kit (Pierce). Protein concentration was measured using the BCA Protein assay and 15–25 µg of protein was used for immunoblotting, done as described above. Membranes were probed overnight with anti-H3K9me3 antibody (1:1000) (Abcam Inc., Cambridge, MA, USA, cat# ab8898). Anti-PARP-1 antibody (1:1000) (Santa Cruz Biotechnology, cat# sc-8007) was used as a nuclear loading control.
Chromatin immunoprecipitation (ChIP)
The EZ-ChIP Chromatin Immunoprecipitation Kit (Millipore, Billerica, MA, USA) protocol was followed. Chromatin was sheared either with a Sonic Dismembrator Model 100 (Thermo Fisher Scientific) or the S220 Focused-ultrasonicator (Covaris, Woburn, MA, USA). Immunoprecipitation of cross-linked protein–DNA was carried out with the following antibodies: ANTI-FLAG M2 affinity gel (Sigma Aldrich, cat# A2220), normal mouse IgG (Millipore), normal rabbit IgG (Santa Cruz, Dallas, TX, USA) and anti-Histone H3 (tri methyl K9) (Abcam, Cambridge, MA, USA). Complexes were eluted, DNA-protein cross-links reversed and the DNA purified and collected. The following primers were used to assess DNA purified from the ChIP protocol: chr.21:42093265–42093385: F 5′-ACATAAATGAGAGATGATAC-3′, R 5′-TCTCCATTTCTCTCATCAATG-3′ and Sat2: F 5′-CTGCACTACCTGAAGAGGAC-3′, R 5′-GATGGTTCAACACTCTTACA-3′. Input chromatin DNA and DNA from pull-down with an IgG antibody were used as controls with each primer pair.
ChIP-Seq library preparation and data analysis
Following transfection with 3x FLAG-KLLN and chromatin immunoprecipitation, 10 ng of DNA from both input control and ChIP-enriched DNA from two replicates was used as starting material for the ChIP-Seq DNA Sample Prep Kit (Illumina, San Diego, CA, USA). Libraries were evaluated for appropriate size using a High Sensitivity DNA Bioanalyzer kit (Agilent) and quantitated with the dsDNA High Sensitivity Qubit Assay (Life Technologies). Single end 50-bp sequencing on the IlluminaHiSeq 2000 was performed at the Génome Québec Innovation Centre.
Data quality of sequences from Génome Québec Innovation Centre was evaluated using FastQC before further downstream analysis. Along with the data generated for KLLN binding, ChIP-seq ENCODE data for histone modifications in MCF7 cells including H3K9me3, H3K27ac, H3K27me3, and H3K36me3, as well as input FASTQ files were downloaded from UCSC Genome Browser. All files were aligned to Hg19 using Bowtie2 and then further analyzed for peak identification with input control by Model-based Analysis for ChIP-Seq (MACS) with significance for called peaks set at FDR < 0.05. The R Bioconductor package ChIPpeakAnno was used to graph frequency of peaks compared to transcription start sites (TSS) and percent of binding sites compared to genomic features. ChIP-seq peak annotation and identification of overlapping and differentially bound peaks was done using the HOMER software package. Ingenuity Pathway Analysis (IPA) was used to determine the functions and upstream regulators of the genes annotated as closest to KLLN binding peaks. Motif calling by HOMER was done using a 200-bp region associated with each peak compared to over 50 000 corresponding GC matched extracted background sequences. The KLLN ChIP-seq peaks were divided into different chromatin states based off of the chromatin states defined for mammary epithelial cells (HMEC) described in Ernst et al. 2011 for which the data were downloaded from the ENCODE project at UCSC (10).
