


Abstract
Enzymatic effectors targeting nucleic acids, proteins and other cellular components are the mainstay of conflicts across life forms. Using comparative genomics we identify a large class of eukaryotic proteins, which include effectors from oomycetes, fungi and other parasites. The majority of these proteins have a characteristic domain architecture with one of several N-terminal ‘Header’ domains, which are predicted to play a role in trafficking of these effectors, including a novel version of the Ubiquitin fold. The Headers are followed by one or more diverse C-terminal domains, such as restriction endonuclease (REase), protein kinase, HNH endonuclease, LK-nuclease (a RNase) and multiple distinct peptidase domains, which are predicted to carry their toxicity determinants. The most common types of these proteins appear to have originated from prokaryotic transposases (e.g. TN7 and Mu) and combine a CDC6/ORC1-STAND clade NTPase domain with a C-terminal REase domain. Other than the so-called Crinkler effectors of oomycetes and fungi, these effectors are encoded by other eukaryotic parasites such as trypanosomatids (the RHS proteins) and the rhizarian Plasmodiophora, and symbionts like Capsaspora. Remarkably, we also find these proteins in free-living eukaryotes, including several viridiplantae, fungi, amoebozoans and animals. These versions might either still be transposons or function in other poorly understood eukaryote-specific inter-organismal and inter-genomic conflicts. These include the Medea1 selfish element of Tribolium that spreads via post-zygotic killing. We present a unified mechanism for the recombination-dependent diversification and action of this widespread class of molecular weaponry deployed across diverse conflicts ranging from parasitic to free-living forms.
INTRODUCTION
Biological systems are locked in conflicts at all levels of their organization (1–4). Such conflicts lie at the heart of interactions ranging from symbiotic mutualism to parasitism or predation. These conflicts span a wide spectrum: those between intra-genomic selfish elements and their host genomes, between multiple genomes in the same cell (e.g. between plasmids and viruses and the cellular genome), and between organisms belonging to the same or different species (1,2,4–6). The ubiquity of conflict in biological systems has selected for a diverse panoply of armaments and defenses, which most commonly manifest at the molecular level as toxins and cognate immunity molecules that neutralize them (1,7).
A dominant theme across all biological conflicts is the use of protein toxins that incapacitate different subcellular machineries (8–10). In the past decade there have been tremendous advances in our understanding in terms of the evolution, structure and function of protein toxins deployed in prokaryotic conflict systems (11–13). One major area of investigation has been the toxin–antitoxin systems, which are intra-genomic selfish elements (14–16). They enforce their maintenance in plasmids and cellular genomes by means of antagonistic interactions between cell-killing toxins and cognate antitoxins that neutralize them (15,17). Another class of toxins and effectors, which have received much attention, are those deployed by bacterial pathogens against their eukaryotic hosts (8,9,18,19). More recently it has become apparent that prokaryotes also deploy protein toxins in intra-specific conflicts with non-kin bacteria (20). These are paralleled by earlier-described proteinic bacteriocins, which are produced by plasmids and target those cells in bacterial population that lack the plasmid (21–23).
Studies on the domain architectures of protein toxins from these prokaryotic conflict systems have revealed several common themes: they often utilize specialized secretory machinery, such as the type-VI (T6SS), type-VII (T7SS) or the so-called PVC systems to specifically deliver the toxin to their target cell (11–13). Another pervasive feature is the enzymatic activity of toxins to which their toxicity is usually entirely attributable. Most frequently they catalyse covalent modifications or cleavage of DNA, RNA, proteins and lipids to either alter their behaviour or render them biologically dysfunctional (8,9,11). Toxins, in particular their catalytic domains, from across these systems rampantly display different forms of polymorphism: they might rapidly diverge in sequence and, in certain cases, structure even between related strains or species (11–13). They might also show evolutionary displacement by structurally divergent and sometimes even catalytically unrelated toxins (11). This phenomenon is particularly striking in a system of toxins deployed primarily in bacterial intra-specific conflicts. Here the C-terminal (usually enzymatic) toxin domains tend to be highly polymorphic while retaining constant N-terminal regions, which contain domains related to secretion, processing and formation of secretion-related superstructures. This polymorphism appears to arise from repeated recombination of the N-terminal constant region with distinct C-terminal toxin-encoding cassettes (12,24). Polymorphism is the hallmark of an arms-race situation in a biological conflict and indicates constant compensatory evolution between the toxin and resistance mechanisms directed against it (3,25).
In contrast to prokaryotic toxins, our understanding of toxins and effectors used by eukaryotic pathogens and those deployed in inter-eukaryotic conflicts is less advanced. Nevertheless, studies indicate that enzymatic effectors, comparable to the bacterial versions, are also used by eukaryotic pathogens. For instance, the apicomplexan parasites, Plasmodium and Toxoplasma, secrete protein kinases into their host cells to alter their behaviour (26–28). Interestingly, these apicomplexan kinases display lineage-specific expansions (LSEs) suggesting certain functional polymorphism and/or diversification to counter evolving host resistance and immunity. Like apicomplexans, pathogenic and mycorrhizal symbiotic fungi (29–31), stramenopile oomycetes (32–34), and the rhizarian Plasmodiophora (35) also display intimate interactions with their eukaryotic hosts, which feature complex but poorly understood biological conflicts involving secretion of numerous effectors, often numbering in the hundreds, into cells of their host. This has been extensively studied in plant pathogenic oomycetes, which cause a characteristic leaf-crinkling phenotype upon establishment of infection (36). This is attributed to the Crinkler (Crn) effectors, which cause cell death and pave the way for the final stage of infection by these pathogens, where they grow on the dead host tissue (necrotrophy) (37). Recently, it was shown that one of the Crinkler effectors possesses a kinase domain, which on being disrupted reduces the cell-death-causing capacity of the effector and its stability in the host cell (32,38). In addition, other enzymatic domains delivered into host cells have been identified in effectors of eukaryotic pathogens, such as a metallopeptidase domain in Magnaporthe oryzae (39) and a Nudix domain in Phytophthora sojae (40).
Crn effectors are of particular interest because they have been distributed by lateral transfer across phylogenetically distant eukaryotes, such as oomycetes and different fungal lineages such as pathogenic chytrids (32,41,42). They have been implicated in destructive oomycete diseases of tomato (36,43,44), potato (potato blight) (32), soybean and forest trees (45) and also chytridiomycosis of frogs caused by Batrachochytrium dendrobatidis (30). The repertoire of Crn genes and pseudogenes is greatly expanded in these organisms (32,36) and their products have been shown to translocate into the host nucleus in a process mediated by their N-terminal region (46). They have also been shown to display both distinct intra-nuclear distribution and levels of cell-death induction (37,45–47). Crn effectors are further recognized to display apparent modularity in their domain structure, and potentially undergo variation via recombination of N- and C-terminal regions (32,37,48). Despite these advances, beyond the report of the kinase domain, there is little objective understanding of the actual domain architectures, potential catalytic diversity of their toxin modules and the relationship of the recombination-driven diversification to their domain architectures.
We had earlier extensively characterized prokaryotic toxins by identifying numerous previously unknown catalytic domains and delivery mechanisms, in addition to clarifying the mechanisms generating polymorphism (11–13). We became interested in the Crn effectors because they showed features paralleling polymorphic prokaryotic effectors in terms of diversification via recombination (48). Given that they are rather poorly understood in terms of their domain composition and provenance we sought to use sensitive sequence analysis, structure comparison and comparative genomics to better understand them. Consequently, we show that Crn effectors are not restricted to oomycetes and pathogenic fungi but are widely distributed across eukaryotes and likely to be used in a variety of conflict systems. We systematically characterize all of the major toxin and delivery-related domains in these systems. Moreover, we also show that their dominant domain architectural theme arose from bacterial transposons, which in turns allows us to explain several of their key evolutionary and functional features.
MATERIALS AND METHODS
We started with an initial sequence library of known Crn homologs extracted from the Genbank Non-Redundant (NR) protein database. Upon identification of new homologs these were then integrated into the initial library for further large-scale sequence analysis as described below. We iterated this procedure for several rounds, and eventually generated an exhaustive collection of CRN homologs and identified the conserved domains found in them (Supplementary data). To detect distant relationships iterative sequence profile searches were conducted using the PSI-BLAST (49) and JACKHMMER (50) programs with profile-inclusion threshold of expect (e)-value at 0.005 against the NR database at National Center for Biotechnology Information (NCBI). Clustering of proteins based on bit score density and length of aligned sequence was performed using the BLASTCLUST program (ftp://ftp.ncbi.nih.gov/blast/documents/blastclust.html). Remote homology searches were performed using profile–profile comparisons with HHpred program (51) against profile libraries comprised of the PFAM and PDB databases as well as an in-house library of profiles of conserved domains. Multiple sequence alignments were built using the Kalign (52), Muscle (53) and PROMALS3D (54) programs with default parameters followed by manual adjustments based on profile–profile alignment, secondary structure prediction and structural alignment.
Secondary structures were predicted using the JPred program (55). Additionally, we generated predicted topologies based on homologs with known structures. Structural visualization and manipulations were carried out using the PyMOL program. Phylogenetic analysis was conducted using an approximately maximum-likelihood method implemented in the FastTree program under default parameters (56). The PhyML program (57) was also used to determine the maximum-likelihood tree using the Jones–Taylor–Thornton model for amino acids substitution with a discrete gamma model (four categories with gamma shape parameter = 2). The trees were rendered using the FigTree program (http://tree.bio.ed.ac.uk/software/figtree/). The analysis of the Shannon entropy (H) for a given multiple sequence alignment was performed using the equation:
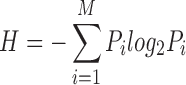 |
P is the fraction of residues of amino acid type i and M is the number of amino acid types. The Shannon entropy for the ith position in the alignment ranges from 0 (only one residue at that position) to 4.32 (all 20 residues equally represented at that position). Analysis of the entropy values which were thus derived was performed using the R language.
Species abbreviations used in the figures are as follows: Aand: Anditalea andensis; Aast: Aphanomyces astaci; Abut: Arcobacter butzleri; Acan: Albugo candida; Acas: Acanthamoeba castellanii; Adea: Angomonas deanei; Aeut: Aphanomyces euteiches; Afum: Aspergillus fumigatus; Akas: Advenella kashmirensis; Anam: Aureobasidium namibiae; Aoli: Arthrobotrys oligospora; Aory: Aspergillus oryzae; Ariv: Aeromonas rivuli; Arub: Aspergillus ruber; Alic.: Alicycliphilus sp.; Asub: Auricularia subglabra; Ater: Aspergillus terreus; Atha: Arabidopsis thaliana; Bact: Bacteroides sp.; Bbog: Bacillus bogoriensis; Bbot: Botryobasidium botryosum; Bcac: Bacteroides caccae; Bden: Batrachochytrium dendrobatidis; Bder: Blastomyces dermatitidis; Begg: Beggiatoa sp.; Bvin: Bartonella vinsonii; CJet: Candidatus Jettenia; CMet: Candidatus Methanomethylophilus; Calb: Candida albicans; Ccht: Coleofasciculus chthonoplastes; Ccin: Coprinopsis cinerea; Cgig: Crassostrea gigas; Cglo: Colletotric hum gloeosporioides; Clan: Curvibacter lanceolatus; Clut: Chryseobacterium luteum; Cowc: Capsaspora owczarzaki; Crei: Chlamydomonas reinhardtii; Chry.: Chryseobacterium sp.; Csub: Coccomyxa subellipsoidea; Ddis: Dictyostelium disco ideum; Dfas: Dictyostelium fasciculatum; Dacr.: Dacryopinax sp.; E: Enterobacteriaceae; Ebac: Erysipelotrichaceae bacterium; Ecol: Escherichia coli; Famn: Ferriphaselus amnicola; Fmed: Fomitiporia mediterranea; Foxy: Fusarium oxysporum; FsV: Feldmannia species virus; Gbre: Geobacter bremensis; Gfer: Geothrix fermentans; Gmar: Galerina marginata; Gmax: Glycine max; Gnip: Gregarina niphandrodes; Gsul: Galdieria sulphuraria; Gthe: Guillardia theta; Hirr: Heterobasidion irregulare; Lbic: Laccaria bicolor; Mabs: Mycobacterium abscessus; Mbre: Monosiga brevicollis; Meth: Methylobacterium sp.; Mhun: Methanospirillum hungatei; Mmob: Methylotenera mobilis; Mneg: Monoraphidium neglectum; Mpie: Marinitoga piezophila; Mpro: Moorea producens; Mver: Mortierella verticillata; Ncra: Neurospora crassa; Ngru: Naegleria gruberi; Orhi: Ochrobactrum rhizosphaerae; Pseu: Pseudoalteromonas; Pbra: Plasmodiophora brassicae; Pgra: Puccinia graminis; Pinf: Phytophthora infestans; Ppal: Polysphondylium pallidum; Ppar: Phytophthora parasitica; Ppat: Physcomitrella patens; Ppin: Paenibacillus pini; Ppol: Paenibacillus polymyxa; Psoj: Phytophthora sojae; Ptri: Pyrenophora tritici-repentis; Rall: Rozella allomycis; Rbic: Ruminococcus bicirculans; Rirr: Rhizophagus irregularis; Rlim: Runella limosa; Rmar: Rubritalea marina; Rsol: Rhizoctonia solani; Rhod.: Rhodococcus sp.; Sbac: Succini vibrionaceae bacterium; Scer: Saccharomyces cerevisiae; Scul: Strigomonas culicis; Sfle: Shigella flexneri; Shim: Streptomyces himastatinicus; Smoe: Selaginella moellendorffii; Sneg: Simkania negevensis; Snov: Starkeya novella; Spom: Schizosaccharomyces pombe; Sele.: Selenomonas sp.; Tbru: Trypanosoma brucei; Tcac: Theobroma cacao; Tcas: Tribolium castaneum; Tcru: Trypanosoma cruzi; Tfle: Thiothrix flexilis; Tjen: Tetrasphaera jenkinsii; Tlit: Thermococcus litoralis; Tmar: Talaromyces marneffei; Tsib: Thermococcus sibiricus; Tton: Trichophyton tonsurans; Ttur: Teredinibacter turnerae; Tviv: Trypanosoma vivax; Vcar: Volvox carteri; Vvin: Vitis vinifera; Yfre: Yersinia frederiksenii; Zbre: Zymoseptoria brevis.
