


Abstract
Trimethylated histone H3 lysine 27 (H3K27me3) is linked to gene silencing, whereas H3K4me3 is associated with gene activation. These two marks frequently co-occupy gene promoters, forming bivalent domains. Bivalency signifies repressed but activatable states of gene expression and can be resolved to active, H3K4me3-prevalent states during multiple cellular processes, including differentiation, development and epithelial mesenchymal transition. However, the molecular mechanism underlying bivalency resolution remains largely unknown. Here, we show that the H3K27 demethylase UTX (also called KDM6A) is required for the resolution and activation of numerous retinoic acid (RA)-inducible bivalent genes during the RA-driven differentiation of mouse embryonic stem cells (ESCs). Notably, UTX loss in mouse ESCs inhibited the RA-driven bivalency resolution and activation of most developmentally critical homeobox (Hox) a–d genes. The UTX-mediated resolution and activation of many bivalent Hox genes during mouse ESC differentiation were recapitulated during RA-driven differentiation of human NT2/D1 embryonal carcinoma cells. In support of the importance of UTX in bivalency resolution, Utx-null mouse ESCs and UTX-depleted NT2/D1 cells displayed defects in RA-driven cellular differentiation. Our results define UTX as a bivalency-resolving histone modifier necessary for stem cell differentiation.
INTRODUCTION
Trimethylated histone H3 lysine 27 (H3K27me3) is a mark of gene repression, whereas H3K4me3 is a mark of gene activation. These two marks representing opposite gene expression states co-localize at many differentiation-specific gene promoters in stem cells to form bivalent promoters in which they are located at different H3 tails in nucleosomes (1,2). It has been widely accepted that bivalent promoters (1000–6000), including most promoters of the developmentally critical homeobox (Hox) a–d cluster genes, are in a poised (i.e. repressed but activatable) state of gene expression (2–7). Since its discovery, bivalency has been considered a key epigenetic signature associated with gene regulation in mouse and human embryonic stem cells (ESCs), hematopoietic stem cells, epithelial mesenchymal transition and developing embryos (2–5,8–11).
During cellular differentiation, 14–32% of bivalent promoters are resolved to transcriptionally active H3K4me3-prevalent monovalent states (4–5,12), although some bivalent domains are newly formed (12). Notably, the bivalent promoters of many critical differentiation-specific genes, including most Hoxa–d cluster genes, are repressed in mouse ESCs but resolved and activated during cellular differentiation (2–5). Therefore, bivalency resolution is believed to be important to cellular differentiation (10).
The establishment of bivalency has been well studied. The H3K4 methyltransferase mixed-lineage leukemia 2 (MLL2; also known as KMT2B) is required for the establishment of H3K4me3 in bivalent domains (13), and the H3K4 methyltransferase MLL1 plays a redundant role in depositing H3K4me3 to generate bivalent domains (14). In addition, two other H3K4 methyltransferases, SET1A and SET1B, and the H3K27 methyltransferase complex PRC2 are associated with the generation of bivalency (10). However, little is known about which histone methylation modifier is responsible for the resolution of bivalent domains into active monovalent states (10).
Bivalency resolution requires H3K27me3 demethylation that is catalyzed by a H3K27 demethylase. We previously showed that the H3K27 demethylase UTX (ubiquitously transcribed tetratricopeptide repeat, X chromosome; also called KDM6A) may mediate the RA-induced activation of the HOXA1–3 and HOXB1–3 genes during RA-driven differentiation of human NT2/D1 embryonal carcinoma cells (15). In addition, UTX has been shown to be essential for several developmental and biological processes, including embryogenesis (16), cardiac development (17), muscular development (18) and animal aging (19). For these reasons, we tested whether UTX plays an important role in bivalency resolution during cellular differentiation. Specifically, we assessed the effect of UTX loss or UTX knockdown on bivalency resolution during RA-driven cellular differentiation. Our results provide evidence that UTX is a bivalency-resolving modifier necessary for RA-driven cellular differentiation.
MATERIALS AND METHODS
Antibodies
Anti-UTX antibodies were obtained as described previously (15). Anti-H3K4me3 (17–614) and anti-H3K27me3 (07–449) antibodies were from Millipore; anti-H3 (ab1971) antibodies were from Abcam; and anti–β-actin (A5441) antibodies were from Sigma-Aldrich.
Mouse ESC culture
Wild type (WT) and Utx-null mouse V6.5 ESCs were maintained with or without irradiated feeder cell layers (less than passage 5) in complete knockout Dulbecco's modified Eagle's medium (Life Technologies) containing 4.5 mg/ml glucose, 20% ESC-grade fetal bovine serum (Life Technologies), 2 mM L-glutamine, 50 μg/ml penicillin and 50 μg/ml streptomycin (Life Technologies), 0.1 mM β-mercaptoethanol, 0.1 mM minimum essential medium with nonessential amino acids, and 1000 U/ml leukemia inhibitory factor (LIF). Glutamine was freshly added and the medium replaced daily. ESCs were trypsinized, transferred onto 10 cm petri dishes and cultured in ESC media without LIF for 5 days to induce embryoid bodies (EBs). EBs were transferred onto gelatin-coated tissue culture dishes, cultured in ESC media without LIF and treated with the indicated RA concentrations for 5 days to induce cellular differentiation. Cells were incubated at 37ºC in a 5% CO2 atmosphere with 95% relative humidity.
NT2/D1 cell differentiation and shRNA knockdown
The NT2/D1 embryonic carcinoma cell line was maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. Cells were treated with 10 μM all trans-RA to induce cellular differentiation. UTX knockdown was performed using small hairpin RNAs (shRNAs) against human UTX. Two different shRNAs cloned into a pLKO.1 vector were used: shUTX-4 (GCATGAACACAGTTCAACTAT) and shUTX-6 (AAGACCACTCAGATAGTGAAT). A lentivirus-mediated infection method was used to deliver shUTX-4, shUTX-6, or control shRNA (shLuciferase, shLuc) to the cells as described previously (20).
RNA isolation and quantitative reverse transcription polymerase chain reaction
Total RNA was isolated using RNeasy kits (Qiagen) according to the manufacturer's instructions. cDNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad) according to the manufacturer's instructions. Quantitative reverse transcription polymerase chain reaction (RT-PCR) was performed in triplicate using SYBR-green and gene-specific primers (Supplementary Table S1). Messenger RNA levels were quantified using the CFX Manager software program (Bio-Rad) and normalized to β-actin mRNA levels.
Microarray experiments and gene ontology analysis
The RNeasy Mini Kit (Qiagen) was used to extract RNA from mouse ESCs. The RNA samples were subjected to Illumina's Direct Hybridization whole-genome expression assay. Labeled complementary RNAs were detected using the Bead Chip and scanned on the iScan or Bead Array Reader. The Illumina MouseWG-6 v2.0 Expression BeadChip was used to analyze mRNA expressions. We used the Web-based Database for Annotation, Visualization and Integrated Discovery (DAVID) program (21) to analyze all bivalent gene sets that were upregulated upon RA treatment (≥1.5-fold change) in the microarray. Function enrichment analysis for each gene set was carried out at the DAVID Web site (http://david.abcc.ncifcrf.gov) as described previously (22). For gene ontology analysis, Q-values (Benjamini) for gene groups were determined using DAVID software program. Q < 0.05 indicates statistically significant changes.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were performed using the Millipore ChIP protocol with minor modifications as described previously (20,23). Chromatin immunoprecipitates for proteins and methyl marks were amplified by quantitative PCR using gene-specific primers (Supplementary Table S1), normalized to input and calculated as relative change in normalized PCR values from day 0 to day 4 after RA treatment.