Histone methyltransferase (HMT) activity assay
Histone methyltransferase (HMT) activity for specific HMTs that target H3K9 was assayed after KLLN overexpression or knockdown. Following transfection with plasmids or siRNA, nuclear protein was collected with the NE-PER Nuclear and Cytoplasmic Extraction kit (Pierce) and protein concentration measured using the BCA Protein assay. HMT activity was measured using the EpiQuik Histone Methyltransferase Activity Assay Kit (H3K9) (Epigentek, Farmingdale, NY, USA) according to manufacturers instructions. For each sample, 3–4 μl of nuclear extract was used for the assay and 1 μl of control enzyme was used as a positive control. Absorbance was read at 450 nm on a microplate reader and HMT activity was calculated as below:
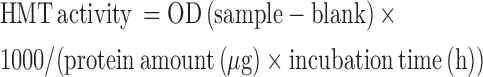 |
To assay HMT activity after KLLN pulldown, 200-300 μg of nuclear protein was incubated overnight with ANTI-FLAG M2 affinity gel (Sigma Aldrich). The protein–protein complexes were washed and eluted by competition with 100 μl of 3x FLAG peptide. HMT activity was measured as described above and calculated as below:
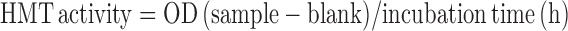 |
Cell staining
For immunofluorescence, cells were transfected with KLLN plasmid and grown on cover slips in a 12-well plate. Forty-eight hours after transfection, cells were fixed for 15 min with 3.7% formaldehyde. After washing with PBS (Phosphate buffered saline), cover slips were permeabilized in 0.3% triton PBS for 5 min and blocked in normal goat serum for 1 h at room-temperature before being incubated overnight at 4°C with primary antibodies diluted in blocking solution: CENP-A at 1:400 (Cell Signaling, cat# 2186) and FLAG M2 at 1:100 (Sigma-Aldrich, cat# F-1804). Coverslips were again washed with PBS and incubated for 1 h with secondary antibodies in 0.3% triton PBS: Alexa Fluor 488 goat anti-rabbit IgG (cat# A11008) at 1:1000 and Alexa Fluor 568 goat anti-mouse IgG (cat# A11031) at 1:1000 (Life Technologies). After washing, coverslips were mounted onto slides with DAPI-containing mounting media (Vector Laboratories, Burlingame, CA, USA). Images were analyzed using upright confocal microscopy (Leica Microsystems, Buffalo Grove, IL, USA) and Leica Confocal Software for image analysis.
Assessment of chromosomal instability
Micronuclei formation
For micronuclei analysis, cells were seeded on cover slips 24 h after KLLN or control knockdown. At the end of an additional 24 h, the cells were fixed with 100% methanol, dried and mounted onto slides using DAPI-containing mounting media (Vector Laboratories). For each of the three replicates per condition, approximately 2000 cells were analyzed for micronuclei using an upright fluorescence microscope (Leica Microsystems). Results were represented as frequency of abnormal versus normal nuclei based on presence or absence of micronuclei, respectively, in each of the treatment conditions. For the assessment of micronuclei frequency in LCLs, cells grown in culture were spun onto a slide using a Cytospin centrifuge (Thermo Fisher Scientific) and stained and analyzed as described above.
Numerical chromosomal aberration
For assessing numerical chromosomal aberration after KLLN knockdown, cells transfected with siRNA were cultured for 48 h. At the end of this time period, cells were treated with 0.1 µg/ml colchicine for 5 h. Cells were then harvested, treated with a hypotonic solution (0.075 M KCl) and fixed in Carnoy's fixative (3:1, Methanol:acetic acid). Cells were then dropped onto slides and stained with DAPI-containing mounting media (Vector Laboratories). For each of the treatment conditions 20 metaphase nuclei spreads were captured using an upright fluorescence microscope (Leica Microsystems). Total number of chromosomes per spread was determined by counting and the average number of chromosomes per treatment condition was plotted on a graph. Results were also represented as number of nuclei with either a normal (46) chromosome complement or an abnormal (>46) chromosome complement for MCF 10A cells.
Co-immunoprecipitation (Co-IP)
For co-immunoprecipitation (Co-IP), cells were transfected with KLLN plasmid and harvested 48–72 h post-transfection. Protein lysates were prepared using MPER as previously described. Protein concentration was measured using the BCA assay and a 1 mg/ml solution of protein lysate was prepared. Protein pull down was done using anti-FLAG M2 affinity gel (20 µl) (Sigma-Aldrich) and anti-DBC1 (10 µg/ml of lysate) (Bethyl Laboratories, Inc., Montgomery, TX, USA, cat# A300). Western blotting was performed as previously described to assess the expression of the bait and prey proteins using anti-FLAG M2 (1:2000) and anti-DBC1 (1:10 000) antibodies. Input lysate removed before pull down was used as a control for equal loading of bait protein.