RESULTS AND DISCUSSION
Identification of Crn and related proteins across eukaryotes
To date the Crn effectors have only been reported in fungal and oomycete pathogens. To better understand their phyletic distribution in eukaryotes, we first used sequence-based clustering to select a diverse library of known Crn effectors. We next used these to seed sequence profile searches with the PSIBLAST and JACKHMMER programs against a library of completely sequenced genomes representing all major eukaryotic lineages with available genomic data. Through these searches, we identified several homologous proteins outside of the taxa previously known to have Crn effectors. We then ran similar searches with the newly identified versions to verify their relationship to the Crn effectors on the basis of reciprocal recovery of representatives from the starting library. Thus, we identified a comprehensive collection of over 7000 homologs including fragmentary genes (pseudogenes or cassettes; see below) in our panel of diverse eukaryotes (Supplementary material). The newly identified versions were found not only in fungal lineages where they were not previously reported but also in several pathogenic and free-living eukaryotes (Figure 1). Among pathogens/symbionts we found LSEs in unrelated lineages like the apicomplexan Gregarina niphandrodes, rhizarian Plasmodiophora, various kinetoplastids like Trypanosoma cruzi, T. brucei, and Angomonas deanei, and the snail symbiont Capsaspora owczarzaki which is related to the animal lineage. Among free-living forms we recovered Crn homologs in the heterolobosean Naegleria, several members of Viridiplantae including chlorophytes and land plants, cryptophyte algae, choanoflagellates, certain animals (e.g. the flour-beetle Tribolium, several hemipteran insects and Hydra) and amoebozoans (Figure 1; see Supplementary material for detailed phyletic overview). Interestingly, versions from trypanosomes were proteins previously known as members of the LSE of RHSPs (Retrotransposon Hot Spot proteins), which are encoded in the vicinity of other pathogenesis and immune-evasion genes such as the variant surface glycoprotein (VSG) cassettes (58,59). Accordingly, we hereinafter refer to this class of proteins united by their sharing of characteristic conserved regions as the CR (Crinkler-RHS-type) proteins. One of the paralogs in Tribolium corresponded to the only active gene from the Medea1 locus, a selfish locus with maternal transmission, which is involved in post-zygotic killing of non-Medea individuals (60).
Figure 1.

(A) Domain architecture network of eukaryotic CR proteins. Domains linked in the same polypeptide are connected by arrows, with the arrow head pointing to the C-terminal domain. The arrow thickness reflects the number of associations found in the total dataset. (B–M) Representative domain architectures of CR proteins in different eukaryotic species. Proteins are labelled by the species abbreviation of organisms in which they are found followed by the NCBI Genbank id (gi). Species abbreviations are provided in the figure and also available in the ‘Materials and Methods’.
Characterization of domain architectures of CR proteins
Given that previous studies on Crn effectors have not objectively determined their domain architectures, we set up a protocol to systematically achieve this. First, we queried our above collection of CR proteins with an extensive library of sequence profiles, which included the Pfam database (61), and a custom collection prepared by augmenting Pfam alignments and adding new domains not in the Pfam database. Second, we initiated sequence searches with diverse CR proteins as starting points to identify regions of sequence similarity that are shared across distantly related organisms. Having isolated such regions, we then used them to run profile searches with PSIBLAST and HMM searches with JACKHMMER to comprehensively delineate new conserved domains. We also used the profiles in profile–profile comparisons with the HHpred program to detect even more distant relationships. As a result, we identified several novel domains and previously unknown divergent versions of characterized domain superfamilies. Finally, collating these results, we were able to arrive at a comprehensive characterization of the domain architectures of CR proteins (Figure 1; see Supplementary material for a complete listing of domain architectures).
Notably, analysis of the domain architectures thus obtained revealed the following common ‘syntax’ for the CR proteins: CR-NTD[i] + CR-toxin[j,k,l…]; i.e. one of several CR-NTD (Crinkler-RHSP-type N-terminal domain) followed by one or more Crinkler-RHSP-type toxin domains (Figure 1). We also term the CR-NTD domains as ‘Header’ domains as they always occupy the N-terminal position relative to other domains in the protein. The Header domains can be unified into a relatively small set of superfamilies that are unrelated to each other despite occupying a similar position in the protein (Figure 1). Analysis of the C-terminal domains revealed that they are likely to carry the toxicity determinants (see below); hence, we collectively refer to these as CR-toxin domains. The majority of these are enzymatic domains belonging to several distinct superfamilies, thereby allowing us to predict the potential catalytic mechanism by which they execute their toxicity. A minor subset of CR-toxin domains are predicted to potentially breach membranes to form pores. Furthermore, the majority of CR proteins display either of two architectural types (Figure 1), both featuring a pair of distinct enzymatic CR-toxin domains C-terminal to the Header domain: (i) a P-loop NTPase domain coupled with a nuclease domain of the restriction endonuclease (REase) superfamily (62,63). This architectural type accounts for a little over one-fourth of the total CR proteins in our dataset. (ii) A REase superfamily domain combined with a eukaryote-type protein kinase domain. This type accounts for a little over one-sixth of the total CR proteins. While a few further CR proteins display other dyads of C-terminal enzymatic domains like the above, the majority of the remaining CR proteins display a single C-terminal toxin domain. These are usually in fewer numbers than the above in any given genome.
The same type of toxin domains might occur with any of the different types of Header domains in the same or different organisms (Figure 1). Hence, we first analyze the toxins followed by a survey of the diversity of Header domains. As the two types of architectures with paired C-terminal enzymatic domains constitute the majority of CR proteins we describe those domains first followed by other types of toxin domains classified as per their predicted mode of action.
The P-loop NTPase domains coupled to REase domains
Our analysis of the CR-toxin domains recovered nine well-defined clades of P-loop NTPase domains (named CR-NTPase1-9; Figure 2A), all of which are coupled with one of several distinct clades of REase domains (see below). While these clades are highly divergent from each other in sequence (Figure 2A), profile–profile comparisons revealed that they are related to each other and also specifically to the STAND-CDC6/ORC1 clade within the AAA+ class of P-loop NTPases (64–66). Secondary structure predictions based on alignments of the individual clades conclusively confirmed that the nine CR-NTPase clades lie within the STAND-CDC6/ORC1 clade because all of them contain its synapomorphy in the form of a helix-extension-helix (HEH) insert after strand-2 of the conserved AAA+ core (Figure 2B and C) (64,66). This element resembles the HEH domains such as SAP and LEM, several of which are known to bind nucleic acids (67), and has been shown to bind the major groove of DNA in proteins like ORC1 (Figure 2D) (66). Another characteristic feature, which confirms this evolutionary affinity of the CR-NTPases, is the presence of a GxP motif at the junction between the second and third helices of the C-terminal helical module of the AAA+ NTPase domain (Figure 2A) (65).
Figure 2.

(A) Multiple sequence alignment of various eukaryotic CR-NTPase families and related AAA+ ATPases. Protein sequences are labelled by their species abbreviation followed by the NCBI gis. For species abbreviations refer to the Supplementary data. (B) Topology diagram depicting the conserved core of the STAND-CDC6/ORC1-like AAA+ ATPases highlighting family-specific and overall features. (C) 3-D cartoon of representative CDC6 structure (pdb: 2QBY) illustrating its DNA-contacting interface. (D) Representative structure of HEH domains depicting their DNA-binding modes. (E) Phylogenetic tree depicting the inter-relationships between various members of the STAND-CDC6/ORC1-like AAA+ATPases.
To further narrow down the provenance of the CR-NTPases, we utilized the wealth of sequence and structure data, which have accumulated since the original definition of the STAND clade of P-loop NTPases and their subsequent unification with the CDC6/ORC1 AAA+ NTPases (68). The STAND-CDC6/ORC1 clade includes the basal clades of CDC6/ORC1 ATPases involved in assembly of the pre-replication complex at replication origins and a crown group of classic STAND NTPases (68,69). The latter clade includes numerous signaling NTPases from bacteria and eukaryotes that are coupled to several super-structure forming domains (e.g. WD40 and tetratricopeptide repeats). Eukaryotic representatives (e.g. APAF1, the plant resistance proteins and the NACHT proteins) are central mediators of apoptosis and anti-pathogen responses (65). Between CDC6/ORC1 and the classic STANDs are several basal lineages of STANDs, which include the so-called MNS clade of NTPases that are lineage-specifically expanded in several archaeal and bacterial genomes (Figure 2E) (65). We discovered that the MNS clade is joined by the bacteriophage Mu-like transposase B subunits (70), the TN7-like transposase TnsC subunits (71,72) and bacterial peptidoglycan-remodelling GspA/ExeA proteins (73) with diverse C-terminal peptidoglycan-binding domains as part of a vast radiation that is basal to the classic STAND clade. Based on sequence comparisons we could confidently place the nine CR-NTPase clades within this basal radiation along with above-named NTPase domains (Figure 2E).
A C-terminal fusion to a REase domain was first reported in lineage-specifically expanded MNS-clade NTPases from Pyrococcus horikoshii, and was subsequently widely observed in other MNS NTPases (62,65). Based on this it was proposed that the LSEs of MNS NTPase genes possibly result from the encoded protein facilitating their transposon-like proliferation and mobility by means of the NTPase and REase domains (65,68). Following up on this, current observations indicate that the entire basal radiation of STAND NTPases is unified by frequent physical and functional coupling with one or more endoDNase domains, which might be either fused to the NTPase C-terminus or are encoded by separate adjacent genes in the operon. This proposal is now strongly supported by our unification of the TN7 and Mu family of transposase subunits with this basal radiation of STAND NTPases (Figure 2E). Extensive studies on the Mu transposase have revealed that the ATPase subunit MuB associates with the RNase H fold (74) endoDNase subunit MuA that cuts the target DNA for transposition (75,76). Similar studies on the TN7 transposase have revealed that its ATPase subunit TnsC associates with two subunits, TnsA and TnsB, which contain REase and RNase H fold endoDNase domains, respectively (71,77). In both Mu and TN7 the ATP-bound STAND ATPase is required for association with the target DNA and activates the endoDNase subunits to cut the donor DNA (78,79). The STAND subunit binds DNA only when bound to ATP but not ADP; thus, it serves as the critical switch for completing the transposition process (80). Given that the TN7 transposase complex has two endoDNase subunits that respectively cut 5′ and 3′ ends, it introduces double-strand (ds) breaks to usually work as a cut-and-paste transposon (4), whereas the Mu transposase with a single endoDNase domain functions as a single-strand (ss) DNA cutter and operates as a replicative transposon (81).
