

Abstract
A highly rapid and sensitive liquid chromatographic–electrospray ionization tandem mass spectrometric (LC–ESI-MS/MS) method was developed and validated for the determination of trans-δ-viniferin (Rs-1) in rat plasma, urine and feces. All biological samples were prepared by liquid–liquid extraction and hesperetin was included as an internal standard (IS). Chromatographic separation was achieved on a shim-pack XR-ODS column using a gradient mobile phase. MS/MS detection was performed by negative ion electrospray ionization. The method was sensitive with a lower limit of quantification of 1.42 ng/mL and linear over the range of 1.42–2172 ng/mL in all matrices. The method was applied to study the pharmacokinetics, bioavailability, metabolism, and excretion of Rs-1 in rats following a single oral or intravenous dose. Two metabolites, Rs-1 glucuronide and Rs-1 sulfate, were detected in plasma and in urine after administration of Rs-1. The absolute oral bioavailability of Rs-1 was 2.3%, and the total absorption rose to 31.5% with addition of its glucuronide and sulfate metabolites. Only 0.09% of the gavaged dose, including Rs-1 and metabolites, was excreted in the urine, while 60.3% was found in the feces in unchanged form. The results indicate that both poor absorption and extensive metabolism were the important factors that led to the poor bioavailability of Rs-1, which can provide a basis for further studies on structural modification and dosage form design.
KEY WORDS: Viniferin, Pharmacokinetics, Excretion, Metabolism, Bioavailability, LC–MS/MS
Graphical abstract
An LC–ESI-MS/MS assay for δ-viniferin (Rs-1) and its metabolites in rat plasma, urine and feces was applied in pharmacokinetics of Rs-1 in rats. The absolute oral bioavailability of Rs-1 was 2.3%. 60.3% of the gavage dose Rs-1 was excreted through feces in unchanged form.
1. Introduction
Resveratrol (trans-3,5,4′-trihydroxystilbene) and its oligomers, including their glucosides, are widely distributed in families, such as Dipterocarpaceae, Vitaceae, Cyperaceae, Leguminosae and Gnetaceae. Monomeric resveratrol displays a broad range of pharmacological activities including cardioprotection, antioxidation, protection against brain damage and prevention against cancers1, 2, 3, 4. Resveratrol can be polymerized into resveratrol oligomers (so-called viniferins) in fungi or plants5, 6, 7, 8, 9 and those resveratrol oligomers have more extensive pharmacological activities, such as anti-HIV and antitumor effects10, 11, 12, 13. However, as with resveratrol and other stilbenes, the pharmacokinetic properties of those compounds are not favorable due to their poor bioavailability, extensive metabolism and rapid elimination14, 15.
Trans-δ-viniferin (Rs-1, Fig. 1), a resveratrol-trans-dehydrodimer, was first found in grapevine (Vitis vinifera L.) in 197716, and many studies have demonstrated that Rs-1 has numerous pharmacological properties, such as anti-inflammation6, 17, antimicrobial action18, 19, antioxidation7, cardioprotection8 and antidiabetes20.
Figure 1.

Chemical structures of Rs-1 and hesperetin (IS).
It has been shown that resveratrol has high absorption but very low bioavailability after oral administration in humans. At least 70% of oral dose was absorbed based on the urinary excretion data, and the relatively poor bioavailability was mainly owing to rapid metabolism14. A previous study has shown that the bioavailability of Rs-1 in rats was low21, but whether poor absorption or rapid metabolism or both were responsible for the poor bioavailability was not determined. The bioavailability, metabolism and excretion of Rs-1 were investigated in the present study. A highly rapid and sensitive LC–MS/MS method was developed and validated for the determination of Rs-1 in rat plasma, urine and feces. This method yielded shorter sample turnover rate per sample and higher sensitivity compared with a previous study21. The method was applied to the assessment of the pharmacokinetics, bioavailability, metabolism and excretion of Rs-1 in healthy rats, and LC–MS/MS was used to identify Rs-1 metabolites.
2. Materials and methods
2.1. Materials and reagents
Rs-1 (purity >98%) was provided by the Institute of Materia Medica, Chinese Academy of Medical Sciences (Beijing, China). Hesperetin (Fig. 1), the internal standard (IS), was purchased from Sigma (St. Louis, MO, USA). Acetonitrile and methanol were purchased from ANPEL (Shanghai, China) and acetic acid was from OE Scientific Inc. (Newark, NJ, USA). All solvents were HPLC grade. Other chemicals used were analytical grade and obtained from Sigma–Aldrich (St. Louis, MO, USA). Water used in this study was prepared in a Milli-Q water purification system (Billerica, MA, USA).