Statistical analysis
All experiments in this study were performed at least in duplicate and where required, in triplicate. All data collected from these in vitro studies were analyzed by two-tailed Students t-tests. Significance of results was determined based on calculated P-values and a P-value less than 0.05 was considered statistically significant. All data were represented with error bars signifying standard error of mean for population mean and standard deviation for sample mean.
RESULTS
DNA binding pattern of KLLN
ChIP-seq on MCF7 breast cancer cells after overexpression of KLLN identified 1422 binding peaks including 1819 sub-peaks for KLLN binding (Supplementary Table S1). KLLN binding peak locations were classified into exonic, intronic, intergenic, 5′UTR, 3′UTR or promoter regions. The majority of the KLLN-binding peaks fell within intergenic regions (Figure 1A), but a substantial proportion of peaks fell within introns (∼10%) and proximal promoters (∼7%), defined as 1 kb from the transcription start site. KLLN binding within introns and promoters (∼17%) represent a disproportionate amount compared to the distribution of transcribed genes, with only ∼2% of the human genome made up of protein coding genes and another ∼4% being transcribed genes and conserved regulatory sequences. Top upstream regulators of the gene set that KLLN binds nearest to, are the HDAC family (P = 2.8 × 10−5) and SETDB1 (P = 5.59 × 10−5), which are both chromatin modifiers.
Figure 1.

Global analysis of KLLN DNA binding pattern and association with histone modifications. (A) Graph of percent of KLLN binding site locations relative to exons, introns, 5′UTRs, 3′UTRs, proximal promoters, immediate downstream region from a transcription start site and intergenic regions. KLLN binding is the highest in intergenic regions but with a substantial amount in the proximal promoters. (B) Graph of enrichment of KLLN binding in regions of H3K9me3, H3K27me3, H2K27ac and H3K36me3. KLLN binding sites in MCF7 cells have substantially more overlap with H3K9me3 compared to H3K27me3, H2K27ac and H3K36me3. (C) Graph of histone modifications centered around KLLN binding sites. There is a regional increase in the peak density for H3K9me3 around KLLN binding sites. (D) IGV screen shots of peaks from ChIP-seq data for the histone mark H3K9me3 and KLLN. The upper panel represents the entire genome, the middle panel is chromosome 1 and the bottom panel is a zoomed in region adjacent to the centromere. Overlap of peaks is observed between KLLN and H3K9me3 throughout the genome. (E) ChIP analysis of a region of overlap, chr. 21:42093265–42093385, identified in ChIP-seq data for KLLN and H3K9me3. Both FLAG-KLLN and H3K9me3 are found to bind the same region in MCF7 and ZR-75-30 cell lines. ChIP with non-specific IgG antibody was used as a control along with a PCR reaction with no DNA as another control.
KLLN binding peaks are associated with H3K9 trimethylation
Since KLLN peaks are associated with genes that are related to chromatin regulation, we compared KLLN peaks to histone marks for two repressive chromatin, H3K9me3 and H3K27me3, and two actively transcribed regions, H2K27ac and H3K36me3. We observed enrichment of KLLN binding peaks overlapping with H3K9me3 marks compared to other analyzed histone marks (Figure 1B–C). When KLLN binding peaks and regions on the genome with high concentration of H3K9me3 residue were compared using the Integrated Genome Viewer (IGV), the enrichment of overlap between KLLN binding and H3K9me3 could be easily visualized (Figure 1D). To corroborate the ChIP-seq results, we utilized ChIP to show that both KLLN and H3K9me3 bind to the same region chr. 21:42093265–42093385 in both MCF7 and ZR-75-30 cell lines (Figure 1E). EGR1, a nuclear protein with DNA-binding capability, was used as a negative control to confirm specific overlap between KLLN and H3K9me3. Comparison of binding peaks for EGR1, generated using ChIP-seq data downloaded from ENCODE, with regions of H3K9me3 using IGV did not show any overlap (Supplementary Figure S1). Therefore, we can conclude that KLLN specifically localizes to regions with high density of H3K9me3 modifications.