The CR-NTPase domains from all nine clades are fused to a single C-terminal endoDNase domain of the REase fold (Figures 1 and 3). Thus, they specifically resemble the MuB ATPase coupled with the MuA REase fold endoDNAse rather than the TnsCAB system with two endoDNase domains. Hence, the CR-NTPase-REase dyads are predicted to function similar to MuBA and target ss-nucleic acids. Examination of the NTPase domain from the nine clades indicates that they possess an intact Walker A motif and a Walker B aspartate (Figure 2A), suggesting that they are likely to bind a nucleoside triphosphate (NTP). This implies that as in the case of the Mu and TN7 transposase ATPase subunits their DNA-binding activity is likely to be regulated by their binding of a NTP (79,80). Taken together, these observations suggest that the effector activity of CR proteins with these CR-NTPase-REase domain dyads is potentially activated by sensing of a specific NTP by their CR-NTPase domain, which then allows them to bind nucleic acids (likely DNA) followed by a single stranded cut catalyzed by the C-terminal REase domain. However, several of the clades of the CR-NTPases lack one or both of the arginine fingers and the conserved glutamate associated with the Walker B motif (Figure 2A), which are hallmarks of highly active AAA+ ATPases including the transposase subunits (64,65). This suggests that at least versions lacking these features might sense NTP but lack multicycle NTPase activity. This might be sufficient, particularly in the context of effector function, where the strong NTPase activity might not be as critical as in the case of active transposases. Additionally, transposase NTPase domains also prevent reinvasion at the same site by another copy of the transposon (target immunity) by active NTP hydrolysis upon interaction with the endoDNase component (82–84). Loss of the above-mentioned residues in multiple CR-NTPase clades might also reflect relaxed selection due to the lack of a need to maintain a highly active NTPase for target immunity in effector versions. Nevertheless, multiple eukaryotic CR-NTPases do possess the features corresponding to AAA+ NTPases with high activity suggesting they could sustain transposase activity either exclusively or in addition to their effector function (see below section: functional diversity of the CR proteins).
Figure 3.

Multiple sequence alignment and predicted topologies for widely present CR-REase families. The conserved catalytic site residues are highlighted in both the alignment and the topology figure. The equivalence of core secondary elements (α1–β1–β2–β3–α2) between these families is also illustrated. Protein sequences are labelled by their species abbreviation followed by the NCBI gis. For species abbreviations refer to the ‘Materials and Methods’.
Kinase domains coupled to REase domains
The Phytophthora infestans CR protein CRN8 was previously identified as having a eukaryote-type protein kinase domain (38), which was thought to belong to the ‘RD type’ based on the presence of an RD signature in the so-called kinase motif VIB that is associated with the active site (85,86). Our analysis revealed a more extensive presence of kinase domains among the CR proteins, which could be classified into five major clades using similarity-score density-based clustering and phylogenetic analysis (Figure 1; Supplementary material). Moreover, only ∼19% of the CR-kinase domains had an RD signature in the VIB motif, while ∼60% of the representatives instead had a GD signature. Further, in at least one clade we found an unusual conserved W two residues upstream of the highly conserved active site N (which is downstream of the nearly invariant D in motif VIB). Despite this diversity our analysis strongly suggested that these CR-kinase clades are monophyletic to the exclusion of other kinases because they display characteristic conserved features, in particular in motif VI and VII (Supplementary material) (85,86). This pattern of sequence diversity of the CR-kinases closely resembles that of the rhoptry protein kinases, which are secreted effectors of apicomplexan parasites like Toxoplasma and Eimeria (26). Together these observations suggest that the CR-kinases are probably under selection for rapid diversification like the apicomplexan kinases. In both cases the sequence variability includes positions close to the invariant active site residues (26,85,86), suggesting that they have specialized to modify several distinct substrates.
We observed that in all complete CR proteins the kinase domain is C-terminal to a REase domain (Figure 1; see below). Thus, they mirror the dyad domain organization of the CR-ATPases which are also obligately coupled to REase domains. This suggests that the action of the kinase domain is likely to be functionally linked to the associated REase domain. Consistent with this proposal, studies on P. infestans CRN8 have shown that disruption of the kinase activity destabilizes the protein and reduces effector activity, but does not result in loss of the effector's ability to cause cell death (38). In contrast, deletion of the region which corresponds to what we have identified in this study as a REase domain resulted in loss of the effector's cell-death activity (38). Therefore the kinase domain is not required for the cell-death induction by the effector; rather it appears to play an auxiliary role in stabilizing the effector protein in the host cell. Based on this we propose that the CR-kinases act as partners for the associated REase domain, which displays the actual toxin activity. The phosphorylation of specific proteins by the kinase domain probably protects the effectors from host intracellular immunity that targets them for degradation.
REase domains found in above systems
We identified a total of 18 distinct clades of REase domains in CR proteins, the largest of which we named CR-REase1-13, with five other minor clades (Figures 1 and 3). Barring members of the CR-REase2 and CR-REase5 clades, all REase domains are obligately associated with either the kinase or CR-NTPase domains described above. In the case of the NTPase-REase association the same CR-REase clade might sometimes come with two distinct NTPases clades (e.g. CR-REase3 with both CR-NTPase5 and CR-NTPase9) or vice versa (e.g. CR-NTPase4 with REase6 and REase11) (Figure 1A). In the case of the kinase the same clade of CR-REase is always coupled with its cognate CR-kinase clade (Figure 1A). Thus, in general this suggests a strong functional interplay between the two domains, which selects for their persistence in a linked dyad. In the case of the coupling with the NTPase this is consistent with the above-described functional linkage, wherein NTP-binding by the NTPase is critical for association of the protein with the nucleic acid substrate, thereby allowing the cognate CR-REase domain to hydrolyze it. In the case of the coupling with the kinase, the tendency for a strong linkage with the cognate REase might result from the role of the kinase in neutralizing intracellular immunity mechanisms directed at the effector (see below for further details). However, in both these dyads, as well as the CR proteins which have just the REase domain, the CR-REase domain is likely to be the primary toxic moiety of the effector. This proposal is consistent with (i) the known endoDNase activity of the transposase REase domains, which as noted above are related to the CR-REases (Figure 3); (ii) the cell-death inducing function mapping to the region corresponding to the REase in the P. infestans CRN8 (i.e. CR-REase4) (38); (iii) the host defense-countering activity of the P. sojae effectors CRN63 and CRN115 (45), which have only CR-REase2 domains not coupled to any other domains.
Examination of the alignment of the CR-REase domains revealed striking sequence diversification between the distinct clades (Figure 3): (i) some clades like CR-REase1, 2, 6, 9 and 12 are marked by inserts of one or more β-hairpins after strand-1 of the core domain, which are likely to play a role in recognition of substrates (62,63). (ii) The core motif associated with the REase active site also assumes several forms beyond the ancestral [ED]xK signature (where x is any amino acid) in different clades (e.g. QxT in CR-REase1). (iii) In certain clades like CR-REase11 a neomorphic glutamine is acquired to replace the typical acidic residue from the [ED]xK signature. In the REase fold this pattern of extensive diversification is otherwise only seen in the restriction endonucleases from the prokaryotic restriction-modification (R-M) systems (62,63,87). This suggests, like their counterparts in the R-M systems, the CR-REases are likely to be rapidly evolving different target sequence specificities probably due to the target cell genome evolving to evade effector action. Consistent with this hypothesis, we observed that several of the REase domains from each of the clades appear to have lost their active site residues suggesting that they have undergone inactivation (Figure 3 and Supplementary material). This inactivation is possibly the result of the effectors being on the way to ‘extinction’ due to development of target-cell immunity against them. Given the parallels to REase domains from R-M (87), and the evolutionary links of at least those REase domains coupled with NTPase domains to transposase endoDNase domains, we propose that CR-REase domains primarily act on target cell DNA. In support of this idea, multiple effectors from Phytophthora (e.g. PiCRN8 and PsCRN63) need to localize to the target cell nucleus to cause cell death (45,46). However, some like PsCRN115 do not seem to require nuclear localization for action (45); hence, we cannot rule out that a subset of the CR-REases have evolved to target RNA. Indeed, the cell-death-causing capacity of PiCRN8 and PsCRN63 could be a direct consequence of their damaging the target-cell genome, whereas the immune-suppression related function attributed to PsCRN115 might result from degradation of specific mRNAs.
Other effector domains found in CR proteins
HNH nucleases
We found two distinct clades of the treble-clef fold HNH endonuclease domains (CR-HNH1 and 2) among the CR-toxin domains (Figure 4A) (12,62). Both these versions of CR-HNH domains have lost the ancestral metal-chelating cysteine residues but retain the characteristic active site histidine (88). In this regard they are reminiscent of the HNH endonuclease domains we had earlier described in bacterial polymorphic toxins (12); however, neither of the versions found in CR proteins show a specific relationship to these HNH domains. CR-HNH1 is primarily found in parasitic oomycetes, the fungal frog pathogen Batrachochytrium and the Feldmannia virus which belongs to the assemblage of Nucleo-cytoplasmic large DNA viruses (89). CR-HNH2 has a wider phyletic spread and is found in several free-living chlorophytes, certain land plants like the clubmosses, parasitic oomycetes, and both free-living and parasitic fungi. While in most CR proteins either clade of HNH domain occurs as the sole toxin domain (Figure 1B), in a few cases, like in the alga Volvox, it might occur fused to other domains: (i) with a C-terminal protein kinase in a configuration similar to the REase-kinase dyad described above; (ii) with an N-terminal REase domain (Figure 1M).
Figure 4.

(A–B) Multiple sequence alignment of CR-HNH nuclease families. (C) Multiple sequence alignment and topology of CR-ubiquitin-like (CR-Ubl) Header domains. (D–F) Multiple sequence alignment of other lineage-specific Header domains including VP-NTD, Mnag-NTD and Caps-NTD domains. Protein sequences are labelled by their species abbreviation followed by the NCBI gis. For species abbreviations refer to the Supplementary data.
LK-nuclease domain
The LK-nuclease is an RNase domain found in proteins from the three superkingdoms of life, including those associated with the germ-cell or nuage RNA–protein complexes of eukaryotes (90). It is structurally related to the PIN endoRNase domain (91), which is frequently found in RNA cleaving toxins of prokaryotic toxin–antitoxin systems as well as enzymes involved in eukaryotic rRNA processing and nonsense mediated decay systems (14). We found the LK-nuclease as the toxin domain in multiple CR proteins from the symbiotic fungus Rhizophagus (Figure 1C). We predict that this nuclease domain is likely to target specific host RNAs via metal-dependent endoRNase activity similar to known reactions catalyzed by the PIN domain (92).
Peptidase domains
We found three unrelated types of peptidase domains among the C-terminal toxin domains of CR proteins. The most widespread of these are the CR-trypsin domains (Figures 1 and 4B), which are an assemblage of previously unrecognized divergent serine peptidase domains with the trypsin fold (93). Such effectors are found in fungi, oomycetes and free-living chlorophytes. We recognized at least three distinct clades of CR-trypsin domains, all of which occur as the sole toxin domain in multiple CR proteins suggesting that this domain by itself is sufficient for effector function (Figure 1B, C and M). However, it is infrequently fused to an N-terminal protein kinase domain (gi: 595490980, Mkk1p in Rhizophagus irregularis; Figure 1C), which could perform an auxiliary function as proposed above.
We also detected a zincin-like metallopeptidase domain in multiple CR proteins from the oomycete P. infestans (Figure 1B). While this toxin domain is reminiscent of the metallopeptidase effector of the parasitic fungus Magnoporthe oryzae (39), it does not show any specific relationship to it. In the free-living slime mold Dictyostelium fasciculatum we uncovered over 10 CR proteins with an Ulp1-like peptidase domain of the papain-like fold (Figure 1H) (94,95). This domain always occurs to the C-terminus of a CR-NTPase+REase dyad (described above). Given its specific relationship to Ulp1, we propose that it acts as a deSUMOylating or deubiquitinating enzyme (95) that might incapacitate the ubiquitin/Ubl-dependent target-cell immune response that degrades effectors (96).
GIMAP/AIG1-like GTPase domains
GIMAP/AIG1 GTPases, which are related to the septins, are a widespread clade of GTPases previously implicated in immunity in both animals and plants (97). A unifying biochemical feature of these GTPases appears to be their GTP-dependent-remodelling of membranes. We found multiple CR proteins from the fungus Rhizophagus (Figure 1C), where this GTPase domain is followed by a novel cysteine-rich C-terminal domain, which we predict to be stabilized via chelated metal atoms. Given the membrane-associated role of these GTPases (97), this C-terminal domain might play a role in further interactions with the membrane. Interestingly, we had previously reported effectors with GIMAP/AIG1 GTPase domains in Amoebophilus, the intracellular bacterial symbiont of amoebae, and in unrelated viruses like the Duck hepatitis A virus and the Anguillid herpesvirus 1 (1). Moreover, in several eukaryotes, related GTPases are found fused to the stonustoxin-like domain (gi: 465958095), which is also involved in membrane interactions (98). This indicates that the GIMAP/AIG1-like GTPases are used as effectors in a wide range of biological conflicts. We propose that in the Rhizophagus CR proteins these GTPase domains are likely to function in membrane remodelling, which is known to occur as part of the symbiont-plant-root-cell interface (99).