Chromatin template preparation, ChIP assays and ChIP-Seq library construction
Chromatin preparation, ChIP assays and ChIP-Seq library construction were performed using modified protocols of our own (24,25) and a modified published protocol (http://www.hudsonalpha.org/myers-lab/protocols). Briefly, cells were grown to log phase and fixed with 1% formaldehyde for 10 min at room temperature. The fixed cells were sonicated directly or snap-frozen in liquid nitrogen and stored at −80°C until use. For sonication, the cells were first incubated in 0.5% Triton X-100 in 1× phosphate-buffered saline with 1× protease inhibitor (Roche Life Science) for 10 min on ice and then re-suspended in ice-cold Tris–ethylenediaminetetraacetic acid (TE) buffer, pH 8.0, with 1× protease inhibitor and fresh phenylmethylsulfonyl fluoride (in a final concentration of 0.5 mM) to a final concentration of 25 million cells/ml. Sonication was performed with a Sonic Dismembrator Model 550 (Fisher Scientific) in 22 cycles with intensity set at 4. Each sonication cycle consisted of a 30 s pulse sonication followed by a 1 min incubation in ice-cold water. The sonicated samples were centrifuged at 14,000 rpm at 4°C for 15 min to pellet insoluble material, and the soluble part (i.e. sheared chromatin) was used for ChIP assays or supplemented with 5% glycerol for storage at −80°C. Exactly 10% of the chromatin was used for the extraction of input DNA using phenol:chloroform:isoamyl alcohol.
For ChIP, 4 μg of antibodies, including anti-H3K4me3 or anti-H3K27me3 antibodies, was bound to protein A or protein G magnetic beads depending on the antibody origin. Freshly made or stored chromatin templates from about 20 million cells were added to the magnetic beads with the associated antibodies and incubated at 4°C overnight. The beads were then washed five times with LiCl wash buffer and one time with TE buffer, re-suspended in TE buffer containing 1% sodium dodecyl sulfate and 50 μg/ml proteinase K, and incubated at 65°C overnight. The precipitated DNA was extracted using phenol:chloroform:isoamyl alcohol. Both precipitated DNA and input DNA were re-suspended in 40 μl of re-suspension buffer from the Illumina kit. The ChIP-Seq libraries were constructed using the TruSeq DNA sample prep kit v2 (Illumina) according to the manufacturer's protocol, except that the gel purification of the products after adaptor ligation was performed with 2% E-gel agarose gel (Thermo Fisher Scientific). DNA fragments within a range of 200–500 bp were purified. Libraries were enriched with PCR for 18 cycles. The libraries were sequenced on the Illumina HiSeq 2500 platform with a single-read 54-cycle sequencing run.
Microarray data analysis
We downloaded 20,221 well-annotated mouse genes (genome version mm9) from http://bejerano.stanford.edu/help/display/GREAT/Genes/. Probes were mapped to the official gene symbols on the basis of annotation documentation provided by Illumina. The gene expression value was calculated as the mean of its associated probe values. The expression values of the whole gene set were normalized between samples based on the quantile normalization function in the R package Limma (http://www.bioconductor.org/packages/release/bioc/html/limma.html) as described previously (26).
ChIP-Seq data analysis
The Bowtie software program was used to map ChIP-Seq reads to the reference mouse mm9 genome as described previously (27). The Dregion function in DANPOS2 (https://sites.google.com/site/danposdoc/) was used to calculate reads densities and call peaks as described previously (28). The length of each read was extended to the nucleosome size (200 bp). The reads count for each sample was normalized to 10 million. ChIP-Seq peaks were defined using a Poisson test P-value of 1e-10. The Profile function in DANPOS2 was used to calculate the heatmap and average density plot values. The GEO accession number for ChIP-Seq data is GSE76692.
Statistical analysis
Each experiment was repeated at least three times. Data are presented as the mean ± standard error of the mean (SEM). Statistical significance was assessed using the two-tailed Student t-test. *P < 0.05, **P < 0.01 and ***P < 0.001 indicate statistically significant differences. The Prism software program (GraphPad) was used for statistical analyses.
RESULTS
UTX loss inhibits RA-driven differentiation of mouse ESCs
To assess the role of UTX in the resolution of bivalent genes during mouse ESC differentiation, we employed WT V6.5 (Utx+/y) and Utx-null V6.5 (UtxΔ/y) mouse ESCs, both of which are pluripotent as described previously (29). We used RA to induce cellular differentiation because RA is frequently used to stimulate the neuronal lineage commitment of mouse ESCs (30–33). It was reported that UTX loss has no significant effect on morphological changes during differentiation induced with 1 μM RA (17,34). However, physiological RA concentrations are 2–100 nM (35,36). For example, mouse embryo has an RA concentration of about 25 nM (37). Therefore, although 1 μM RA has been often used for mouse ESC differentiation, 1 μM RA may be too high to assess the effect of UTX loss on mouse ESC differentiation. For this reason, we used three different RA concentrations (0.2 μM, 0.5 μM and 1 μM) to induce the differentiation of EBs derived from WT and Utx-null V6.5 mouse ESCs.
Our results showed that the degree of differentiation between WT and Utx-null V6.5 mouse ESCs at RA concentrations of 1 or 0.5 μM was not obviously different (Figure 1A), consistent with the previous reports (16,34). However, at a RA concentration of 0.2 μM, UTX loss inhibited the RA-driven differentiation of mouse ESCs (Figure 1A). Importantly, such an inhibitory effect is in agreement with the complete embryonic lethality of Utx-deficient female mice and the high percentage of lethality of Utx-deficient male mice (16). Similar to our results, deficiency of the histone acetyltransferase MOZ, a physiological activator of Hox genes, interferes with RA-driven Hox gene expression in mouse ESCs at a low RA level (20 nM) but not high RA concentrations (0.5 μM and 1 μM) (38). Therefore, we chose 0.2 μM RA to differentiate mouse ESCs in all the subsequent experiments.
Figure 1.

UTX loss impedes RA-driven mouse ESC differentiation (A) Microscopy images of WT and Utx-null V6.5 mouse ESCs during RA-induced differentiation. WT or Utx-null cells were plated on petri dishes and cultured to form EBs by withdrawing LIF for 5 days. EBs were treated with all-trans RA (0.2 μM, 0.5 μM or 1 μM) to induce differentiation for 5 days. AP, alkaline phosphatase; Scale bar, 100 μm. (B) Quantitative RT-PCR analysis showed that UTX loss had no significant effect on the RA-induced reduction of Nanog, Oct4 and Sox2 mRNA levels. (C) Quantitative RT-PCR analysis showed that UTX loss had a negative effect on the RA-induced increases of Notch1, Pax6 and Pax3 mRNA levels. Data are presented as the mean ± SEM (error bars) of at least three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 indicate statistically significant changes.