While specific histone marks can be used as a surrogate for active and repressed regions of the genome, multiple histone marks make up signatures which can more precisely define a given regions activity. Of the seven chromatin states defined in HMECs using a combinatorial histone modification code (10), KLLN was found to bind in regions defined as heterochromatin (60%) and regions of repetitive CNV (15%) that are primarily marked by H3K9me3 (Supplementary Figure S2). The closest genes to areas of co-occurrence between KLLN and H3K9me3 were used to identify GO pathways potentially regulated by this interaction. The top enriched pathways are broad and include cell process (P = 1 × 10−14.7) and cell communication (P = 1 × 10−13.3) but specific pathways integral to cancer development and progression such as regulation of protein kinase B (AKT) signaling (P = 1 × 10−11.4) and epidermal growth factor-activated receptor activity (P = 1 × 10−10.8) were also enriched (Supplementary Table S2).
KLLN is associated with increased H3K9 methyltransferase activity and increased global H3K9 trimethylation
Since KLLN binding peaks overlap consistently with H3K9me3 residues, we assessed H3K9me3 specific histone methyltransferase (HMT) activity specifically to determine if KLLN protein and potential binding partners are able to directly regulate this methylation. Overexpression of KLLN in MCF7 and MCF10A cells resulted in significant increases in HMT activity (Figure 2A–B). siRNA-based interference of KLLN was associated with decreased HMT activity in both cell lines (Figure 2A–B). Our observations suggest that KLLN and/or its binding partners are directly involved in regulation of H3K9-trimethylation. To assess whether variations in HMT activity altered global H3K9-trimethylation, we immunoblotted for H3K9me3 and found that when KLLN was overexpressed, there was a concomitant increase in H3K9me3 in MCF7 and MCF10A cells (Figure 2C–D). Knockdown of KLLN expression resulted in decreased H3K9me3 (Figure 2C–D). Therefore, KLLN and its binding partners are strongly associated with the maintenance of H3K9me3 by regulating the activity of histone methyltransferases.
Figure 2.

H3K9 methyltransferase activity and global H3K9me3 levels. (A) Graph of H3K9-specific methyltransferase activity of native protein isolated from the nuclear fraction of MCF7 cells after transfection with FLAG-KLLN or vector alone, and with and without siRNA-based knockdown of KLLN. H3K9 specific methyltransferase activity is increased with overexpression of KLLN in MCF7 cells (P = 0.017). Knockdown of KLLN decreased methyltransferase activity (P = 0.012). (B) Similar graph of H3K9 methyltransferase activity in MCF10A cells. KLLN overexpression increased methyltransferase activity (P = 0.023) whereas knockdown of KLLN decreased the same. (C) Immunoblot analysis of native protein isolated from the nuclear fraction of MCF7 cells for global H3K9 trimethylation levels after transfection with FLAG-KLLN or vector alone, and with and without siRNA-based knockdown of KLLN. Overexpression of KLLN increased global H3K9me3 levels whereas knockdown of KLLN decreased these levels. (D) Similar immunoblot analysis of global H3K9me3 levels in MCF10A cells. Increased global H3K9me3 after overexpression of KLLN while knockdown of KLLN decreased the same.
KLLN localizes to the pericentric region
H3K9me3 is the primary mark of pericentric heterochromatin regions and important for centromere stability (11). Corroborating previous observations (11), H3K9me3 is seen here to mark the pericentric Sat2 sequence (Figure 3A). We additionally demonstrate that KLLN binds to the Sat2 sequence in the pericentric region (Figure 3A). To further confirm KLLN localization at the pericentric region, we used a centromere-binding protein, CENP-A, as a marker. Confocal analysis shows overlap between FLAG-KLLN and CENP-A (Figure 3B), thereby confirming the enrichment of KLLN binding in pericentric regions.
Figure 3.