Non-enzymatic effector domains
We recovered both globular and membrane-spanning non-enzymatic domains among the C-terminal toxin domains of CR proteins. Examples of the former are found in rare CR proteins from Rhizophagus that display C-terminal POZ and TLD domains (gi: 595477572; Figure 1C). The POZ domain might function as a substrate adaptor for the cullin E3 ubiquitin ligases (100) with the C-terminal β-sandwich TLD domain engaging the actual substrate (101). Thus, these effectors could coopt the plant host ubiquitin system to modify specific proteins. We found certain other globular effector domains in chlorophyte CR proteins (Chl-Toxin1, 2, 3; Supplementary material), which we were unable to unify with any previously known domain and their functions remain enigmatic. In chlorophyte CR proteins we also found two distinct CR-toxin domains, which we predict to form membrane spanning domains, both with two TM helices (Figure 1M; Supplementary material). Based on the precedence offered by pore-forming toxins (102,103), we propose that these domains affect their toxicity by breaching membranes.
Miscellaneous transposon-derived domains
In several cases we found the Header domains of CR proteins or entire CR proteins fused to transposon-derived domains. As we found multiple copies of such fusions in the genomes in which they are present we interpret these as genuine associations. Most striking of these are fusions of the entire open reading frame (ORF) of a mobile element to complete CR proteins in P. sojae (Figure 1B). These mobile element ORFs contain two DNA polymerase modules (104) fused to an OTU-like papain fold peptidase domain (105) and an HNH endoDNase domain distinct from those found in CR-toxins (Figure 1B). While this mobile element occurs by itself in multiple copies in P. sojae, it is fused to distinct CR proteins with different toxin domains in multiple instances. In the heterolobosean amoeboflagellate Naegleria, the CR protein Header domains are occasionally fused to active TRANSIB transposase domains (gi: 290971552) (106). In both these instances it is conceivable that these fusions to active mobile elements play a role in the recombination or proliferation of the CR proteins (see below). Finally, in multiple oomycetes (Figure 1B) we found helix-turn-helix (HTH) DNA-binding domains derived from transposases (68) as the C-terminal effector domains of CR proteins. It is plausible that in these cases the C-terminal domains bring about their action by binding DNA in the target cells.
CR-NTD or Header domains
Previous studies on Crn effectors from oomycetes had emphatically demonstrated that the N-termini of these proteins contain the determinants for their localization into plant cells (46). It was proposed that these N-termini contained a secretory signal peptide and that a further downstream motif termed ‘FLAK’ (after its conserved amino acid sequence) was responsible for the actual translocation into the target cell. Our analysis using two distinct sensitive sets of hidden Markov models for eukaryotic signal peptides categorically ruled out the presence of a signal peptide at the N-termini of these proteins (107,108). Our analysis also showed that the so-called FLAK peptide is part of a globular domain that had hitherto not been correctly defined. Moreover, the FLAK motif is not conserved across all exemplars of this domain (Figure 4C). Further, our analysis showed that even the extended domain family that encompasses versions with the so-called FLAK motif is only one of numerous Header domains belonging to several distinct unrelated structural classes. Importantly, we showed that the same types of C-terminal CR-toxin domains might be combined with practically every type of N-terminal Header domain and in some organisms the same type of CR-toxin domains might be combined with different sets of unrelated Header domains (Figure 1). This observation indicates that despite their structural diversity, most Header domains are likely to have generally comparable functions in translocation of the effector protein into the target cell. While some Header domains are shared between organisms across a wide phylogenetic range, others are restricted to single lineages. This implies that despite their general functional equivalence there might be notable differences in terms of the specific proteins with which the Header domains interact in the translocation process. Given that our analysis has systematically dissected the Header domains in an objective manner for the first time, we describe in detail their different classes below.
The Ubiquitin-like Header domain (CR-Ubl)
Using sequence-profile searches we showed that this family encompasses practically all Header domains of oomycete Crn proteins, as well as the majority of those from fungi (Figures 1 and 4C). This family includes all those which were previously identified as having the FLAK motif (46) as well as numerous others which lack this motif. Our sequence profile searches also established that this Header domain is statistically significantly related to the N-terminal domain of the SSK1/Mcs4 signaling proteins from fungi (PSI-BLAST query P. sojae Crn protein, gi: 348686210 recovers SSK1/Mcs4 orthologs with e = 10−5 to 10−7 in iteration 7 against NR database). Profile–profile comparisons revealed that this Header domain and the SSK1/Mcs4 NTD contain an ubiquitin-like (Ubl) domain specifically related to the Rad23-N clade of Ubls (Figure 4C). All these Ubls are united by a highly variable region inserted in the ‘connector arm’ that links strand-4 to strand-5 (109). Accordingly, we named this domain the CR-Ubl Header domain. Given that SSK1/Mcs4 orthologs are more widely distributed in fungi than CR proteins with the CR-Ubl Header, it is possible that this Header domain was derived from the ancestral SSK1/Mcs4 NTD.
The so-called signal peptide and the FLAK motif that were claimed for the oomycete Crn effectors respectively map to the conserved strand-1 and strand-3 of the CR-Ubl domain (Figure 4C). These observations suggest that, consistent with previous experimental results (46), the CR-Ubl domain as a whole is likely to be the primary determinant of translocation into the target cell, rather than just the mispredicted signal peptide or the FLAK motif. Fungal SSK1/Mcs4 proteins contain C-terminal Receiver domains, which are phosphorylated on an aspartate residue as part of histidine-kinase-dependent signaling cascades (24). SSK1/Mcs4 orthologs in different fungi play important roles in responses to stresses, such as oxidative and osmotic shock, in both a phosphorylation-dependent and independent manner (24,110). They mediate this action by means of the N-terminal Ubl domain that interacts with the MAPKKK heteromer. Based on this precedence we suggest that the oomycete and fungal CR proteins gain their entry into target cells by specific interactions mediated by the Ubl domain, analogous to those in stress signaling by SSK1/Mcs4. Furthermore, interactions of the CR-Ubl domain with nuclear-localized proteins in the target cell could also enable them to access the nucleus by translocating in a complex with the latter (37,38,45,47). CR-Ubl domains are found in both oomycete and fungal pathogens irrespective of whether they target animal or plant hosts (Figure 4C). This suggests that CR-Ubl domain is versatile enough to mediate interactions with proteins from vastly different systems, which is also consistent with the variability seen in the ‘connector arm’ region of these domains (Figure 4C).
α-helical Header domains
Unlike the CR-Ubl domain all the remaining Header domains are various unrelated, entirely α-helical domains (Figures 1 and 4D–F; Supplementary material). Two of them show a wide phyletic spread: The first of these is found in CR proteins across the Viridiplantae lineage, ranging from diverse chlorophyte algae to certain land plants like the club moss Selaginella. Accordingly, we term it the Viridiplantae (VP) Header domain (VP-NTD, Figure 4D) and predict that it adopts a globular fold, likely in the form of an α-helical bundle. The second widespread Header is found across distantly related photosynthetic eukaryotes, such as chlorophyte algae, land plants, phaeophycean (brown) algae and cryptophyte algae, and we accordingly term it the photosynthetic (PS) Header domain (PS-NTD; Figure 1 and Supplementary material). This Header assumes a coiled-coil structure with at least 10 heptad repeats of which the C-terminal 4–5 heptads have a characteristic QLR motif (Supplementary material). The presence of the coiled-coil raises the possibility that effectors with the PS-NTD might undergo dimerization. The remaining Header domains show successively more restricted phyletic spreads. The Ascomycete Header (Asco-NTD) is restricted to CR proteins from certain ascomycetes making it the second type of Header domain found in fungi (Figure 1C). The CR proteins from diverse eudicot plants are characterized by a Header domain in the form of an HTH domain of the Myb family (Figure 1M). The remaining α-helical Header domains are restricted to particular genera, such as the chlorophyte Monoraphidium neglectum (Mneg-NTD, Figure 1M), Capsaspora (Caps-NTD1,2,3, Figure 1I) and Naegleria (Figure 1F). Of these the Header from Naegleria is predicted to adopt an α-helical fold similar to the SAM domain (Figure 1F) (111). The VSGA Header, so named due it being encoded in chromosomal proximity to the VSG genes, is found only in the RHSPs from trypanosomes (Figure 1D) and is predicted to adopt an α-helical bundle fold. Unlike their trypanosome homologs, CR proteins from the related kinetoplastid Angomonas, a gut parasite of insects, are typified by a Header domain that is unrelated to the VSGA domain. Notably, the C-terminal region of the Angomonas Header (Ango-NTD) has an α-helical element with 10 successive hydrophobic residues, suggesting that it might directly interact with membranes (Figure 1D and Supplementary material).
This diversity of Header domains suggests that several distinct structural scaffolds, especially of the α-helical type, can effectively perform an equivalent function. This reinforces the idea that most Header domains primarily function through mediating specific interactions with other proteins, which are potentially from the target cell, to bring about their translocation. Thus, as long as a structure can mediate such interactions, there is no constraint on the particular fold the Header domain might adopt. However, we suspect that certain Header domains are likely to have unique mechanisms of action. Interestingly, despite the fact that most CR proteins are likely to be used as secreted effectors, the Angomonas Header (Ango-NTD; Figure 1D) is the only one that shows a hydrophobic region suggestive of membrane interaction. This suggests that its mode of action is likely to be different from that of VSGA domain of the related trypanosomes, and might involve direct interactions with the target cell membrane. In contrast, Myb domains have been implicated in DNA-binding (68); hence, the eudicot CR proteins (Figure 1M) with this domain might not be secreted but perhaps function to directly target intracellular invasive DNA.
Reconstructing the evolutionary history of the CR proteins
The dominant CR proteins have evolved from prokaryotic transposons
Our identification of the CR-NTPase domains as members of the STAND-CDC6/ORC1 clade of AAA+ NTPases helps elucidate the provenance of the CR proteins (Figure 2E). The STAND-CDC6/ORC1 probably emerged in archaea, where CDC6/ORC1 proteins are central players in recognition of origins of replication (68). From the prototypical CDC6/ORC1 enzyme a version emerged wherein the ancestral NTPase was coupled to one or more endonuclease domains, either in the same polypeptide or in a distinct polypeptide encoded by the same operon, which allowed the mobility of these units (Figure 2E). These versions then spread throughout prokaryotes and diversified into various elements, ranging from transposons typified by TN7 to bacteriophages such as Mu (75,83). One further version appears to have emerged in proteobacteria, namely, the GspA/ExeA proteins, wherein a similar STAND-CDC6/ORC1 NTPase domain is combined with diverse C-terminal peptidoglycan-binding domains (Supplementary material). Consistent with this architecture, the GspA/ExeA proteins have been shown to remodel peptidoglycan to facilitate protein export via the type-II secretion system (73). However, our analysis revealed that several NTPases close to the GspA/ExeA proteins are operonically linked to Mu-type transposases (Supplementary material), suggesting that even the GspA/ExeA proteins are likely to be a proteobacteria-specific derivation from ancestral mobile versions linked to transposases. Given that the CR-NTPases are coupled to C-terminal REase domains they present an architecture that is identical to the ancestral mobile version seen in prokaryotes. Moreover, they represent only one among a more diverse array of architectural/operonic themes in prokaryotes (Figure 2E) supporting the idea that the CR-NTPase-REase dyad of eukaryotes was derived from a prokaryotic mobile element with the same combination of domains.
Eukaryotic diversification of CR proteins: multiple acquisitions from prokaryotes and lineage-specific expansions
Consistent with their primary role as transposases of intragenomic elements or viruses, there is no evidence that the prokaryotic versions have equivalents of the Header (N-terminal) domains seen in eukaryotes. It is possible that some of the prokaryotic versions are deployed against intracellular invasive DNA but they are unlikely to be deployed as effectors against other cells. Thus, their deployment in eukaryotes as effectors delivered into target cells marks a notable functional shift. This seems to have proceeded via combination of the CR-NTPase+CR-REase dyads with diverse N-terminal domains, which enabled their export in eukaryotes. Given that the CR-Ubl domain is the most frequently encountered Header in the CR proteins, it is possible that this was one of the early combinations that allowed their deployment as effectors in fungi followed by their acquisition by oomycetes by lateral transfer. In eukaryotes a major trend in the subsequent evolution of the CR proteins appears to have been LSE (Figure 5A–F). Pulses of LSEs with relatively low levels of inter-paralog sequence divergence were accompanied by occasional spurts of major sequence divergence resulting in founding of new clades of CR-NTPases. Given the transposon ancestry of at least the CR-NTPase+CR-REase dyad, it would be of interest to investigate if some of them retain their capacity for mobility, thereby facilitating their own proliferation in eukaryotic genomes. Multiple clades of CR-NTPase+CR-REase dyads as well as other CR protein domains are shared between distantly related eukaryotes (Figures 2 and 3). Thus, in addition to LSEs, CR-effector domains in particular might also be prone to lateral transfers between distant branches of the eukaryotic tree.
Figure 5.

(A–F) Phylogenetic trees illustrating LSEs of eukaryotic CR protein domains and specific gene transfers from bacterial homologs (indicated by the curved arrows). LSEs are shown as coloured triangles/sectors in the tree. Bootstrap values are shown for the major branches only. The bacterial branches are coloured black. The complete trees from which these were derived can be retrieved from the Supplementary data. For species abbreviations refer to ‘Materials and Methods’. (G) Positional entropy comparison between CR-NTD and CR toxin domains. (H) Entropy plot for CR-Ubl 1+ CR-REase 2 type proteins in P. infestans.