UTX loss in mouse ESCs did not affect RA-induced decreases in the expression of several major pluripotent genes (Oct4, Nanog and Sox2) but reduced the RA-driven activation of some ectoderm marker genes (e.g. Notch1 and Pax6) (Figure 1B and C). UTX loss also decreased the RA-driven activation of the mesoderm marker gene Pax3 with increased basal expression in RA-untreated Utx-null cells (Figure 1C). These results suggest that UTX promotes ectoderm and mesoderm lineage during RA-driven differentiation, consistent with previous reports (34,39).
UTX is necessary for the RA-induced resolution and activation of many RA-inducible bivalent genes in mouse ESCs
To assess the effect of UTX loss on bivalent gene expression in mouse ESCs at the genome-wide level, we first identified all bivalent genes using ChIP-Seq experiments. Most bivalent promoters identified using our ChIP-Seq data highly overlapped with those from other publicly available data set (Supplemental Figure S1) (3). Our genome-wide expression analysis demonstrated that in mouse WT ESCs, RA treatment significantly induced at least by 1.5-fold expression of 637 bivalent genes (hereafter referred to as ‘RA-inducible bivalent genes’), including most Hoxa-d cluster genes (Figure 2A). This number of RA-inducible bivalent genes was close to 772 RA-inducible bivalent genes reported by Mohn et al. (12). Consistent with previous reports (4–5,12), our gene ontology analysis showed that these genes were linked to several important biological processes, including body patterning, cellular differentiation and developmental processes (Figure 2A; Supplementary Table S2).
Figure 2.

UTX loss inhibits RA-mediated activation of many RA-inducible bivalent genes. (A) Gene ontology analysis of 637 RA-inducible bivalent genes using the program DAVID. RA-inducible bivalent genes were genes whose expression levels were at least 1.5-fold and significantly increased by RA treatment (i.e. WT [RA] / WT ≥ 1.5). The mRNA levels were measured using the expression microarray Mouse WG-6 v2 Expression Bead Chip. Statistical significance was determined by a Q value (< 0.05). (B) Scatter plots of RA-induced fold changes (log2 scale) in gene expression in WT and Utx-null mouse ESCs. Each dot in plots represents an individual gene. Correlation coefficients were calculated in the log2 scale. (C) Gene ontology analysis of RA-inducible genes (316) that were significantly downregulated by UTX loss using the program DAVID. RA-inducible bivalent genes downregulated by UTX loss were genes whose RA-induced increases in gene expression were significantly reduced in Utx-null cells compared to WT cells (i.e. Utx-null [RA] / WT [RA] < 1). Statistical significance was determined by a Q value (< 0.05).
We then compared gene expression between WT and Utx-null V6.5 mouse ESCs. UTX loss caused more changes in the expression levels of RA-inducible bivalent genes than in those of non-bivalent genes or the rest of bivalent genes—the rest of bivalent genes denotes bivalent genes that are not RA-inducible bivalent genes (Figure 2B). In particular, UTX loss significantly decreased the expression levels of 316 RA-inducible genes. These genes, which include many Hoxa-d cluster genes, were also associated with body patterning, differentiation and developmental processes (Figure 2C; Supplementary Tables S3 and S4). These results indicate that UTX regulates a substantial number of RA-inducible bivalent genes but not the rest of bivalent genes or non-bivalent genes during RA-driven differentiation of mouse ESCs.
To assess the overall effect of UTX loss on the resolution of all RA-inducible bivalent genes, we compared the ChIP-Seq signals of H3K27me3 and H3K4me3 between WT and Utx-null V6.5 mouse ESCs treated with or without RA. In WT cells, RA treatment decreased average H3K27me3 levels and slightly increased average H3K4me3 levels in RA-inducible bivalent genes, but had negligible effects on average H3K27me3 and H3K4me3 levels in the rest of bivalent genes and non-bivalent genes (Figure 3A and B). Interestingly, UTX loss impeded both RA-induced decreases of average H3K27me3 levels and RA-induced increases of average H3K4me3 levels in RA-inducible bivalent genes (Figure 3C and D). The effects of UTX loss on the RA-induced changes of average H3K27me3 and H3K4me3 levels were similar between 637 RA-inducible bivalent genes and 316 UTX-loss-downregulated, RA-inducible bivalent genes, suggesting that UTX-mediated modification of bivalency occurs mainly in these 316 genes (Figure 3E). In the absence of RA, average H3K27me3 levels of both RA-inducible bivalent genes and the rest of bivalent genes were lower in Utx-null mouse ESCs than in WT V6.5 mouse ESCs (Supplementary Figure S2A and B). However, the overall expression levels of RA-inducible bivalent genes and the rest of bivalent genes between WT and Utx-null cells were not obviously changed, suggesting that reduced H3K27me3 levels in Utx-null cells did not increase expression levels (Supplementary Figure S3A–C). Decreased H3K27me3 levels in Utx-null mouse ESCs might result from reduced levels of the H3K27 methyltransferase Ezh1 (Supplementary Figure S3D). Nevertheless, these results indicate that UTX is essential for the resolution and activation of RA-inducible bivalent genes during RA-driven differentiation of mouse ESCs.
Figure 3.

UTX resolves the bivalency of many RA-inducible bivalent genes during RA-driven differentiation of mouse ESCs. (A and C) Comparison of average ChIP-Seq reads densities for H3K4me3 and H3K27me3 between RA-treated and untreated WT cells (A) and between RA-treated and untreated Utx-null cells (C). The average values of three biological replicates of the 637 RA-inducible bivalent genes (top), the rest of bivalent genes (middle) and non-bivalent genes (bottom) were plotted from -5K to 5K around the transcription start site (TSS). (B and D) ChIP-Seq enrichment profiles for H3K4me3 (first column) and H3K27me3 (second column) levels and for RA-induced changes in H3K4me3 (third column) and H3K27me3 (fourth column) levels in WT (B) and Utx-null (D) mouse ESCs. (E) Comparison of ChIP-Seq reads densities for H3K4me3 and H3K27me3 between RA-treated and untreated WT cells and between RA-treated and untreated Utx-null cells. The 316 RA-inducible bivalent genes downregulated by UTX loss were used.