2.2. Liquid chromatography and tandem mass spectrometry conditions
Chromatographic separations were performed on a Shimadzu UFLC-20AD XR system (Shimadzu, Tokyo, Japan) with a shim-pack XR-ODS column (75 mm×3 mm, 2.2 μm, Shimadzu, Tokyo, Japan) maintained at a temperature of 40 °C and a flow rate of 0.3 mL/min. The mobile phase was composed of A (0.1% formic acid–water, v/v) and B (0.1% formic acid–acetonitrile, v/v). A gradient elution program was as follow: 0–0.3 min, 45%–75% B; 0.3–1.8 min, 75% B; 1.8–2.1 min, 75%–45% B; 2.1–4 min, 45% B. The injection volume was 10 μL.
The Shimadzu UFLC-20AD XR system was coupled on-line to an Applied Biosystems Sciex Qtrap 5500 (MDS-Sciex, Concord, Canada) with an electrospray ionization (ESI) interface. The mass spectrometry was carried out using multiple reaction monitoring (MRM) and operated by electrospray ionization in negative mode for Rs-1 and IS. Transitions of precursor→product ion were monitored at m/z 453.2→411.2 for Rs-1 with a declustering potential (DP) of –178 V, collision energy (CE) of –35 V, and m/z 301.1→163.9 for IS with a DP of –100 V, CE of –32 V. Their collision exit potential (CXP) and entrance potential (EP) were both –10 V. The source parameters were optimized as follows: ion spray voltage, –4500 V; temperature, 550 °C; curtain gas (nitrogen), 28 psi; nebulizer and turbo gases (nitrogen), 50 and 50 psi, respectively. MRM transition was monitored with a dwell time of 200 ms. Quantification was determined from the peak area ratio of Rs-1 to IS. Analyst 1.6 software was used for control of equipment, data acquisition and analysis. The full scan product mass spectra of Rs-1 and IS are shown in Fig. 2.
Figure 2.

Product ion spectra (MS/MS) of (A) Rs-1 and (B) hesperetin (IS).
2.3. Preparation of stock and working solutions
Stock solutions of Rs-1 and hesperetin (IS) at 0.2 mg/mL were prepared by dissolving the proper amount of the accurately weighed reference material in methanol. A series of working solutions for calibration standards (CS) in a concentration range of 14.2–21,720 ng/mL and quality control (QC) at 14.2, 35.6, 3480 and 17,380 ng/mL were prepared by subsequently diluting Rs-1 stock solution with methanol. The IS working solution at 200 ng/mL was prepared by diluting IS stock solution with methanol. All of the solutions were stored at –20 °C.
2.4. Preparation of standard and quality control samples
CS and QC samples of Rs-1 were prepared freshly by spiking the appropriate amount of the working solution into 100 μL of blank plasma, urine, feces suspension (1 g feces was homogenized with 20 mL of saline for 2 min). CS samples were prepared at 9 concentrations of 1.42–2172 ng/mL and QC samples were at 4 levels of 1.42 (lower limit of quantification QC), 3.56 (low QC), 348 (middle QC) and 1738 (high QC) ng/mL.
2.5. Sample preparation
A liquid–liquid extraction (LLE) method was used to extract the analyte from biological samples (plasma, urine and feces). For Rs-1 analysis, an aliquot of 100 μL of each plasma, urine or feces suspension sample was mixed with 20 μL of IS working solution and the mixture was vortex-mixed for 30 s. For Rs-1 in plasma and urine, 600 μL ethyl acetate was added, and in feces, 600 μL diethyl ether was added. After vortex-mixing for 3 min and centrifugation at 12,000 rpm for 10 min at 4 °C, the supernatant fraction was transferred to a new tube and evaporated to dryness under a gentle nitrogen stream at 40 °C. The residue was reconstituted in 200 μL methanol, vortex-mixed for 3 min and subjected to centrifugation at 12,000 rpm for 10 min at 4 °C. The supernate was injected for LC–MS/MS analysis. To avoid photochemical isomerization of Rs-1 to the cis form all laboratory procedures were performed in dim light.
2.6. Hydrolysis of Rs-1 glucuronide and sulfate with β-glucuronidase and sulfatase
For quantification of Rs-1 glucuronide metabolites and sulfate metabolites, plasma samples and urine samples were incubated with β-glucuronidase (Type B-1, Sigma–Aldrich) or sulfatase (Type VIII, Sigma–Aldrich). The content difference of Rs-1 between samples with and without enzymolysis was used to calculate the concentration of Rs-1 glucuronide or sulfate. For Rs-1 glucuronide analysis, 5000 U of enzyme dissolved in 20 μL of enzyme buffer (0.17 mol/L ammonium acetate, pH 5.0) was added to 10 μL plasma or 20 μL urine. The mixture was vortex-mixed for 30 s and incubated with agitation at 37 °C for 2 h. For sulfate metabolites of Rs-1, 10 μL of plasma or 20 μL of urine was incubated with 20 U of sulfatase dissolved in 40 μL of the above enzyme buffer. The plasma samples were incubated with agitation at 37 °C for 36 h, while the urine samples for 12 h. After incubation, 90 μL of blank plasma or 80 μL of blank urine was added to the corresponding samples before analysis. The following procedures were carried out to determine Rs-1, as described in Section 2.5.