Our phylogenetic analysis also revealed a second notable process in the evolution of eukaryotic CR proteins—the repeated acquisition of CR-NTPase and REase domains via lateral transfer of prokaryotic transposons into eukaryotes (Figure 5D–F). The eukaryotic CR-NTPase-5+REase3 and CR-NTPase9+REase3 dyads form a higher order grouping, which in turn group with bacterial versions to the exclusion of all other CR-NTPase+CR-REase dyads (Figure 5E). These observations suggest that while there was an ancient acquisition of CR-NTPase+REase dyads followed by their proliferation and dissemination across eukaryotes, some others were acquired due to subsequent lateral transfers. Further, the eukaryotic CR-NTPase8+REase7 dyads are interspersed within the radiation of their bacterial cognates pointing to at least four independent transfers of this dyad from bacteria. At least one of these, seen in animals, can be clearly linked to bacterial versions from intracellular symbiotic rickettsiae suggesting that such associations might have served as conduits for transfers (Figure 5D). Interestingly, these dyads are seldom combined with any Header domains. This raises the possibility that they are relatively recent transfers that still retain their ancestral transposon state and are yet to be incorporated as full-fledged effectors. In the case of the CR-REase+Kinase dyads we found that the kinase domains are clearly eukaryotic in origin. However, the REase domains are ultimately related to prokaryotic versions from which they are likely to have been derived. In at least one case, CR-REase-5, the REase domain shows clear affinities to a bacterial REase domain to the exclusion of other CR-REase domains (Figure 5F). This suggests that on at least one occasion the REase domain of the REase+kinase dyad was displaced by an independently acquired version from bacteria while preserving the overall domain architecture. Similarly, the two CR-HNH and the CR-trypsin domains appear to have also been originally acquired from bacteria (Figure 4A and B).
The modular evolution of CR proteins
While the architectural modularity of Crn effectors have been recognized and discussed in several previous studies (32,37,48), until this study their constituent domains had never been correctly defined and analyzed in functional terms. This allows us to obtain a proper understanding regarding the role of modularity in evolution of the larger class of CR proteins as defined here. First, the principle of combination of different C-terminal effector domains with unrelated Header domains appears to be preserved across major eukaryotic lineages suggesting that this organizational principle enforces a strong selective pressure to repeatedly select for similar architectures (Figure 1). Thus, the CR proteins as a class are united by two orthogonal features: (i) shared Header and/or CR-toxin domains and (ii) similarity in domain architectural organization (Figure 1). In this respect they resemble the prokaryotic polymorphic and related toxins, which also strongly preserve a certain domain architectural template (11). Second, the finding that catalytic effector domains have been repeatedly acquired from bacteria or laterally transferred between distantly related eukaryotes implies that the system of CR proteins provides a niche for diverse catalytic domains with effector capability. Hence, selection for varied means of attacking target cells appears to be a notable driver that has allowed CR proteins to incorporate a range of effector domains from diverse sources. The newly acquired effector domains are likely incorporated by displacement of the original effector domain thereby retaining the characteristic CR protein architectural template. However, the total variety of toxin domains thus far found in the CR proteins appears to be lower than what is found in the prokaryotic polymorphic toxins and related systems (11). This is probably related to the ability of eukaryotes to diversify their effector repertoire within their larger genomes via LSEs. This probably allows them to use a diversified repertoire of the same type of effector domain as opposed to multiple unrelated domains. The presence of multiple paralogs also relates to the results of multiple studies (32,37,48), which have demonstrated recombination between paralogous Crn effector genes in oomycetes and fragments thereof. Mapping the results of these studies on to our domain definitions suggests that these recombination events, likely driven by gene conversion, result in CR proteins with distantly related CR-toxin domains having very similar Header domains and vice versa.
While the above principles provide a general explanation for modularity of CR proteins, we used the newly obtained domain definitions to map sequence variability on to the different domains to understand other selective pressures that might be influencing the modularity of these proteins. To do this, we first created sets of lineage-specifically expanded CR proteins, which had 15 or greater representatives in an organism with homology spanning the entire length of the protein. We created multiple alignments for each of these sets and computed Shannon entropy (‘Materials and Methods’) for each position. Plots of these values provide a measure of the sequence variability across a given domain (Figure 5G). Finally, we used these values to compute mean entropy per domain. Comparison of mean entropy per domain reveals interesting features in different organisms. In the case of CR proteins with a CR-Ubl+CR-REase2 combination from P. infestans we found that the N-terminal CR-Ubl domain is significantly more variable than the CR-REase2 domain (Figure 5H). In the case of CR proteins with the CR-Ubl+CR-NTPase1+CR-REase1 combination from Batrachochytrium the pattern was the opposite: the CR-Ubl domain was significantly less variable compared to the two C-terminal catalytic domains from the effector part of the protein (Figure 5G). In the case of CR proteins with an Asco-NTD+CR-REase8+CR-kinase architecture from Claviceps purpurea we observed that the Asco-NTD and REase domains were significantly more variable than the C-terminal kinase domain (Figure 5G). Finally, in the case of the clubmoss Selaginella we found that in CR proteins with CR-REase-4 and the CR-Kinase domains, both domains were similarly variable with no significant difference in their per domain mean entropy (Figure 5G).
These observations were intriguing because, though they indicate a general propensity for differential variability of the domains within lineage-specifically expanded CR proteins, there was no common tendency with respect to the differential variability of the domains in the CR proteins. This is in sharp contrast to the prokaryotic polymorphic toxins and related effectors, wherein the C-terminal effectors as a rule show much greater variability and/or polymorphism relative to their N-terminal regions associated with secretion and/or trafficking (11,12). This suggests that there are notable differences in selective pressures faced by the eukaryotic effectors. One possibility could be differences in the type of immune response deployed against these effectors. It is possible that CR proteins of oomycete plant pathogens like P. infestans are countered by specific intracellular defense mechanisms (e.g. F-Box-E3 Ubiquitin ligases or TIR-AP-ATPase-LRR family resistance proteins (112,113)) before or as they are trafficked to the nucleus. Thus, these could face greater pressure for variability of the N-terminal CR-Ubl domain for evading interactions with such immune mechanisms. In these proteins it appears there is little diversification of the effector domain; hence, once these CR proteins evade immune detection, which appears to primarily target the CR-Ubl Header, there is little role for development of resistance against the actual effector domain as a counter-mechanism (Figure 5H). In the CR proteins of the plant pathogenic fungus Claviceps purpurea (Cpur in Figure 5G) the lower variability of the CR-kinase domain might reflect its above-proposed auxiliary role in targeting of a conserved cellular protein to ward off destabilization of the effector. However, in these proteins the primary effector domain (CR-REase-8) is as comparably variable as the Header (Asco-NTD; Figure 5G). This pressure for diversification of the effector domain, unlike in the P. infestans example, might reflect its nucleic target sequences being more prone to variability and thereby development of resistance. A similar situation is seen in the chytrid animal pathogen Batrachochytrium (Bden in Figure 5G), suggesting that here too the effector domains might face similar pressures for diversification as in the example from Claviceps. However, in this case the low variability of the CR-Ubl Header domain suggests that these effectors might not be the target of an intracellular immune response comparable to what may be inferred in the plant pathogens.
Functional diversity of the CR proteins
Possible transposase-effector duality of CR proteins in eukaryotic pathogens and symbionts
The archetypal CR proteins were defined as effectors of certain fungi and oomycetes (30,37,44–46). In this work we have detected such CR proteins more widely across not just fungal pathogens but also pathogens/symbionts belonging to multiple distant clades of eukaryotes: Plasmodiophora from the rhizarian lineage (Figure 1G), Capsaspora which is a basal branch of the animal lineage (Figure 1I) and the kinetoplastids from the euglenozoan lineage (Figure 1D). Plasmodiophora mimics the pathogenic behaviour of fungal and oomycete pathogens in general terms and has undergone multiple lateral transfers with them (114). Hence, their presence here is unsurprising and the CR proteins are likely to function as effectors. Capsaspora is a symbiont/parasite that lives in the snail blood and kills trematodes like Schistosoma mansoni that infect the snail (115). The exact mechanism by which Capsaspora attacks the trematode parasites of the snail has not been elucidated. Given the presence of CR proteins with CR-NTPase+CR-REase and CR-REase+CR-kinase architectures combined with distinct Headers (Caps-NTD1-3), we propose that these might be potential effectors used by Capsaspora either in interactions with snail host or the nematode parasites of the snail.
Detection of CR proteins in both trypanosomes that infect humans and other mammals (agents of sleeping sickness, Trypanosoma brucei; Chagas disease, T. cruzi; Nagana disease, T. vivax) and their insect-pathogenic relatives (Angomonas and Strigomonas) is more surprising because this raises questions regarding the possible significance of these proteins for the pathogenesis of these organisms (Figure 1D). CR proteins in Trypanosoma cruzi and T. brucei (RHSP) are encoded in subtelomeric regions alongside members of several multigene families, including trans-Sialidase, MASP, Mucin, VSG and dispersed gene family-1, which have been proposed to have a role in pathogenesis (58,59,116). Previous studies have also shown that trypanosome CR proteins have several pseudogenes and their complements show considerable variability between strains due to the rampant recombination events in subtelomeric regions (58,59). These studies on T.brucei RHSPs have suggested that they are produced in the procyclic stage in the Tsetse fly and human bloodstream forms and are likely expressed throughout the lifecycle of the parasite (58). Based on generic antisera the RSHPs were detected as being present in the parasite nucleus and perinuclear region (58). Given that all CR proteins from kinetoplastids are architecturally equivalent to the prokaryotic transposases, this expression pattern is more consistent with them having a transposase function in the parasite nucleus. Indeed, their active CR-REase1 domain could catalyze DNA breaks to facilitate the high frequency of recombination in subtelomeric regions. This could have a potential role in generating diversity in the neighbouring genes which might play a role in parasite-host interactions. On the other hand, the presence of a distinct Header domain (the VSGA domain) raises the possibility that at least a subset of RHSPs are delivered as effectors into host cells. If this were the case it would imply that kinetoplastid RHSPs could function as effectors just as in the case of fungi. Interestingly, our analysis suggests that the insect-pathogenic forms like Angomonas and Strigomonas have acquired their CR proteins (Ango-NTD+CR-NTPase7+CR-REase6) independently of the versions in the trypanosomatids (VSGA+CR-NTPase1+CR-REase1) (Figure 1D). This suggests there might have been strong selection to acquire CR proteins, which in turn might favour the hypothesis of at least some of them functioning as effectors.
Conversely, even in the case of other eukaryotic pathogens, where the effector role appears more likely, at least some CR proteins with the dominant CR-NTPase+CR-REase architectures could retain their ancestral transposase-like activity to drive recombination via introduction of single-strand breaks. This could possibly additionally help explain the widespread recombination observed in the CR protein genes of oomycete and fungal pathogens and mycorrhizal symbionts (32,37,41,48) comparable to what is seen in the kinetoplastids (58,59,116). However, the CR proteins with architectures other than the CR-NTPase+CR-REase combination are likely to function solely as effectors.
Role for CR proteins in free-living organisms
One of the striking results of this study is the recovery of CR proteins in phylogenetically diverse free-living eukaryotes including chlorophyte algae, land plants, amoebozoans, free-living fungi (e.g mushrooms), choanoflagellates, animals and Naegleria gruberi (Figure 1 and Supplementary material). The simplest explanation for the occurrence of CR proteins in these lineages could be that they retain the ancestral state of being transposons and are not deployed as secreted effectors. Such an explanation could be valid for those CR proteins showing the transposase-like architectures (i.e. CR-NTPase+CR-REase without Header domains). Given the low inter-paralog sequence divergence, this appears plausible for the CR-NTPase+CR-REase dyad in the beetle Tribolium (Figure 1K) and a subset of those in land plants like Selaginella and Physcomitrella (Figure 1M), all lacking N-terminal Header domains and showing signs of recent proliferation. In the beetle Tribolium, genes coding for the CR-NTPase8+REase7 dyad have dispersed across the genome upon recent proliferation (Figure 5D). Further, at least one of these copies, corresponding to the Medea1 element, shows evidence for insertion into a preexisting Tc1 transposon (60), thereby providing evidence for the mobility of these genes within eukaryotic genomes. However, in an interesting twist, the Medea1 element exhibits unusual behaviour (117): (i) its product, either transcript or protein, is maternally transmitted via the egg; (ii) the Medea1 product post-zygotically kills all developing animals that lack at least one copy of the Medea1 locus inherited from their parents. We propose that this behaviour represents an intermediate condition between being just a pure transposon or a pure effector. The killing of the Medea1-locus-lacking offspring is likely caused by the maternally derived Medea1-encoded CR-NTPase8-REase7 dyad endonucleolytically cleaving their genome. This cleavage probably happens in the neuro-muscular tissues during development as death is preceded by paralysis (117). In contrast, we posit that a phenomenon similar to target-immunity likely rescues the genome of those offspring with a functional Medea1 locus. In light of this it is conceivable that the other uncharacterized Medea loci (Medea2-4) in Tribolium (2,60,117) are the additional copies of this element.