UTX mediates the RA-induced resolution and activation of most bivalent Hoxa–d genes in mouse ESCs
Several reports have reproducibly shown that most Hoxa–d cluster genes have RA-inducible bivalent promoters (2–6). Therefore, we chose the Hoxa–d cluster genes to determine the effect of UTX loss on bivalency resolution at specific promoters during RA-induced differentiation of mouse ESCs. We compared H3K4me3 and H3K27me3 ChIP-Seq peaks between WT and Utx-null V6.5 mouse ESCs in the presence or absence of RA. For the majority of Hoxa–d cluster genes in RA-treated (i.e. differentiated) WT cells, the H3K27me3 peaks were obviously reduced, and the H3K4me3 peaks were slightly enhanced (Figure 4A and B). However, this bivalency resolution was impeded by UTX loss. Consistent with these ChIP-Seq results, the expression of the majority of the Hoxa–d cluster genes was at least 4-fold RA-induced in WT mouse ESCs but were much less RA-induced in Utx-null mouse ESCs (Figure 4C and D). For example, the expression of Hoxa2, a3, a4, a5, a6, a7, a9, b1, b2, b4, b5, b6, b8 and b9 among Hoxa and Hoxb cluster genes was highly RA-induced, and these RA-inducible genes had reduced H3K27me3 levels and slightly augmented H3K4me3 levels during the RA-driven differentiation. UTX loss drastically downregulated the RA-driven activation of these RA-inducible genes and inhibited their bivalency resolution (Figure 4A and C).
Figure 4.

UTX loss blocks the RA-induced bivalency resolution and activation of most bivalent Hoxa and Hoxb genes in mouse ESCs. (A and B) ChIP-Seq density profiles for H3K27me3 (red) and H3K4me3 (blue) at the mouse Hoxa/Hoxb (A) and Hoxc/Hoxd (B) cluster genes. H3K4me3 and H3K27me3 levels at the Hoxa–d promoters in WT, RA-induced WT, Utx-null and RA-induced Utx-null V6.5 mouse ESCs were analyzed using ChIP-Seq. The vertical axis shows ChIP-Seq signals at a maximal value of 100. (C and D) Effect of UTX knockdown on RA-induced changes in expression levels of Hoxa/Hoxb (C) and Hoxc/Hoxd (D) cluster genes. mRNA levels of Hoxa–d cluster genes in WT, RA-induced WT, Utx-null and RA-induced Utx-null V6.5 mouse ESCs were analyzed using quantitative RT-PCR. Cells were treated with 0.2 μM RA. Data are presented as the mean ± SEM (error bars) of at least three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 indicate statistically significant changes.
To confirm the above ChIP-Seq results, we assessed the effect of UTX loss on H3K27me3 and H3K4me3 levels at 4 RA-inducible bivalent genes (Hoxa3, Hoxb2, Hoxc4 and Hoxd4) and 4 RA-non-inducible bivalent genes (Hoxa10, Hoxb13, Hoxc10 and Hoxd10) by performing quantitative ChIP experiments. UTX loss in mouse ESCs significantly increased H3K27me3 levels at the 4 RA-inducible bivalent Hox genes but not the 4 RA-non-inducible bivalent genes (Figure 5A–H). In contrast, UTX loss in mouse ESCs significantly decreased H3K4me3 levels at 2 RA-inducible bivalent Hox genes (Hoxa3 and Hoxb2) (Figure 5A and B) but did not affect those at the other two bivalent Hox genes (Hoxc4 and Hoxd4) and the 4 RA-non-inducible bivalent genes (Figure 5C–H). These ChIP results are consistent with the aforementioned ChIP-Seq results.
Figure 5.

UTX loss interferes with the RA-induced bivalency resolution in the RA-inducible bivalent genes Hoxa3, Hoxb2, Hoxc4 and Hoxd4 in mouse ESCs. Quantitative ChIP analysis was performed to analyze the effect of UTX loss on the RA-induced changes of H3K4me3, H3K27me3 and H3 levels at the Hoxa3 (A), Hoxb2 (B), Hoxc4 (C), Hoxd4 (D), Hoxa10 (E), Hoxb13 (F), Hoxc10 (G) and Hoxd10 (H) in RA-untreated and RA-treated WT V6.5 mouse ESCs as well as RA-untreated and RA-treated Utx-null V6.5 mouse ESCs. Cells were treated with 0.2 μM RA for 0 or 5 days.
Taken together, these results indicate that UTX is necessary for the resolution and activation of most RA-inducible bivalent Hox genes during the RA-driven differentiation of mouse ESCs.
UTX is required for the RA-induced resolution and activation of most bivalent HOXA–D genes in human NT2/D1 cells
We previously showed that UTX occupies HOXA1–3 and HOXB1–3 during RA-driven differentiation in human NT2/D1 cells (15), a well-studied neuron-committed cell line that undergoes neuronal differentiation upon RA treatment (40,41). Thus, we sought to determine whether UTX is also required for the resolution of bivalent HOXA–D cluster genes in NT2/D1 cells. Our ChIP-Seq results showed that of 39 HOX cluster genes, 38, except HOXA6, had bivalent domains in NT2/D1 cells (Figure 6A; Supplementary Figure S4A and B). Then, we determined the effect of UTX knockdown on H3K4me3 and H3K27me3 levels in bivalent HOX genes (Supplementary Figure S4C and D). Most bivalent HOX genes were resolved during the RA-induced differentiation of NT2/D1 cells, but their resolution was blocked by UTX knockdown (Figure 6B; Supplementary Figure S5A). In line with this, UTX knockdown drastically inhibited the RA-driven activation of most RA-inducible HOXA–D cluster genes (Figure 6C; Supplementary Figure S5B). In addition, UTX knockdown impeded the RA-induced neuronal differentiation of NT2/D1 cells and the RA-induced expression of the neuron-related genes NESTIN and N-OCT3 in NT2/D1 cells (Figure 6D and E). To assess the importance of UTX's catalytic activity during the RA-induced differentiation of NT2/D1 cells and the targeting specificity of shUTXs, we performed a rescue experiment for UTX-depleted NT2/D1 cells. As shown in Supplementary Figure S6A–F, ectopic expression of WT UTX but not its catalytic mutant significantly restored differentiation morphologies and gene expression (HOXA1, HOXA2, NESTIN and N-OCT3) during RA-induced differentiation of UTX-depleted NT2/D1 cells. These results indicate that UTX and its catalytic activity are essential for the resolution and activation of most RA-inducible bivalent HOXA–D genes in NT2/D1 cells.
Figure 6.