KLLN localization at pericentric regions and chromosomal instability. (A) ChIP analysis of the pericentric satellite sequence for both KLLN binding and H3K9me3. ChIP with non-specific IgG antibody and a PCR reaction with no DNA used as negative controls. Overlap of KLLN and H3K9me3 seen at pericentric satellite (Sat2) regions. (B) Confocal analysis of FLAG-KLLN transfected MCF7 cells stained with DAPI (blue), nuclear staining, CENP-A (green), a centromere-binding protein and FLAG (red), for FLAG-KLLN fusion protein. In the nucleus with a successful transfection of FLAG-KLLN indicated by the red staining, co-localization of CENP-A and FLAG-KLLN can be visualized as yellow staining. KLLN localizes at the pericentric region. (C) Graph of micronuclear frequency after siRNA-based knockdown with KLLN in MCF7 cells. Micronuclei are significantly increased (P = 0.014) with KLLN knockdown. The white arrowhead indicates a micronucleus next to a regular nucleus. (D) Graph of micronuclear frequency versus KLLN expression in 11 LCL samples. KLLN expression for sample CCF00769 was set at 1 and expression of all other samples were expressed as a value relative to this sample. Micronuclei frequency is significantly increased (P = 0.003) with reduced KLLN expression (<5). (E) Graph for analysis of chromosome number as a marker of chromosomal instability after siRNA-based knockdown of KLLN in MCF10A cells. Increased number of nuclei with an abnormal chromosome number indicates chromosomal instability after KLLN knockdown (P = 0.0001).
KLLN localization to pericentric region is required for maintenance of H3K9me3 and chromosomal stability
Since altered regulation of pericentric heterochromatin is known to contribute to chromosomal instability (CIN) (12), we sought to assess the effects of decreased KLLN expression on CIN by micronuclear enumeration and chromosome number analysis. We found that there was a significant increase of micronuclei after KLLN knockdown compared to control in MCF7 cells (Figure 3C). We also assessed micronuclei frequency relative to KLLN expression in 11 LCL samples. We found that relative KLLN expression of <5 on our adjusted scale resulted in increased numbers of micronuclei (Figure 3D). Therefore, loss of KLLN expression can lead to increased CIN.
We also tested the effect of KLLN loss on another marker of CIN, abnormal chromosomal number. The mean number of chromosomes per nucleus tended to increase after KLLN knockdown in MCF7 and MCF10A cells (Supplementary Figure S3). Since every nuclei counted could have varied chromosome numbers, observed increase in chromosomal number was not statistically significant (due to high standard deviation), but the trend indicates chromosomal instability. In MCF10A cells, where more than half the nuclei had a normal complement of 46 chromosomes, we found that KLLN knockdown resulted in a significant increase in nuclei with an abnormal chromosome number (>46 chromosomes, Figure 3E). Hence, loss of KLLN induces chromosomal instability through its requirement for H3K9 trimethylation and the maintenance of pericentric heterochromatin.
KLLN interaction with DBC1 abrogates inhibitory effect of DBC1 on H3K9 histone methyltransferase, thereby promoting H3K9 trimethylation
To test whether KLLN was directly involved with increased H3K9me3, we assessed HMT activity of the supernatant after pulling down interacting partners of KLLN, and found that HMT activity increased (Figure 4A). This suggested that KLLN has an inhibitory effect on one of its interacting partners that regulated H3K9me3. DBC1, identified as an interactor of KLLN through mass spectrometry (data not shown), is a known inhibitor of SUV39H1, an H3K9 histone methyltransferase essential for maintenance of pericentric heterochromatin and mitotic fidelity (12,13). We confirmed this interaction by co-immunoprecipitation in control-transfected breast cancer cells as well as those transfected with KLLN plasmid (Figure 4B–C). The supernatant used for HMT activity assay also had decreased DBC1 (Figure 4D). Mass spectrometry (MS) results could not identify any other interactors of KLLN that had a direct effect on HMT activity.
Figure 4.

KLLN interaction with DBC1. (A) Graph of H3K9-specific methyltransferase activity using supernatant of nuclear fraction after IP with FLAG antibody in MCF7 cells transfected with FLAG-KLLN or vector alone. H3K9 methyltransferase activity was increased in supernatant without KLLN or its interacting proteins. (B) Immunoblot analysis of co-immunoprecipitation (Co-IP) with FLAG or DBC1 antibody. KLLN and DBC1 shown to interact with each other. (C) Immunoblot analysis of lysates immunoprecipitated using DBC1 antibody in untreated MCF7 cells. Endogenous KLLN was successfully pulled down by DBC1 antibody. (D) Immunoblot analysis of nuclear fraction lysates from FLAG-KLLN transfected MCF7 cells and supernatant lysate after IP with FLAG antibody to pull down KLLN and its interacting partners. The supernatant after IP had reduced DBC1 protein levels. (E) Schematic of mechanism for KLLN regulation of H3K9me3 (modified from Li et al., 2009). KLLN interaction with DBC1 abrogates DBC1 inhibition of SUV39H1, an H3K9 trimethylation-specific histone methyltransferase.