A transposon role is less likely for CR-NTPase+CR-REase dyads, which display a clear Header domain, and those architectures with other effector domains, such as the REase+Kinase dyad, the GIMAP/AIG1-like GTPase, the peptidase CR-trypsin or potential pore-forming toxins. Additionally, the more extensive inter-paralog divergence observed in some of the CR-NTPase+CR-REase combinations lacking the Header domain (e.g. in amoebozoans) suggests that these forms are unlikely to have arisen from recent transposition events; this raises the possibility that they might have been fixed due to recruitment for other functions. Based on the existing precedence offered by the prokaryotic polymorphic toxins and related systems we propose that one possible function for the CR proteins (in particular those with distinct Header domains) in free-living eukaryotes is in conflict with other free-living forms. Just as prokaryotes deploy polymorphic toxins in conflicts between non-kin of the same species (11,20), it is conceivable that at least the microbial eukaryotes deploy CR proteins as effectors in comparable conflicts arising from competition with non-kin cells for common limiting resources and niches.
However, other functions are possible, especially for those with a transposase-like architecture, and lacking a Header domain. They could be deployed in intracellular conflicts involving parasitic nucleic acids, either in restriction of viral nucleic acids or as ‘policing’ agents that help restrict other transposons by specifically targeting them prior to or during integration. Such functions are particularly attractive for the versions from multicellular land plants (e.g. those from Arabidopsis, Vitis, Solanum and Theobroma) which have an N-terminal Myb domain (Figure 1M). Alternatively, in these multicellular organisms CR proteins could be used as counter-effectors against pathogens and mycorrhizal symbionts, such as oomycetes and fungi, which form intimate contacts with the plant cells. A further possibility is suggested by comparisons with expansions related to STAND-CDC6/ORC1 NTPases in prokaryotes. The prototypical members of this clade, the multiple paralogs of the archaeal CDC6/ORC1 proteins, recognize distinct origins of replication to either recruit the pre-initiation complex or in some cases negatively regulate the assembly of this complex (64,66,69). In both phage Mu and TN7, the NTPase subunit (respectively MuB and TnsC) of the transposase functions not only in recruitment of the endonuclease subunit(s) but also in target inhibition, i.e. inhibition of integration of another transposon at the same site (76,82,83). This indicates that a common functional denominator across this clade of NTPases is the recruitment of or the inhibition of assembly of protein complexes relating to replication and transposition. Hence, it is conceivable that such a role is more generally exploited across members of this clade. Several expansions related to the MNS clade STAND NTPases in archaea such as Methanococcus and Sulfolobus lack the associated endonuclease domain (either as a C-terminal domain or an adjacent gene in an operon). Such versions could potentially serve as a defensive strategy to block invasion of the genome by transposons by means of a target immunity-like activity. Likewise, the CR proteins with the CR-NTPase domains could also function similarly, especially in free-living organisms, to inhibit the action of effectors deployed by parasites, or the integration of transposons and viruses.
CONCLUSIONS
The above analysis of CR proteins suggests that they represent a wide-spread phenomenon in eukaryotes that encompasses and goes beyond host-pathogen interactions. The systematic delineation of domains in CR proteins along with the establishment of their basic architectural logic helps us understand their mechanism of action—both in terms of their effector function and trafficking (Figure 1). Thus, we are able to present a unified model for the action of a major class of eukaryotic effectors with both similarities and unique features when compared to their prokaryotic counterparts. Their effector moieties are predicted to primarily attack nucleic acids in target cells, though we recovered some others which are predicted to target proteins, utilize the target-cell Ubl system or breach membranes. In this respect they are comparable to their prokaryotic counterparts, in particular the polymorphic toxins and related systems (11–13). However, it should be noted that though the eukaryotic and prokaryotic systems share multiple catalytic domain superfamilies that function as effectors, the numerically dominant superfamilies in either system are different. CR proteins are unique in terms of their trafficking mechanisms. While signal peptides were previously claimed for some Crn effectors, we were able to dispel this possibility. Rather, we show that the eukaryotic CR proteins as a group are characterized by numerous distinct Header domains that include a widespread Rad23-like Ubl and several distinct α-helical domains. This pattern along with the absence of signal peptides to secrete these proteins via the general secretory pathway raises a key question regarding their export from the producing cell. Several eukaryotic and prokaryotic parasites have been proposed to secrete microvesicles, which might then fuse with the membrane of target cells to deliver their cargo (118,119). The signal-peptide-less architectures of the CR proteins suggest that this might be the dominant mechanism for the export of these effectors. In this proposal the Header domains could help in mediating specific interactions with other proteins or perhaps the membrane (in the case of the Angomonas Header) that allow them to be sequestered into such microvesicles.
The finding that the dominant architecture of CR proteins is related to transposases brings together multiple disparate areas of biological conflicts, i.e. intragenomic conflicts involving transposons, selfish elements that spread in populations (e.g. Tribolium Medea), and interorganismal conflicts involving secreted effectors. We had earlier noted that certain ‘preferred’ domains tend to be frequently exchanged between different conflict systems (7)—this study provides further evidence in this regard by showing how the CR-NTPase+CR-REase combination is potentially used both as a transposase and as an effector. Parallel to the selfish transmission of Medea, recent studies also suggest that post-recombination distortion of transmission might increase the number of recombinant offspring as part of response to parasites and pathogens (120). It remains to be seen if elements coding for CR proteins might have a role in phenomena such as these in organisms coding for them. Moreover, we also suggest that at least some of these CR proteins might retain their transposase-like function in eukaryotes and potentially facilitate diversification of multigene clusters in repetitive genomic regions by triggering recombination via their endonuclease activity. If this were indeed the case then these CR proteins might join other transposase-derived diversity generating systems, such as the Transib transposon-derived system in generation of diversity in jawed-vertebrate immune receptors (106) and retroelement-derived diversity generating systems in prokaryotes/bacteriophages (121). The identification of bacterial polymorphic toxins and related systems revealed that effectors are not only used in pathogen-host interactions but also in conflicts for resources between free-living forms (11). While in principle such systems should exist in eukaryotes, they have not been characterized at the molecular level. Identification of CR proteins in free-living eukaryotes provides a potential candidate for such systems. Thus, it opens new opportunities for investigating such conflicts that hitherto remained poorly understood in eukaryotes. Further, a subset of the CR proteins, which are not secreted, might provide insights into intracellular immunity mechanisms against invasive nucleic acids or parasites.
In conclusion, this study provides a framework to investigate this widespread class of proteins, which might help clarify multiple poorly understood biological conflicts of eukaryotes. Moreover, the effector domains, in particular the nucleases, characterized here also have the potential for being developed as reagents to target cellular nucleic acids with the objective of engineering specific outcomes.
AVAILABILITY
The supplementary information is also available from the following FTP site: ftp://ftp.ncbi.nih.gov/pub/aravind/CREFFECTORS/CR-effectors.html.
Note added in proof
While this paper was under review a new paper appeared (New Phytol. 2016, 210, 602-617), which described a CRN effector from the oomycete root pathogen Aphanomyces euteiches. This effector was shown to contain a HNH domain belonging to the CR-HNH1 clade described here and was shown to potentially damage DNA, consistent with our predictions.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural Research Program of National Library of Medicine at the National Institutes of Health, USA. NMV's postdoctoral fellowship is funded by a partnership between CNPq (Brazil) and the National Institutes of Health (USA).
Conflict of interest statement. None declared.
REFERENCES
- 1.Aravind L., Anantharaman V., Zhang D., de Souza R.F., Iyer L.M. Gene flow and biological conflict systems in the origin and evolution of eukaryotes. Front Cell Infect. Microbiol. 2012;2:89. doi: 10.3389/fcimb.2012.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burt A., Trivers R. Genes in Conflict: The Biology of Selfish Genetic Elements. Cambridge: The Belknap Press of Harvard University Press; 2006. [Google Scholar]
- 3.Dawkins R., Krebs J.R. Arms races between and within species. Proc. R. Soc. Lond. B. Biol. Sci. 1979;205:489–511. doi: 10.1098/rspb.1979.0081. [DOI] [PubMed] [Google Scholar]
- 4.Hurst L.D., Atlan A., Bengtsson B.O. Genetic conflicts. Q. Rev. Biol. 1996;71:317–364. doi: 10.1086/419442. [DOI] [PubMed] [Google Scholar]
- 5.Smith J.M., Price G.R. The logic of animal conflict. Nature. 1973;246:15–18. [Google Scholar]
- 6.Werren J.H. Selfish genetic elements, genetic conflict, and evolutionary innovation. Proc. Natl Acad. Sci. U.S.A. 2011;108(Suppl. 2):10863–10870. doi: 10.1073/pnas.1102343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang D., Iyer L.M., Burroughs A.M., Aravind L. Resilience of biochemical activity in protein domains in the face of structural divergence. Curr. Opin. Struct. Biol. 2014;26:92–103. doi: 10.1016/j.sbi.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aepfelbacher M., Aktories K., Just I. Bacterial Protein Toxins. Berlin; NY: Springer; 2000. [Google Scholar]
- 9.Alouf J.E., Popoff M.R. The Comprehensive Sourcebook of Bacterial Protein Toxins. 3rd edn. Amsterdam; Boston: Elsevier Academic Press; 2006. [Google Scholar]
- 10.Proft T. Microbial Toxins: Molecular and Cellular Biology. Norfolk: BIOS Scientific; 2005. [Google Scholar]
- 11.Zhang D., de Souza R.F., Anantharaman V., Iyer L.M., Aravind L. Polymorphic toxin systems: Comprehensive characterization of trafficking modes, processing, mechanisms of action, immunity and ecology using comparative genomics. Biol. Direct. 2012;7:18. doi: 10.1186/1745-6150-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang D., Iyer L.M., Aravind L. A novel immunity system for bacterial nucleic acid degrading toxins and its recruitment in various eukaryotic and DNA viral systems. Nucleic Acids Res. 2011;39:4532–4552. doi: 10.1093/nar/gkr036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iyer L.M., Zhang D., Rogozin I.B., Aravind L. Evolution of the deaminase fold and multiple origins of eukaryotic editing and mutagenic nucleic acid deaminases from bacterial toxin systems. Nucleic Acids Res. 2011;39:9473–9497. doi: 10.1093/nar/gkr691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anantharaman V., Aravind L. New connections in the prokaryotic toxin-antitoxin network: relationship with the eukaryotic nonsense-mediated RNA decay system. Genome Biol. 2003;4:R81. doi: 10.1186/gb-2003-4-12-r81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Melderen L. Toxin-antitoxin systems: why so many, what for? Curr. Opin. Microbiol. 2010;13:781–785. doi: 10.1016/j.mib.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 16.Leplae R., Geeraerts D., Hallez R., Guglielmini J., Dreze P., Van Melderen L. Diversity of bacterial type II toxin-antitoxin systems: a comprehensive search and functional analysis of novel families. Nucleic Acids Res. 2011;39:5513–5525. doi: 10.1093/nar/gkr131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buts L., Lah J., Dao-Thi M.H., Wyns L., Loris R. Toxin-antitoxin modules as bacterial metabolic stress managers. Trends Biochem. Sci. 2005;30:672–679. doi: 10.1016/j.tibs.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 18.Backert S., Meyer T.F. Type IV secretion systems and their effectors in bacterial pathogenesis. Curr. Opin. Microbiol. 2006;9:207–217. doi: 10.1016/j.mib.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 19.Galan J.E., Wolf-Watz H. Protein delivery into eukaryotic cells by type III secretion machines. Nature. 2006;444:567–573. doi: 10.1038/nature05272. [DOI] [PubMed] [Google Scholar]