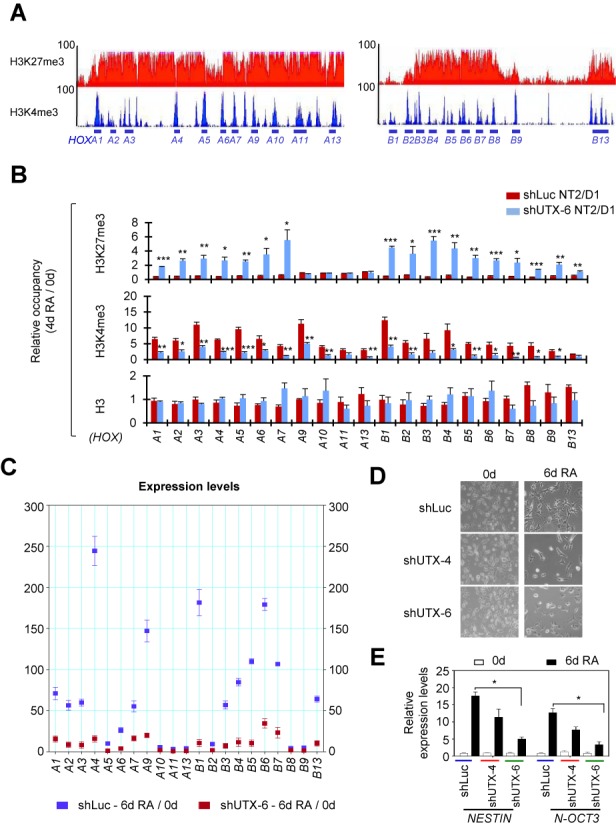
During RA-driven differentiation of human NT2/D1 cells, UTX is responsible for the bivalency resolution and activation of most RA-inducible bivalent HOXA and HOXB genes. (A) ChIP-Seq density profiles of H3K27me3 (red) and H3K4me3 (blue) at the human HOXA and HOXB cluster genes in NT2/D1 cells. The vertical axis shows the coverage for histone methylation set at a maximal value of 100. (B) Effect of UTX knockdown on the RA-induced changes in H3K27me3, H3K4me3 and H3 levels at the HOXA and HOXB cluster promoters in NT2/D1 cells. shLuc- or shUTX-6-treated cells were cultured in media containing 10 μM RA for 0 or 4 days. Chromatin levels for histone marks were analyzed using a quantitative ChIP assay. PCR values for each time point were normalized to input. The relative occupancy represents the fold change in chromatin levels from day 0 to day 4 (4d/0d). (C) Effect of UTX knockdown on the RA-induced changes in the expression levels of HOXA and HOXB cluster genes. shLuc- or shUTX-6-treated NT2/D1 cells were incubated with 10 μM RA for 0 or 6 days. mRNA levels were analyzed using quantitative RT-PCR. Data are presented as the mean ± SEM (error bars) of least three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 indicate statistically significant changes. (D and E) Effect of UTX knockdown on morphological changes (D) and activation of NESTIN and N-OCT3 gene (E) during the RA-induced differentiation of NT2/D1 cells. UTX was depleted using shUTX-4 and shUTX-6 in NT2/D1 cells. Cells (2–5 × 104) were seeded and treated with 10 μM RA for 0 or 6 days. Cell differentiation patterns were monitored using a microscope. Representative microscopy images of three independent experiments are shown (magnification 40X). Expression levels of NESTIN and N-OCT3 gene were measured by quantitative RT-PCR.
UTX is recruited to most RA-inducible bivalent Hox genes during RA-induced differentiation of mouse ESCs and human NT2/D1 cells
As described above, UTX loss in mouse ESCs highly downregulated the RA-driven activation of most RA-inducible bivalent Hox genes. In the cases of the Hoxa and Hoxb cluster genes, the expression of all RA-inducible bivalent genes (i.e. Hoxa2, a3, a4, a5, a6, a7, a9, b1, b2, b4, b5, b6, b8 and b9) was drastically downregulated by UTX loss, whereas expression of RA-non-inducible bivalent genes (Hoxa1, a10, a11, a13, b3, b7 and b13) was not or only weakly downregulated by UTX loss (Figure 4C). Similarly, UTX knockdown downregulated most RA-inducible HOX genes but did not downregulate RA-non-inducible HOX genes in human NT2/D1 cells (Figure 6C). For instance, among human HOXA and HOXB cluster genes, HOXA1, A2, A3, A4, A6, A7, A9, B1, B3, B4, B5, B6, B7 and B13 (but not HOXA5, A10, A11, A13, B2, B8 and B9) were downregulated by UTX depletion.
To understand how UTX selectively activates certain Hox genes, we determined to which Hox genes UTX is recruited during the RA-driven differentiation of both mouse ESCs and human NT2/D1 using quantitative ChIP assay. Our results showed that UTX was recruited to most RA-inducible Hoxa–d genes (e.g. Hoxa2, a3, a4, a5, a6, a7, a9, b2, b4, b5, b6, b8 and b9) during RA-induced differentiation of mouse ESCs (Figure 7A). In addition, UTX occupancy was increased at other 24 RA-inducible bivalent genes, including pattern specification and organ morphogenesis genes (Bmi1, Fgfr2, Irx5, Fst, Kif3a and Foxc2), developmental genes (Wnt4, Wnt6 and Robo3), neuronal genes (Gria2), transcription factor genes (Gata3 and Nr2f2) and homeobox protein genes (Meis1, Onecut2 and Irx5) (Figure 7B). In contrast, UTX occupancy was not changed at RA-non-inducible Hox genes (e.g. Hoxa1, a10, a11, a13, b7 and b13) (Figure 7A). Similarly, UTX was recruited to most RA-inducible HOX genes during RA-driven differentiation of NT2/D1 cells (Figure 8A). These results indicate that the recruitment of UTX to specific RA-inducible bivalent genes, including Hox genes, is primarily responsible for the selective activation and resolution of such genes.
Figure 7.

UTX recruitment is associated with bivalency resolution in most RA-inducible bivalent Hox genes during RA-driven differentiation of mouse ESCs. (A and B) Quantitative ChIP analysis of the promoter occupancy of UTX and H3 at the Hoxa–d cluster genes (A) and at other 24 RA-inducible bivalent genes (B) in RA-untreated and RA-treated WT V6.5 mouse ESCs. Cells were treated with 0.2 μM RA for 0 or 5 days.
Figure 8.

UTX recruitment is linked to bivalency resolution in most RA-inducible bivalent HOX genes during RA-driven differentiation of human NT2/D1 cells. (A) Quantitative ChIP analysis of the promoter occupancy of UTX and H3 at the HOXA–D cluster genes in RA-untreated and RA-treated WT NT2/D1 cells. NT2/D1 cells were treated with 10 μM RA for 0 or 4 days. The relative occupancy is the fold change in chromatin levels at the day 4 over day 0. PCR values for each time point were normalized to input. Data are presented as the mean ± SEM (error bars) of least three independent experiments. *P < 0.05 and **P < 0.01 indicate statistically significant changes. (B) A hypothetical model of UTX as a bivalency-resolving demethylase necessary for RA-induced differentiation of mouse ESC cells.
DISCUSSION
In the present study, we show that UTX plays an essential role in resolving and activating many RA-inducible bivalent genes during the RA-driven differentiation of mouse ESCs treated with a physiologically relevant RA concentration (0.2 μM) (Figure 8B). Moreover, we demonstrated that the UTX-mediated resolution and activation of many RA-inducible bivalent HOX genes occur during RA-driven differentiation of human NT2/D1 cells. Furthermore, we showed that UTX loss and UTX knockdown interfered with the RA-induced differentiation of mouse ESCs and human NT2/D1 cells, respectively. Therefore, our findings indicate that the UTX-mediated resolution and activation of many RA-inducible bivalent genes, including numerous Hoxa–d cluster genes, are required for RA-driven differentiation of mouse ESCs and human NT2/D1 stem cells. These results are in line with our recent study showing that UTX recruitment at p53/OCT4/NANOG target genes, such as bivalent HOXA1 and HOXA2 promoters, is accompanied by gene activation and H3K27me3 reduction during the RA-induced differentiation of human ESCs (42).