DISCUSSION
While it was previously known that KLLN binds to DNA, the random pattern of global KLLN binding was only speculated (7). Our results show conclusively that this binding is not random and that a substantial proportion of the binding peaks were observed within the introns and proximal promoter regions of genes. Intergenic regions housed the majority of the KLLN binding peaks, notably at specific genomic sites such as pericentric heterochromatin and regions of H3K9me3, a marker of constitutive heterochromatin throughout the genome, including at the pericentric regions. Intriguingly, we found that KLLN directly or through its binding partners were able to regulate H3K9me3 by altering HMT activity specific for this modification. Furthermore, we were able to demonstrate that disruption of KLLN function leading to loss of H3K9me3 resulted in increased chromosomal instability. Therefore, we were able to conclusively show that KLLN maintains pericentric H3K9me3 and thereby preserving genomic stability. The role played by KLLN in the maintenance of chromatin organization is therefore critical toward its function as a tumor suppressor gene.
By exploring KLLN's binding pattern, we have been able to demonstrate KLLN's role in maintenance of pericentric heterochromatin through the regulation of H3K9 trimethylation. H3K9me3 is primarily involved in the maintenance of pericentric heterochromatin and is associated with transcriptional repression, and centromere and chromosomal stability (11,13,14). We have conclusively shown that there is significant overlap between KLLN binding peaks and regions of H3K9me3 but also that KLLN regulates H3K9-specific HMT activity and global H3K9me3 in the genome. Particularly, KLLN and H3K9me3 are co-localized at the pericentric chromosomal regions. It has previously been demonstrated that reduced H3K9 trimethylation especially at the regions of pericentric heterochromatin results in chromosomal instability, a hallmark of cancer (12,13,15). Genomic instability due to heterochromatin relaxation has been demonstrated in cancers with an overexpression of alpha-satellite pericentric regions observed in pancreatic, lung, kidney, ovarian and prostate cancer (16). In our studies, we are able to show that decreased KLLN expression resulted in genomic instability characterized by an increase in micronuclei frequency and increase in numerical chromosomal aberrations in breast cancer cells. Therefore, KLLN must have a significant role in the maintenance of H3K9me3 and thereby pericentric heterochromatin and when KLLN is altered or lost, genomic instability ensues that could lead to tumorigenesis.
KLLN, alone or potentially as part of a complex, methylates histone 3 at lysine 9 and has a sentinel role in maintaining the H3K9me3 mark. We also identified DBC1, a binding partner of KLLN, which directly regulates the activity of SUV39H1. SUV39H1, an H3K9me3 methyltransferase, is primarily responsible for the establishment of H3K9me3 at the pericentric region and activity of this methyltransferase activity is essential for heterochromatin formation and maintenance of mitotic fidelity and chromosomal stability (12,13). When the function of SUV39H1 is abrogated, there is increased expression of pericentric alpha-satellites, a marker of altered heterochromatin and results in genomic instability (17). DBC1, first identified in a homozygously deleted region on chromosome 8 in breast cancers, is a nuclear protein that is known to bind to some nuclear receptors including androgen receptor and several enzymes including SUV39H1, histone deacetylases (SIRT1 and HDAC3) and transcriptional factor BRCA1 (18–21). Binding of DBC1 to SUV39H1 and SIRT1 results in the inhibition of the binding of the latter two proteins, thereby inhibiting their functions (21,22). Our mass spectrometry analysis did not reveal any other interactors of KLLN that directly regulated HMT activity. Our results indicate that KLLN regulation of H3K9me3 is achieved by the inhibitory effect of KLLN on one of its interacting partners. Therefore, a potential mechanism for KLLN's regulation of H3K9 trimethylation is that the KLLN-DBC1 interaction abrogating SUV39H1 inhibition by DBC1 (Figure 4E).