- 20.Aoki S.K., Diner E.J., de Roodenbeke C.T., Burgess B.R., Poole S.J., Braaten B.A., Jones A.M., Webb J.S., Hayes C.S., Cotter P.A., et al. A widespread family of polymorphic contact-dependent toxin delivery systems in bacteria. Nature. 2010;468:439–442. doi: 10.1038/nature09490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cascales E., Buchanan S.K., Duche D., Kleanthous C., Lloubes R., Postle K., Riley M., Slatin S., Cavard D. Colicin biology. Microbiol. Mol. Biol. Rev. 2007;71:158–229. doi: 10.1128/MMBR.00036-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Daw M.A., Falkiner F.R. Bacteriocins: nature, function and structure. Micron. 1996;27:467–479. doi: 10.1016/s0968-4328(96)00028-5. [DOI] [PubMed] [Google Scholar]
- 23.Konisky J. Colicins and other bacteriocins with established modes of action. Annu. Rev. Microbiol. 1982;36:125–144. doi: 10.1146/annurev.mi.36.100182.001013. [DOI] [PubMed] [Google Scholar]
- 24.Morigasaki S., Shiozaki K. Phosphorelay-dependent and -independent regulation of MAPKKK by the Mcs4 response regulator in fission yeast. Commun. Integr. Biol. 2013;6:e25020. doi: 10.4161/cib.25020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thompson J.N. The Coevolutionary Process. Chicago: University of Chicago Press; 1994. [Google Scholar]
- 26.Saeij J.P., Coller S., Boyle J.P., Jerome M.E., White M.W., Boothroyd J.C. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature. 2007;445:324–327. doi: 10.1038/nature05395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baum J., Gilberger T.W., Frischknecht F., Meissner M. Host-cell invasion by malaria parasites: insights from Plasmodium and Toxoplasma. Trends Parasitol. 2008;24:557–563. doi: 10.1016/j.pt.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 28.Oakley M.S., Kumar S., Anantharaman V., Zheng H., Mahajan B., Haynes J.D., Moch J.K., Fairhurst R., McCutchan T.F., Aravind L. Molecular factors and biochemical pathways induced by febrile temperature in intraerythrocytic Plasmodium falciparum parasites. Infect. Immun. 2007;75:2012–2025. doi: 10.1128/IAI.01236-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Jonge R., Bolton M.D., Thomma B.P. How filamentous pathogens co-opt plants: the ins and outs of fungal effectors. Curr. Opin. Plant Biol. 2011;14:400–406. doi: 10.1016/j.pbi.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 30.Rosenblum E.B., Poorten T.J., Joneson S., Settles M. Substrate-specific gene expression in Batrachochytrium dendrobatidis, the chytrid pathogen of amphibians. PLoS One. 2012;7:e49924. doi: 10.1371/journal.pone.0049924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stergiopoulos I., de Wit P.J. Fungal effector proteins. Annu. Rev. Phytopathol. 2009;47:233–263. doi: 10.1146/annurev.phyto.112408.132637. [DOI] [PubMed] [Google Scholar]
- 32.Haas B.J., Kamoun S., Zody M.C., Jiang R.H., Handsaker R.E., Cano L.M., Grabherr M., Kodira C.D., Raffaele S., Torto-Alalibo T., et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature. 2009;461:393–398. doi: 10.1038/nature08358. [DOI] [PubMed] [Google Scholar]
- 33.Wawra S., Belmonte R., Lobach L., Saraiva M., Willems A., van West P. Secretion, delivery and function of oomycete effector proteins. Curr. Opin. Microbiol. 2012;15:685–691. doi: 10.1016/j.mib.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 34.Jiang R.H., Tyler B.M. Mechanisms and evolution of virulence in oomycetes. Annu. Rev. Phytopathol. 2012;50:295–318. doi: 10.1146/annurev-phyto-081211-172912. [DOI] [PubMed] [Google Scholar]
- 35.Hwang S.F., Strelkov S.E., Feng J., Gossen B.D., Howard R.J. Plasmodiophora brassicae: a review of an emerging pathogen of the Canadian canola (Brassica napus) crop. Mol. Plant Pathol. 2012;13:105–113. doi: 10.1111/j.1364-3703.2011.00729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Torto T.A., Li S., Styer A., Huitema E., Testa A., Gow N.A., van West P., Kamoun S. EST mining and functional expression assays identify extracellular effector proteins from the plant pathogen Phytophthora. Genome Res. 2003;13:1675–1685. doi: 10.1101/gr.910003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stam R., Jupe J., Howden A.J., Morris J.A., Boevink P.C., Hedley P.E., Huitema E. Identification and characterisation CRN effectors in Phytophthora capsici shows modularity and functional diversity. PLoS One. 2013;8:e59517. doi: 10.1371/journal.pone.0059517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Damme M., Bozkurt T.O., Cakir C., Schornack S., Sklenar J., Jones A.M., Kamoun S. The Irish potato famine pathogen Phytophthora infestans translocates the CRN8 kinase into host plant cells. PLoS Pathog. 2012;8:e1002875. doi: 10.1371/journal.ppat.1002875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jia Y., McAdams S.A., Bryan G.T., Hershey H.P., Valent B. Direct interaction of resistance gene and avirulence gene products confers rice blast resistance. EMBO J. 2000;19:4004–4014. doi: 10.1093/emboj/19.15.4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dong S., Yin W., Kong G., Yang X., Qutob D., Chen Q., Kale S.D., Sui Y., Zhang Z., Dou D., et al. Phytophthora sojae avirulence effector Avr3b is a secreted NADH and ADP-ribose pyrophosphorylase that modulates plant immunity. PLoS Pathog. 2011;7:e1002353. doi: 10.1371/journal.ppat.1002353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Joneson S., Stajich J.E., Shiu S.H., Rosenblum E.B. Genomic transition to pathogenicity in chytrid fungi. PLoS Pathog. 2011;7:e1002338. doi: 10.1371/journal.ppat.1002338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin K., Limpens E., Zhang Z., Ivanov S., Saunders D.G., Mu D., Pang E., Cao H., Cha H., Lin T., et al. Single nucleus genome sequencing reveals high similarity among nuclei of an endomycorrhizal fungus. PLoS Genet. 2014;10:e1004078. doi: 10.1371/journal.pgen.1004078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zuluaga A.P., Vega-Arreguin J.C., Fei Z., Ponnala L., Lee S.J., Matas A.J., Patev S., Fry W.E., Rose J.K. Transcriptional dynamics of Phytophthora infestans during sequential stages of hemibiotrophic infection of tomato. Mol. Plant Pathol. 2015;17:29–41. doi: 10.1111/mpp.12263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mafurah J.J., Ma H., Zhang M., Xu J., He F., Ye T., Shen D., Chen Y., Rajput N.A., Dou D. A virulence essential CRN effector of Phytophthora capsici suppresses host defense and induces cell death in plant nucleus. PLoS One. 2015;10:e0127965. doi: 10.1371/journal.pone.0127965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu T., Ye W., Ru Y., Yang X., Gu B., Tao K., Lu S., Dong S., Zheng X., Shan W., et al. Two host cytoplasmic effectors are required for pathogenesis of Phytophthora sojae by suppression of host defenses. Plant Physiol. 2011;155:490–501. doi: 10.1104/pp.110.166470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schornack S., van Damme M., Bozkurt T.O., Cano L.M., Smoker M., Thines M., Gaulin E., Kamoun S., Huitema E. Ancient class of translocated oomycete effectors targets the host nucleus. Proc. Natl Acad. Sci. U.S.A. 2010;107:17421–17426. doi: 10.1073/pnas.1008491107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stam R., Howden A.J., Delgado-Cerezo M., TM M.M.A., Motion G.B., Pham J., Huitema E. Characterization of cell death inducing Phytophthora capsici CRN effectors suggests diverse activities in the host nucleus. Front Plant Sci. 2013;4:387. doi: 10.3389/fpls.2013.00387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shen D., Liu T., Ye W., Liu L., Liu P., Wu Y., Wang Y., Dou D. Gene duplication and fragment recombination drive functional diversification of a superfamily of cytoplasmic effectors in Phytophthora sojae. PLoS One. 2013;8:e70036. doi: 10.1371/journal.pone.0070036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eddy S.R. A new generation of homology search tools based on probabilistic inference. Genome informatics. International Conference on Genome Informatics. 2009;23:205–211. [PubMed] [Google Scholar]
- 51.Soding J., Biegert A., Lupas A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lassmann T., Sonnhammer E.L. Kalign–an accurate and fast multiple sequence alignment algorithm. BMC Bioinform. 2005;6:298. doi: 10.1186/1471-2105-6-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pei J., Kim B.H., Grishin N.V. PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008;36:2295–2300. doi: 10.1093/nar/gkn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Drozdetskiy A., Cole C., Procter J., Barton G.J. JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 2015;43:W389–W394. doi: 10.1093/nar/gkv332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Price M.N., Dehal P.S., Arkin A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PloS One. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guindon S., Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 58.Bringaud F., Biteau N., Melville S.E., Hez S., El-Sayed N.M., Leech V., Berriman M., Hall N., Donelson J.E., Baltz T. A new, expressed multigene family containing a hot spot for insertion of retroelements is associated with polymorphic subtelomeric regions of Trypanosoma brucei. Eukaryot. Cell. 2002;1:137–151. doi: 10.1128/EC.1.1.137-151.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.El-Sayed N.M., Ghedin E., Song J., MacLeod A., Bringaud F., Larkin C., Wanless D., Peterson J., Hou L., Taylor S., et al. The sequence and analysis of Trypanosoma brucei chromosome II. Nucleic Acids Res. 2003;31:4856–4863. doi: 10.1093/nar/gkg673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lorenzen M.D., Gnirke A., Margolis J., Garnes J., Campbell M., Stuart J.J., Aggarwal R., Richards S., Park Y., Beeman R.W. The maternal-effect, selfish genetic element Medea is associated with a composite Tc1 transposon. Proc. Natl Acad. Sci. U.S.A. 2008;105:10085–10089. doi: 10.1073/pnas.0800444105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Punta M., Coggill P.C., Eberhardt R.Y., Mistry J., Tate J., Boursnell C., Pang N., Forslund K., Ceric G., Clements J., et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aravind L., Makarova K.S., Koonin E.V. Holliday junction resolvases and related nucleases: identification of new families, phyletic distribution and evolutionary trajectories. Nucleic Acids Res. 2000;28:3417–3432. doi: 10.1093/nar/28.18.3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Knizewski L., Kinch L.N., Grishin N.V., Rychlewski L., Ginalski K. Realm of PD-(D/E)XK nuclease superfamily revisited: detection of novel families with modified transitive meta profile searches. BMC Struct. Biol. 2007;7:40. doi: 10.1186/1472-6807-7-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Iyer L.M., Leipe D.D., Koonin E.V., Aravind L. Evolutionary history and higher order classification of AAA+ ATPases. J. Struct. Biol. 2004;146:11–31. doi: 10.1016/j.jsb.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 65.Leipe D.D., Koonin E.V., Aravind L. STAND, a class of P-loop NTPases including animal and plant regulators of programmed cell death: multiple, complex domain architectures, unusual phyletic patterns, and evolution by horizontal gene transfer. J. Mol. Biol. 2004;343:1–28. doi: 10.1016/j.jmb.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 66.Dueber E.L., Corn J.E., Bell S.D., Berger J.M. Replication origin recognition and deformation by a heterodimeric archaeal Orc1 complex. Science. 2007;317:1210–1213. doi: 10.1126/science.1143690. [DOI] [PubMed] [Google Scholar]
- 67.Aravind L., Mazumder R., Vasudevan S., Koonin E.V. Trends in protein evolution inferred from sequence and structure analysis. Curr. Opin. Struct. Biol. 2002;12:392–399. doi: 10.1016/s0959-440x(02)00334-2. [DOI] [PubMed] [Google Scholar]
- 68.Iyer L.M., Aravind L. Insights from the architecture of the bacterial transcription apparatus. J. Struct. Biol. 2012;179:299–319. doi: 10.1016/j.jsb.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dueber E.C., Costa A., Corn J.E., Bell S.D., Berger J.M. Molecular determinants of origin discrimination by Orc1 initiators in archaea. Nucleic Acids Res. 2011;39:3621–3631. doi: 10.1093/nar/gkq1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yamauchi M., Baker T.A. An ATP-ADP switch in MuB controls progression of the Mu transposition pathway. EMBO J. 1998;17:5509–5518. doi: 10.1093/emboj/17.18.5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ronning D.R., Li Y., Perez Z.N., Ross P.D., Hickman A.B., Craig N.L., Dyda F. The carboxy-terminal portion of TnsC activates the Tn7 transposase through a specific interaction with TnsA. EMBO J. 2004;23:2972–2981. doi: 10.1038/sj.emboj.7600311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bainton R.J., Kubo K.M., Feng J.N., Craig N.L. Tn7 transposition: target DNA recognition is mediated by multiple Tn7-encoded proteins in a purified in vitro system. Cell. 1993;72:931–943. doi: 10.1016/0092-8674(93)90581-a. [DOI] [PubMed] [Google Scholar]