Consistent with our findings showing that UTX is necessary for bivalency resolution and proper cellular differentiation, UTX is critical for developmental and aging processes, including sex-specific embryogenesis (16,43), cardio-genesis (17), muscle development (18) and aging (19). For example, Utx-deficient female mice show the complete embryonic lethality, and Utx-deficient male mice display the high percentage of lethality (16), as aforementioned. In addition, UTX is mutated in genetic diseases and multiple types of cancer (44). For instance, haploinsufficiency of UTX, which often results from nonsense and frameshift mutations of UTX, is associated with Kabuki syndrome, a rare congenital syndrome with a multiple malformation disorder and mental retardation (45,46).
During bivalency resolution, H3K27me3 must be removed by an H3K27 demethylase. In addition to UTX, H3K27 demethylases include JMJD3 and UTY (15,47–51). Although it was initially documented that UTY may not have a demethylase function (43), it was later reported that UTY may have a weak H3K27 demethylation activity (52). However, UTY may not be a general bivalency-resolving H3K27 demethylase because it is a male-specific protein whose encoding gene is on the Y chromosome. Similar to UTX, JMJD3 is recruited to specific developmental genes during differentiation (53). Therefore, JMJD3 can be considered to be a candidate to demethylate H3K27me3 in bivalent promoters. Given the notion that UTX loss did not affect the RA-induced activation of bivalent genes in mouse ESCs at a high RA concentration (i.e. 1 μM) (16), it can be speculated that at such a high RA concentration, JMJD3 may be recruited to bivalent genes and compensate for the effects of UTX loss on the RA-induced activation of bivalent genes. However, the RA-induced resolution and activation of most bivalent Hoxa–d genes still occur in UTX- and JMJD3-null mouse female ESCs treated with 1 μM RA (54). Because the known H3K27 demethylases UTX, JMJD3 and UTY are deficient in these cells, it is possible that the resolution and activation of most bivalent Hoxa–d genes at a high RA concentration are mediated by a different mechanism, including a yet-to-be identified H3K27 demethylase that is also suggested by Shpargel et al. (54).
We also found that H3K4me3 levels were slightly increased during the bivalency resolution of RA-inducible bivalent genes. We and others previously provided evidence that UTX and the H3K4 methyltransferase MLL4 (also known as KMT2D and ALR) form a stable multi-protein complex (15,55–56). Our additional study showed that UTX interacts with a C-terminal region of MLL4 (57). We have also shown that MLL4 can catalyze mono-, di- or trimethylation at H3K4 (20), although MLL4 may preferably monomethylate and dimethylate H3K4 (58,59). Interestingly, MLL4 in the presence of its cofactors, such as WDR5, RbBP5, Ash2L and Dpy-30, trimethylates H3K4 (60). Thus, we speculate that during bivalency resolution, MLL4 may generate H3K4me3 while UTX demethylates H3K27me3.
Recently, Marks et al. reported that mouse ESCs maintained in two kinase inhibitors (2i)/LIF-containing media in the absence of serum had about 1000 bivalent genes that are substantially fewer than the 3000–4000 bivalent genes initially reported (6). This difference results from reduced H3K27me3 levels in 2i/LIF-cultured mouse ESCs. Thus, the role of bivalency in pluripotent cells and their differentiation is somewhat controversial (10,61). However, some have argued that because many bivalent genes’ H3K27me3 signals, although higher than those of active genes, fall below the computational threshold, such bivalent genes might be categorized as monovalent genes in 2i/LIF-cultured mouse ESCs (10). In addition, although there may be fewer bivalent promoters than initially reported, numerous critical differentiation-specific genes, including most Hoxa–d cluster genes, still belong to RA-inducible bivalent genes even in 2i/LIF-cultured mouse ESCs (6). Notably, our results indicate that UTX plays a crucial role in the bivalency resolution and activation of most RA-inducible bivalent Hox genes during the RA-driven cellular differentiation. Moreover, UTX-mediated resolution of bivalency, although less critical for early embryo development (54), may be important for other stages of animal development, because many UTX-resolved bivalent genes, including the Hoxa–d cluster genes (62,63), are necessary for proper animal development (Figure 2C). Consistent with this, Utx knockout resulted in developmental defects, such as embryonic lethality (16) and defective cardiogenesis (17). Therefore, we are in favor of the notion that bivalency and its resolution are critical for stem cell differentiation and animal development.
Supplementary Material
Acknowledgments
We thank Dr Zhenbo Han and Mr. Su Zhang for their technical assistance and Joseph Munch for manuscript editing.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute of Health (NIH) [R01 GM095659, R01CA157919 to M.G.L.]; Cancer Prevention and Research Institute of Texas (CPRIT) [RP110183 to M.G.L.]; Center for Cancer Epigenetics at MD Anderson Cancer Center [to M.G.L.]; Center for Cancer Epigenetics at MD Anderson Cancer Center [postdoctoral fellowship to S.S.D.]; NIH [R01 HG007538 to W.L.]; CPRIT [RP150292 to W.L.]; NIH [R01 ES025761 to Z.W.]. Funding for open access charge: NIH [R01 GM095659, R01 CA157919].
Conflict of interest statement. None declared.