Interestingly, PTEN, with which KLLN shares a bidirectional promoter, is also critical for maintenance of chromosomal stability. PTEN has been shown to interact with CENP-C, a kinetochore associated protein, with disruption of this association leading to chromosomal aberrations (23). Recently, it has been reported that PTEN deficiency results in impairment of heterochromatin structure and increase in embedded gene expression (24). PTEN maintains chromatin condensation through its interaction with histone H1 and HP1α (24,25). In this study, we have also been able to generate potential pathways regulated by KLLN based on the DNA binding patterns in combination with histone modification data. Signaling networks quintessential to cell cycle regulation such as AKT and EGFR signaling were extracted in the analysis. Taken together, the involvement of both KLLN and PTEN in maintenance of heterochromatin structure and in the regulation of the AKT pathway could create a functional crosstalk between KLLN and PTEN. Since both genes are known susceptibility genes for CS, their functional interplay could have a significant effect on the cancer risk in CS patients. The interplay of the two genes could have broader implications in sporadic cancers as well as in normal development.
In conclusion, our data establish the global DNA binding capability of KLLN suggestive of its role in regulation of gene expression. Our study demonstrates a clear role for KLLN in maintenance of chromatin organization thereby maintaining genomic stability, which begins to uncover the basis of KLLN as a tumor suppressor and as a susceptibility gene for inherited cancers.
Supplementary Material
Acknowledgments
Author contributions: E.A.N., M.S. and C.E. conceived the study and designed the experiments. E.A.N. and M.S. equally contributed to conducting the experiments. E.A.N and F.N analyzed the next-gen sequencing data. E.A.N., M.S and C.E. interpreted the data, and drafted and critically revised the manuscript. All authors gave final approval of the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Breast Cancer Research Foundation and the National Cancer Institute [P01CA124570 both to C.E. in part]; NCI [F30CA168151 to E.A.N.]; M.S. is an Ambrose Monell Foundation Cancer Genomic Medicine Fellow; C.E. is the Sondra J. and Stephen R. Hardis Endowed Chair in Cancer Genomic Medicine at the Cleveland Clinic and is an ACS Clinical Research Professor.
Conflict of interest statement. None declared.
REFERENCES
- 1.Tan M.H., Mester J., Peterson C., Yang Y., Chen J.L., Rybicki L.A., Milas K., Pederson H., Remzi B., Orloff M.S., et al. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am. J. Hum. Genet. 2011;88:42–56. doi: 10.1016/j.ajhg.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eng C. Will the real Cowden syndrome please stand up: revised diagnostic criteria. J. Med. Genet. 2000;37:828–830. doi: 10.1136/jmg.37.11.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennett K.L., Mester J., Eng C. Germline epigenetic regulation of KILLIN in Cowden and Cowden-like syndrome. JAMA. 2010;304:2724–2731. doi: 10.1001/jama.2010.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bennett K.L., Campbell R., Ganapathi S., Zhou M., Rini B., Ganapathi R., Neumann H.P., Eng C. Germline and somatic DNA methylation and epigenetic regulation of KILLIN in renal cell carcinoma. Genes Chromosomes Cancer. 2011;50:654–661. doi: 10.1002/gcc.20887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nizialek E.A., Peterson C., Mester J.L., Downes-Kelly E., Eng C. Germline and somatic KLLN alterations in breast cancer dysregulate G2 arrest. Hum. Mol. Genet. 2013;22:2451–2461. doi: 10.1093/hmg/ddt097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y., He X., Yu Q., Eng C. Androgen receptor-induced tumor suppressor, KLLN, inhibits breast cancer growth and transcriptionally activates p53/p73-mediated apoptosis in breast carcinomas. Hum. Mol. Genet. 2013;22:2263–2272. doi: 10.1093/hmg/ddt077. [DOI] [PubMed] [Google Scholar]