- 73.Strozen T.G., Stanley H., Gu Y., Boyd J., Bagdasarian M., Sandkvist M., Howard S.P. Involvement of the GspAB complex in assembly of the type II secretion system secretin of Aeromonas and Vibrio species. J. Bacteriol. 2011;193:2322–2331. doi: 10.1128/JB.01413-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Majorek K.A., Dunin-Horkawicz S., Steczkiewicz K., Muszewska A., Nowotny M., Ginalski K., Bujnicki J.M. The RNase H-like superfamily: new members, comparative structural analysis and evolutionary classification. Nucleic Acids Res. 2014;42:4160–4179. doi: 10.1093/nar/gkt1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rice P., Mizuuchi K. Structure of the bacteriophage Mu transposase core: a common structural motif for DNA transposition and retroviral integration. Cell. 1995;82:209–220. doi: 10.1016/0092-8674(95)90308-9. [DOI] [PubMed] [Google Scholar]
- 76.Han Y.W., Mizuuchi K. Phage Mu transposition immunity: protein pattern formation along DNA by a diffusion-ratchet mechanism. Mol. Cell. 2010;39:48–58. doi: 10.1016/j.molcel.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Choi K.Y., Spencer J.M., Craig N.L. The Tn7 transposition regulator TnsC interacts with the transposase subunit TnsB and target selector TnsD. Proc. Natl Acad. Sci. U.S.A. 2014;111:E2858–E2865. doi: 10.1073/pnas.1409869111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.May E.W., Craig N.L. Switching from cut-and-paste to replicative Tn7 transposition. Science. 1996;272:401–404. doi: 10.1126/science.272.5260.401. [DOI] [PubMed] [Google Scholar]
- 79.Maxwell A., Craigie R., Mizuuchi K. B protein of bacteriophage mu is an ATPase that preferentially stimulates intermolecular DNA strand transfer. Proc. Natl Acad. Sci. U.S.A. 1987;84:699–703. doi: 10.1073/pnas.84.3.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gamas P., Craig N.L. Purification and characterization of TnsC, a Tn7 transposition protein that binds ATP and DNA. Nucleic Acids Res. 1992;20:2525–2532. doi: 10.1093/nar/20.10.2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mizuuchi K., Adzuma K. Inversion of the phosphate chirality at the target site of Mu DNA strand transfer: evidence for a one-step transesterification mechanism. Cell. 1991;66:129–140. doi: 10.1016/0092-8674(91)90145-o. [DOI] [PubMed] [Google Scholar]
- 82.Stellwagen A.E., Craig N.L. Avoiding self: two Tn7-encoded proteins mediate target immunity in Tn7 transposition. EMBO J. 1997;16:6823–6834. doi: 10.1093/emboj/16.22.6823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Skelding Z., Queen-Baker J., Craig N.L. Alternative interactions between the Tn7 transposase and the Tn7 target DNA binding protein regulate target immunity and transposition. EMBO J. 2003;22:5904–5917. doi: 10.1093/emboj/cdg551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Greene E.C., Mizuuchi K. Target immunity during Mu DNA transposition. Transpososome assembly and DNA looping enhance MuA-mediated disassembly of the MuB target complex. Mol. Cell. 2002;10:1367–1378. doi: 10.1016/s1097-2765(02)00733-5. [DOI] [PubMed] [Google Scholar]
- 85.Kannan N., Taylor S.S., Zhai Y., Venter J.C., Manning G. Structural and functional diversity of the microbial kinome. PLoS Biol. 2007;5:e17. doi: 10.1371/journal.pbio.0050017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Leonard C.J., Aravind L., Koonin E.V. Novel families of putative protein kinases in bacteria and archaea: evolution of the ‘eukaryotic’ protein kinase superfamily. Genome Res. 1998;8:1038–1047. doi: 10.1101/gr.8.10.1038. [DOI] [PubMed] [Google Scholar]
- 87.Ishikawa K., Fukuda E., Kobayashi I. Conflicts targeting epigenetic systems and their resolution by cell death: novel concepts for methyl-specific and other restriction systems. DNA Res. 2010;17:325–342. doi: 10.1093/dnares/dsq027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Krishna S.S., Majumdar I., Grishin N.V. Structural classification of zinc fingers: survey and summary. Nucleic Acids Res. 2003;31:532–550. doi: 10.1093/nar/gkg161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Iyer L.M., Balaji S., Koonin E.V., Aravind L. Evolutionary genomics of nucleo-cytoplasmic large DNA viruses. Virus Res. 2006;117:156–184. doi: 10.1016/j.virusres.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 90.Anantharaman V., Zhang D., Aravind L. OST-HTH: a novel predicted RNA-binding domain. Biol. Direct. 2010;5:13. doi: 10.1186/1745-6150-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Anantharaman V., Aravind L. The NYN domains: novel predicted RNAses with a PIN domain-like fold. RNA Biol. 2006;3:18–27. doi: 10.4161/rna.3.1.2548. [DOI] [PubMed] [Google Scholar]
- 92.Winther K.S., Brodersen D.E., Brown A.K., Gerdes K. VapC20 of Mycobacterium tuberculosis cleaves the sarcin-ricin loop of 23S rRNA. Nat. Commun. 2013;4:2796. doi: 10.1038/ncomms3796. [DOI] [PubMed] [Google Scholar]
- 93.Nienaber V.L., Breddam K., Birktoft J.J. A glutamic acid specific serine protease utilizes a novel histidine triad in substrate binding. Biochemistry. 1993;32:11469–11475. doi: 10.1021/bi00094a001. [DOI] [PubMed] [Google Scholar]
- 94.Strunnikov A.V., Aravind L., Koonin E.V. Saccharomyces cerevisiae SMT4 encodes an evolutionarily conserved protease with a role in chromosome condensation regulation. Genetics. 2001;158:95–107. doi: 10.1093/genetics/158.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li S.J., Hochstrasser M. The Ulp1 SUMO isopeptidase: distinct domains required for viability, nuclear envelope localization, and substrate specificity. J. Cell Biol. 2003;160:1069–1081. doi: 10.1083/jcb.200212052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang S., Xu J.R. Effectors and effector delivery in Magnaporthe oryzae. PLoS Pathog. 2014;10:e1003826. doi: 10.1371/journal.ppat.1003826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schwefel D., Frohlich C., Eichhorst J., Wiesner B., Behlke J., Aravind L., Daumke O. Structural basis of oligomerization in septin-like GTPase of immunity-associated protein 2 (GIMAP2) Proc. Natl Acad. Sci. U.S.A. 2010;107:20299–20304. doi: 10.1073/pnas.1010322107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ellisdon A.M., Reboul C.F., Panjikar S., Huynh K., Oellig C.A., Winter K.L., Dunstone M.A., Hodgson W.C., Seymour J., Dearden P.K., et al. Stonefish toxin defines an ancient branch of the perforin-like superfamily. Proc. Natl Acad. Sci .U.S.A. 2015;112:15360–15365. doi: 10.1073/pnas.1507622112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gutjahr C., Parniske M. Cell and developmental biology of arbuscular mycorrhiza symbiosis. Annu. Rev. Cell Dev. Biol. 2013;29:593–617. doi: 10.1146/annurev-cellbio-101512-122413. [DOI] [PubMed] [Google Scholar]
- 100.Geyer R., Wee S., Anderson S., Yates J., Wolf D.A. BTB/POZ domain proteins are putative substrate adaptors for cullin 3 ubiquitin ligases. Mol. Cell. 2003;12:783–790. doi: 10.1016/s1097-2765(03)00341-1. [DOI] [PubMed] [Google Scholar]
- 101.Blaise M., Alsarraf H.M., Wong J.E., Midtgaard S.R., Laroche F., Schack L., Spaink H., Stougaard J., Thirup S. Crystal structure of the TLDc domain of oxidation resistance protein 2 from zebrafish. Proteins. 2012;80:1694–1698. doi: 10.1002/prot.24050. [DOI] [PubMed] [Google Scholar]
- 102.Burroughs A.M., Zhang D., Schaffer D.E., Iyer L.M., Aravind L. Comparative genomic analyses reveal a vast, novel network of nucleotide-centric systems in biological conflicts, immunity and signaling. Nucleic Acids Res. 2015;43:10633–10654. doi: 10.1093/nar/gkv1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gilbert R.J. Pore-forming toxins. Cell. Mol. Life Sci. 2002;59:832–844. doi: 10.1007/s00018-002-8471-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Iyer L.M., Abhiman S., Aravind L. A new family of polymerases related to superfamily A DNA polymerases and T7-like DNA-dependent RNA polymerases. Biol. Direct. 2008;3:39. doi: 10.1186/1745-6150-3-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Makarova K.S., Aravind L., Koonin E.V. A novel superfamily of predicted cysteine proteases from eukaryotes, viruses and Chlamydia pneumoniae. Trends Biochem. Sci. 2000;25:50–52. doi: 10.1016/s0968-0004(99)01530-3. [DOI] [PubMed] [Google Scholar]
- 106.Kapitonov V.V., Jurka J. RAG1 core and V(D)J recombination signal sequences were derived from Transib transposons. PLoS Biol. 2005;3:e181. doi: 10.1371/journal.pbio.0030181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kall L., Krogh A., Sonnhammer E.L. Advantages of combined transmembrane topology and signal peptide prediction–the Phobius web server. Nucleic Acids Res. 2007;35:W429–W432. doi: 10.1093/nar/gkm256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nielsen H., Engelbrecht J., Brunak S., von Heijne G. A neural network method for identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Int. J. Neural. Syst. 1997;8:581–599. doi: 10.1142/s0129065797000537. [DOI] [PubMed] [Google Scholar]
- 109.Burroughs A.M., Balaji S., Iyer L.M., Aravind L. Small but versatile: the extraordinary functional and structural diversity of the beta-grasp fold. Biol. Direct. 2007;2:18. doi: 10.1186/1745-6150-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chauhan N., Latge J.P., Calderone R. Signalling and oxidant adaptation in Candida albicans and Aspergillus fumigatus. Nat. Rev. Microbiol. 2006;4:435–444. doi: 10.1038/nrmicro1426. [DOI] [PubMed] [Google Scholar]
- 111.Schultz J., Ponting C.P., Hofmann K., Bork P. SAM as a protein interaction domain involved in developmental regulation. Protein Sci. 1997;6:249–253. doi: 10.1002/pro.5560060128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jones J.D., Dangl J.L. The plant immune system. Nature. 2006;444:323–329. doi: 10.1038/nature05286. [DOI] [PubMed] [Google Scholar]
- 113.Park C.H., Chen S., Shirsekar G., Zhou B., Khang C.H., Songkumarn P., Afzal A.J., Ning Y., Wang R., Bellizzi M., et al. The Magnaporthe oryzae effector AvrPiz-t targets the RING E3 ubiquitin ligase APIP6 to suppress pathogen-associated molecular pattern-triggered immunity in rice. Plant Cell. 2012;24:4748–4762. doi: 10.1105/tpc.112.105429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schwelm A., Fogelqvist J., Knaust A., Julke S., Lilja T., Bonilla-Rosso G., Karlsson M., Shevchenko A., Dhandapani V., Choi S.R., et al. The Plasmodiophora brassicae genome reveals insights in its life cycle and ancestry of chitin synthases. Sci. Rep. 2015;5:11153. doi: 10.1038/srep11153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Owczarzak A., Stibbs H.H., Bayne C.J. The destruction of Schistosoma mansoni mother sporocysts in vitro by amoebae isolated from Biomphalaria glabrata: an ultrastructural study. J. Invertebr. Pathol. 1980;35:26–33. doi: 10.1016/0022-2011(80)90079-8. [DOI] [PubMed] [Google Scholar]
- 116.El-Sayed N.M., Myler P.J., Bartholomeu D.C., Nilsson D., Aggarwal G., Tran A.N., Ghedin E., Worthey E.A., Delcher A.L., Blandin G., et al. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science. 2005;309:409–415. doi: 10.1126/science.1112631. [DOI] [PubMed] [Google Scholar]
- 117.Beeman R.W., Friesen K.S., Denell R.E. Maternal-effect selfish genes in flour beetles. Science. 1992;256:89–92. doi: 10.1126/science.1566060. [DOI] [PubMed] [Google Scholar]
- 118.Mantel P.Y., Hoang A.N., Goldowitz I., Potashnikova D., Hamza B., Vorobjev I., Ghiran I., Toner M., Irimia D., Ivanov A.R., et al. Malaria-infected erythrocyte-derived microvesicles mediate cellular communication within the parasite population and with the host immune system. Cell Host Microbe. 2013;13:521–534. doi: 10.1016/j.chom.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Silverman J.M., Reiner N.E. Exosomes and other microvesicles in infection biology: organelles with unanticipated phenotypes. Cell Microbiol. 2011;13:1–9. doi: 10.1111/j.1462-5822.2010.01537.x. [DOI] [PubMed] [Google Scholar]
- 120.Singh N.D., Criscoe D.R., Skolfield S., Kohl K.P., Keebaugh E.S., Schlenke T.A. Fruit flies diversify their offspring in response to parasite infection. Science. 2015;349:747–750. doi: 10.1126/science.aab1768. [DOI] [PubMed] [Google Scholar]
- 121.Medhekar B., Miller J.F. Diversity-generating retroelements. Curr. Opin. Microbiol. 2007;10:388–395. doi: 10.1016/j.mib.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.