REFERENCES
- 1.Voigt P., LeRoy G., Drury W.J. 3rd, Zee B.M., Son J., Beck D.B., Young N.L., Garcia B.A., Reinberg D. Asymmetrically modified nucleosomes. Cell. 2012;151:181–193. doi: 10.1016/j.cell.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bernstein B.E., Mikkelsen T.S., Xie X., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K., et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 3.Mikkelsen T.S., Ku M., Jaffe D.B., Issac B., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T.K., Koche R.P., et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pan G., Tian S., Nie J., Yang C., Ruotti V., Wei H., Jonsdottir G.A., Stewart R., Thomson J.A. Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell. 2007;1:299–312. doi: 10.1016/j.stem.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 5.Zhao X.D., Han X., Chew J.L., Liu J., Chiu K.P., Choo A., Orlov Y.L., Sung W.K., Shahab A., Kuznetsov V.A., et al. Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell. 2007;1:286–298. doi: 10.1016/j.stem.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 6.Marks H., Kalkan T., Menafra R., Denissov S., Jones K., Hofemeister H., Nichols J., Kranz A., Stewart A.F., Smith A., et al. The transcriptional and epigenomic foundations of ground state pluripotency. Cell. 2012;149:590–604. doi: 10.1016/j.cell.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mikkelsen T.S., Hanna J., Zhang X., Ku M., Wernig M., Schorderet P., Bernstein B.E., Jaenisch R., Lander E.S., Meissner A. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cui K., Zang C., Roh T.Y., Schones D.E., Childs R.W., Peng W., Zhao K. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell. 2009;4:80–93. doi: 10.1016/j.stem.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malouf G.G., Taube J.H., Lu Y., Roysarkar T., Panjarian S., Estecio M.R., Jelinek J., Yamazaki J., Raynal N.J., Long H. Architecture of epigenetic reprogramming following Twist1-mediated epithelial-mesenchymal transition. Genome Biol. 2013;14:R144–R161. doi: 10.1186/gb-2013-14-12-r144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Voigt P., Tee W.W., Reinberg D. A double take on bivalent promoters. Genes Dev. 2013;27:1318–1338. doi: 10.1101/gad.219626.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rugg-Gunn P.J., Cox B.J., Ralston A., Rossant J. Distinct histone modifications in stem cell lines and tissue lineages from the early mouse embryo. Proc. Natl. Acad. Sci. U.S.A. 2010;107:10783–10790. doi: 10.1073/pnas.0914507107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohn F., Weber M., Rebhan M., Roloff T.C., Richter J., Stadler M.B., Bibel M., Schubeler D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell. 2008;30:755–766. doi: 10.1016/j.molcel.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 13.Hu D., Garruss A.S., Gao X., Morgan M.A., Cook M., Smith E.R., Shilatifard A. The Mll2 branch of the COMPASS family regulates bivalent promoters in mouse embryonic stem cells. Nat. Struct. Mol. Biol. 2013;20:1093–1097. doi: 10.1038/nsmb.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Denissov S., Hofemeister H., Marks H., Kranz A., Ciotta G., Singh S., Anastassiadis K., Stunnenberg H.G., Stewart A.F. Mll2 is required for H3K4 trimethylation on bivalent promoters in embryonic stem cells, whereas Mll1 is redundant. Development. 2014;141:526–537. doi: 10.1242/dev.102681. [DOI] [PubMed] [Google Scholar]
- 15.Lee M.G., Villa R., Trojer P., Norman J., Yan K.P., Reinberg D., Di Croce L., Shiekhattar R. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science. 2007;318:447–450. doi: 10.1126/science.1149042. [DOI] [PubMed] [Google Scholar]
- 16.Welstead G.G., Creyghton M.P., Bilodeau S., Cheng A.W., Markoulaki S., Young R.A., Jaenisch R. X-linked H3K27me3 demethylase Utx is required for embryonic development in a sex-specific manner. Proc. Natl. Acad. Sci. U.S.A. 2012;109:13004–13009. doi: 10.1073/pnas.1210787109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee S., Lee J.W., Lee S.K. UTX, a histone H3-lysine 27 demethylase, acts as a critical switch to activate the cardiac developmental program. Dev. Cell. 2012;22:25–37. doi: 10.1016/j.devcel.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seenundun S., Rampalli S., Liu Q.C., Aziz A., Palii C., Hong S., Blais A., Brand M., Ge K., Dilworth F.J. UTX mediates demethylation of H3K27me3 at muscle-specific genes during myogenesis. EMBO J. 2010;29:1401–1411. doi: 10.1038/emboj.2010.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin C., Li J., Green C.D., Yu X., Tang X., Han D., Xian B., Wang D., Huang X., Cao X., et al. Histone demethylase UTX-1 regulates C. elegans life span by targeting the insulin/IGF-1 signaling pathway. Cell Metab. 2011;14:161–172. doi: 10.1016/j.cmet.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 20.Dhar S.S., Lee S.H., Kan P.Y., Voigt P., Ma L., Shi X., Reinberg D., Lee M.G. Trans-tail regulation of MLL4-catalyzed H3K4 methylation by H4R3 symmetric dimethylation is mediated by a tandem PHD of MLL4. Genes Dev. 2012;26:2749–2762. doi: 10.1101/gad.203356.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dennis G., Jr, Sherman B.T., Hosack D.A., Yang J., Gao W., Lane H.C., Lempicki R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:R60.1–R60.11. [PubMed] [Google Scholar]
- 22.Jiao X., Sherman B.T., Huang da W., Stephens R., Baseler M.W., Lane H.C., Lempicki R.A. DAVID-WS: a stateful web service to facilitate gene/protein list analysis. Bioinformatics. 2012;28:1805–1806. doi: 10.1093/bioinformatics/bts251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagner K.W., Alam H., Dhar S.S., Giri U., Li N., Wei Y., Giri D., Cascone T., Kim J.H., Ye Y., et al. KDM2A promotes lung tumorigenesis by epigenetically enhancing ERK1/2 signaling. J. Clin. Invest. 2013;123:5231–5246. doi: 10.1172/JCI68642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z., Zang C., Cui K., Schones D.E., Barski A., Peng W., Zhao K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–1031. doi: 10.1016/j.cell.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Z., Zang C., Rosenfeld J.A., Schones D.E., Barski A., Cuddapah S., Cui K., Roh T.Y., Peng W., Zhang M.Q., Zhao K. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25–R35. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen K., Xi Y., Pan X., Li Z., Kaestner K., Tyler J., Dent S., He X., Li W. DANPOS: dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res. 2013;23:341–351. doi: 10.1101/gr.142067.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mansour A.A., Gafni O., Weinberger L., Zviran A., Ayyash M., Rais Y., Krupalnik V., Zerbib M., Amann-Zalcenstein D., Maza I., et al. The H3K27 demethylase Utx regulates somatic and germ cell epigenetic reprogramming. Nature. 2012;488:409–413. doi: 10.1038/nature11272. [DOI] [PubMed] [Google Scholar]
- 30.Wichterle H., Lieberam I., Porter J.A., Jessell T.M. Directed differentiation of embryonic stem cells into motor neurons. Cell. 2002;110:385–397. doi: 10.1016/s0092-8674(02)00835-8. [DOI] [PubMed] [Google Scholar]
- 31.Kim M., Habiba A., Doherty J.M., Mills J.C., Mercer R.W., Huettner J.E. Regulation of mouse embryonic stem cell neural differentiation by retinoic acid. Dev. Biol. 2009;328:456–471. doi: 10.1016/j.ydbio.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bain G., Kitchens D., Yao M., Huettner J.E., Gottlieb D.I. Embryonic stem cells express neuronal properties in vitro. Dev. Biol. 1995;168:342–357. doi: 10.1006/dbio.1995.1085. [DOI] [PubMed] [Google Scholar]