- 7.Cho Y.J., Liang P. Killin is a p53-regulated nuclear inhibitor of DNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 2008;105:5396–5401. doi: 10.1073/pnas.0705410105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stambolic V., MacPherson D., Sas D., Lin Y., Snow B., Jang Y., Benchimol S., Mak T.W. Regulation of PTEN transcription by p53. Mol. Cell. 2001;8:317–325. doi: 10.1016/s1097-2765(01)00323-9. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y., Radhakrishnan D., He X., Peehl D.M., Eng C. Transcription factor KLLN inhibits tumor growth by AR suppression, induces apoptosis by TP53/TP73 stimulation in prostate carcinomas, and correlates with cellular differentiation. J. Clin. Endocrinol. Metab. 2013;98:E586–E594. doi: 10.1210/jc.2012-3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ernst J., Kheradpour P., Mikkelsen T.S., Shoresh N., Ward L.D., Epstein C.B., Zhang X., Wang L., Issner R., Coyne M., et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schotta G., Lachner M., Sarma K., Ebert A., Sengupta R., Reuter G., Reinberg D., Jenuwein T. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004;18:1251–1262. doi: 10.1101/gad.300704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Slee R.B., Steiner C.M., Herbert B.S., Vance G.H., Hickey R.J., Schwarz T., Christan S., Radovich M., Schneider B.P., Schindelhauer D., et al. Cancer-associated alteration of pericentromeric heterochromatin may contribute to chromosome instability. Oncogene. 2012;31:3244–3253. doi: 10.1038/onc.2011.502. [DOI] [PubMed] [Google Scholar]
- 13.Peters A.H., O'Carroll D., Scherthan H., Mechtler K., Sauer S., Schofer C., Weipoltshammer K., Pagani M., Lachner M., Kohlmaier A., et al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell. 2001;107:323–337. doi: 10.1016/s0092-8674(01)00542-6. [DOI] [PubMed] [Google Scholar]
- 14.Stewart M.D., Li J., Wong J. Relationship between histone H3 lysine 9 methylation, transcription repression, and heterochromatin protein 1 recruitment. Mol. Cell. Biol. 2005;25:2525–2538. doi: 10.1128/MCB.25.7.2525-2538.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 16.Ting D.T., Lipson D., Paul S., Brannigan B.W., Akhavanfard S., Coffman E.J., Contino G., Deshpande V., Iafrate A.J., Letovsky S., et al. Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science. 2011;331:593–596. doi: 10.1126/science.1200801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang D., Zhou J., Liu X., Lu D., Shen C., Du Y., Wei F.Z., Song B., Lu X., Yu Y., et al. Methylation of SUV39H1 by SET7/9 results in heterochromatin relaxation and genome instability. Proc. Natl. Acad. Sci. U.S.A. 2013;110:5516–5521. doi: 10.1073/pnas.1216596110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hiraike H., Wada-Hiraike O., Nakagawa S., Koyama S., Miyamoto Y., Sone K., Tanikawa M., Tsuruga T., Nagasaka K., Matsumoto Y., et al. Identification of DBC1 as a transcriptional repressor for BRCA1. Br. J. Cancer. 2010;102:1061–1067. doi: 10.1038/sj.bjc.6605577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fu J., Jiang J., Li J., Wang S., Shi G., Feng Q., White E., Qin J., Wong J. Deleted in breast cancer 1, a novel androgen receptor (AR) coactivator that promotes AR DNA-binding activity. J. Biol. Chem. 2009;284:6832–6840. doi: 10.1074/jbc.M808988200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chini C.C., Escande C., Nin V., Chini E.N. HDAC3 is negatively regulated by the nuclear protein DBC1. J. Biol. Chem. 2010;285:40830–40837. doi: 10.1074/jbc.M110.153270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Z., Chen L., Kabra N., Wang C., Fang J., Chen J. Inhibition of SUV39H1 methyltransferase activity by DBC1. J. Biol. Chem. 2009;284:10361–10366. doi: 10.1074/jbc.M900956200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim J.E., Chen J., Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008;451:583–586. doi: 10.1038/nature06500. [DOI] [PubMed] [Google Scholar]
- 23.Shen W.H., Balajee A.S., Wang J., Wu H., Eng C., Pandolfi P.P., Yin Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128:157–170. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 24.Gong L., Govan J.M., Evans E.B., Dai H., Wang E., Lee S.W., Lin H.K., Lazar A.J., Mills G.B., Lin S.Y. Nuclear PTEN tumor-suppressor functions through maintaining heterochromatin structure. Cell cycle. 2015;14:2323–2332. doi: 10.1080/15384101.2015.1044174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Z.H., Zhu M., Yang J., Liang H., He J., He S., Wang P., Kang X., McNutt M.A., Yin Y., et al. PTEN interacts with histone H1 and controls chromatin condensation. Cell reports. 2014;8:2003–2014. doi: 10.1016/j.celrep.2014.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.