- 33.Li X.J., Hu B.Y., Jones S.A., Zhang Y.S., Lavaute T., Du Z.W., Zhang S.C. Directed differentiation of ventral spinal progenitors and motor neurons from human embryonic stem cells by small molecules. Stem Cells. 2008;26:886–893. doi: 10.1634/stemcells.2007-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morales Torres C., Laugesen A., Helin K. Utx is required for proper induction of ectoderm and mesoderm during differentiation of embryonic stem cells. PLoS One. 2013;8:e60020. doi: 10.1371/journal.pone.0060020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kane M.A., Chen N., Sparks S., Napoli J.L. Quantification of endogenous retinoic acid in limited biological samples by LC/MS/MS. Biochem. J. 2005;388:363–369. doi: 10.1042/BJ20041867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kane M.A., Folias A.E., Wang C., Napoli J.L. Quantitative profiling of endogenous retinoic acid in vivo and in vitro by tandem mass spectrometry. Anal. Chem. 2008;80:1702–1708. doi: 10.1021/ac702030f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mic F.A., Molotkov A., Benbrook D.M., Duester G. Retinoid activation of retinoic acid receptor but not retinoid X receptor is sufficient to rescue lethal defect in retinoic acid synthesis. Proc. Natl. Acad. Sci. U.S.A. 2003;100:7135–7140. doi: 10.1073/pnas.1231422100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sheikh B.N., Downer N.L., Kueh A.J., Thomas T., Voss A.K. Excessive versus physiologically relevant levels of retinoic acid in embryonic stem cell differentiation. Stem Cells. 2014;32:1451–1458. doi: 10.1002/stem.1604. [DOI] [PubMed] [Google Scholar]
- 39.Wang C., Lee J.E., Cho Y.W., Xiao Y., Jin Q., Liu C., Ge K. UTX regulates mesoderm differentiation of embryonic stem cells independent of H3K27 demethylase activity. Proc. Natl. Acad. Sci. U.S.A. 2012;109:15324–15329. doi: 10.1073/pnas.1204166109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Houldsworth J., Heath S.C., Bosl G.J., Studer L., Chaganti R.S. Expression profiling of lineage differentiation in pluripotential human embryonal carcinoma cells. Cell Growth Differ. 2002;13:257–264. [PubMed] [Google Scholar]
- 41.Simeone A., Acampora D., Arcioni L., Andrews P.W., Boncinelli E., Mavilio F. Sequential activation of HOX2 homeobox genes by retinoic acid in human embryonal carcinoma cells. Nature. 1990;346:763–766. doi: 10.1038/346763a0. [DOI] [PubMed] [Google Scholar]
- 42.Akdemir K.C., Jain A.K., Allton K., Aronow B., Xu X., Cooney A.J., Li W., Barton M.C. Genome-wide profiling reveals stimulus-specific functions of p53 during differentiation and DNA damage of human embryonic stem cells. Nucleic Acids Res. 2014;42:205–223. doi: 10.1093/nar/gkt866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shpargel K.B., Sengoku T., Yokoyama S., Magnuson T. UTX and UTY demonstrate histone demethylase-independent function in mouse embryonic development. PLoS Genet. 2012;8:e1002964. doi: 10.1371/journal.pgen.1002964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suva M.L., Riggi N., Bernstein B.E. Epigenetic reprogramming in cancer. Science. 2013;339:1567–1570. doi: 10.1126/science.1230184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miyake N., Mizuno S., Okamoto N., Ohashi H., Shiina M., Ogata K., Tsurusaki Y., Nakashima M., Saitsu H., Niikawa N., et al. KDM6A point mutations cause Kabuki syndrome. Hum. Mutat. 2013;34:108–110. doi: 10.1002/humu.22229. [DOI] [PubMed] [Google Scholar]
- 46.Miyake N., Koshimizu E., Okamoto N., Mizuno S., Ogata T., Nagai T., Kosho T., Ohashi H., Kato M., Sasaki G., et al. MLL2 and KDM6A mutations in patients with Kabuki syndrome. Am. J. Med. Genet. A. 2013;161A:2234–2243. doi: 10.1002/ajmg.a.36072. [DOI] [PubMed] [Google Scholar]
- 47.De Santa F., Totaro M.G., Prosperini E., Notarbartolo S., Testa G., Natoli G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell. 2007;130:1083–1094. doi: 10.1016/j.cell.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 48.Hong S., Cho Y.W., Yu L.R., Yu H., Veenstra T.D., Ge K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc. Natl. Acad. Sci. U.S.A. 2007;104:18439–18444. doi: 10.1073/pnas.0707292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agger K., Cloos P.A., Christensen J., Pasini D., Rose S., Rappsilber J., Issaeva I., Canaani E., Salcini A.E., Helin K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007;449:731–734. doi: 10.1038/nature06145. [DOI] [PubMed] [Google Scholar]
- 50.Xiang Y., Zhu Z., Han G., Lin H., Xu L., Chen C.D. JMJD3 is a histone H3K27 demethylase. Cell Res. 2007;17:850–857. doi: 10.1038/cr.2007.83. [DOI] [PubMed] [Google Scholar]
- 51.Lan F., Bayliss P.E., Rinn J.L., Whetstine J.R., Wang J.K., Chen S., Iwase S., Alpatov R., Issaeva I., Canaani E., et al. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature. 2007;449:689–694. doi: 10.1038/nature06192. [DOI] [PubMed] [Google Scholar]
- 52.Walport L.J., Hopkinson R.J., Vollmar M., Madden S.K., Gileadi C., Oppermann U., Schofield C.J., Johansson C. Human UTY(KDM6C) is a male-specific N-methyl lysyl demethylase. J. Biol. Chem. 2014;289:18302–18313. doi: 10.1074/jbc.M114.555052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burgold T., Spreafico F., De Santa F., Totaro M.G., Prosperini E., Natoli G., Testa G. The histone H3 lysine 27-specific demethylase Jmjd3 is required for neural commitment. PLoS One. 2008;3:e3034. doi: 10.1371/journal.pone.0003034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shpargel K.B., Starmer J., Yee D., Pohlers M., Magnuson T. KDM6 demethylase independent loss of histone H3 lysine 27 trimethylation during early embryonic development. PLoS Genet. 2014;10:e1004507. doi: 10.1371/journal.pgen.1004507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Issaeva I., Zonis Y., Rozovskaia T., Orlovsky K., Croce C.M., Nakamura T., Mazo A., Eisenbach L., Canaani E. Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Mol. Cell. Biol. 2007;27:1889–1903. doi: 10.1128/MCB.01506-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cho Y.W., Hong T., Hong S., Guo H., Yu H., Kim D., Guszczynski T., Dressler G.R., Copeland T.D., Kalkum M., et al. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J. Biol. Chem. 2007;282:20395–20406. doi: 10.1074/jbc.M701574200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim J.H., Sharma A., Dhar S.S., Lee S.H., Gu B., Chan C.H., Lin H.K., Lee M.G. UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Res. 2014;74:1705–1717. doi: 10.1158/0008-5472.CAN-13-1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee J.E., Wang C., Xu S., Cho Y.W., Wang L., Feng X., Baldridge A., Sartorelli V., Zhuang L., Peng W., et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife. 2013;2:e01503. doi: 10.7554/eLife.01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hu D., Gao X., Morgan M.A., Herz H.M., Smith E.R., Shilatifard A. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol. Cell. Biol. 2013;33:4745–4754. doi: 10.1128/MCB.01181-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Y., Mittal A., Reid J., Reich S., Gamblin S.J., Wilson J.R. Evolving Catalytic Properties of the MLL Family SET Domain. Structure. 2015;23:1921–1933. doi: 10.1016/j.str.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen T., Dent S.Y. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat. Rev. Genet. 2014;15:93–106. doi: 10.1038/nrg3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mallo M., Alonso C.R. The regulation of Hox gene expression during animal development. Development. 2013;140:3951–3963. doi: 10.1242/dev.068346. [DOI] [PubMed] [Google Scholar]
- 63.Pearson J.C., Lemons D., McGinnis W. Modulating Hox gene functions during animal body patterning. Nat. Rev. Genet. 2005;6:893–904. doi: 10.1038/nrg1726. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.