2.7. Method validation
The extraction methods were validated according to the Guidance for Industry, Bioanalytical Method Validation of the Food and Drug Administration (US FDA)22 and European Medical Agency (EMA) guidelines23 of bioanalytical method validation.
Selectivity was investigated by analyzing blank plasma samples, urine, and feces from 6 individual rats that were not administered Rs-1 and comparing with the corresponding spiked biological matrices at the lower limit of quantification (LLOQ) level or the samples collected after intravenous or oral administration of Rs-1. There should be no interference from the endogenous substances at the retention times of both Rs-1 and IS using the proposed pretreatment procedure and LC–MS/MS conditions.
The calibration curve was acquired by comparing the peak area ratio of analyte to IS (Y) to the nominal analyte concentration (X) by weighted least-squares linear regression (1/X2 as a weighting factor). The LLOQ was defined as the lowest concentration of the calibration curve with S/N ratio greater than 10 and acceptable accuracy (RE, %; ±20%) and precision (RSD, %; ≤20).
Accuracy and precision were determined in all matrices at QC concentrations (LLOQ QC, LQC, MQC and HQC; n=5 each) using different analytical batches on the same day and on 3 consecutive days. The acceptance criteria for intra- and inter-day precision (RSD, %) was required to be ≤15% except for LLOQ, whose precision should be ≤20%, and accuracy (RE, %) was limited to ±15% except for LLOQ (±20%).
Extraction recovery was determined by comparing peak area of Rs-1-spiked blank matrix (plasma, urine or feces) with those obtained from the analyte spiked in post-extracted matrix supernate. The matrix effect was evaluated by comparing the peak areas of post-extraction matrix supernate spiked with the analyte versus the standard solution of the analyte at corresponding concentrations. The extraction recovery and matrix effect were evaluated at 4 QC concentrations (n=5). IS (200 ng/mL) was also investigated similarly.
Stability was examined in all matrices at 4 QC concentrations (n=5) under different conditions: at room temperature (25±3 °C) for 24 h (short-term stability); through three freeze–thaw cycles; stored at –80 °C for 20 days (long-term stability); and ready-to-inject samples placed in an autosampler at room temperature for 24 h (post-preparative stability). The analyte was considered stable in the biological matrix when 85%–115% of the initial concentrations were found.
The Rs-1 concentrations of some plasma, urine and feces samples exceeded the upper limit of quantification (ULOQ). In the dilution integrity experiment, the accuracy and precision of QC samples (n=5 each) diluted from a high concentration were assessed to investigate the effect of diluting over-range samples into the linear range with blank matrix.
To evaluate the impact of incubation with β-glucuronidase and sulfatase on the determination of Rs-1, 4 levels of QC samples of Rs-1 in plasma and urine were processed by enzymatic hydrolysis identically to test samples. The requirement for accuracy (RE, %) was within ±15% compared to the nominal concentration.
2.8. Pharmacokinetic studies
2.8.1. Dosing formulations
The suspension formulation for oral dosing was prepared by suspending an appropriate amount of Rs-1 in a vehicle of 0.5% (w/v) sodium carboxymethyl cellulose (CMC-Na). The suspension was prepared freshly and shaken vigorously before oral gavage, and a dosing volume of 10 mL/kg body weight was used for gavage administration. For intravenous injection, the solution was prepared freshly by dissolving Rs-1 in a vehicle of dimethyl sulfoxide (DMSO):polyethylene glycol (PEG-300):physiological saline (5:50:45, v/v/v) and a dosing volume of 5 mL/kg body weight was used for intravenous injection.
2.8.2. Animals and animal husbandry
The pharmacokinetic study was carried out in healthy male and female Wistar rats (body weight 200±20 g, 8 weeks) obtained from Beijing Military Medical Sciences Experimental Animal Co., Ltd. (Beijing, China). All animals were kept in an environmentally controlled room which was maintained at a temperature of 24±2 °C with a relative humidity of 55±10% and an approximately 12 h light/dark cycle. All rats had free access to water and food.
2.8.3. Bioavailability study
Twelve rats (6 female and 6 male) were used for the bioavailability study and were kept in a fasting state with free access to water for 12 h prior to the experiment. All rats were randomly divided into two groups (n=6 per group) for oral and intravenous administration. Rats in groups 1 were administered a single dose of Rs-1 suspension at the dose of 70 mg/kg and serial blood samples (0.4 mL) were collected from the ocular vein into 1.5 mL Eppendorf tubes at 0.167, 0.5, 1, 1.25, 1.5, 1.75, 2, 2.5, 3.5, 4.5, 6, 8, 12 and 24 h. Rats in group 2 received a single intravenous administration of Rs-1 solution at a dose of 1.37 mg/kg via tail vein and blood samples were collected at 0.033, 0.083, 0.167, 0.33, 0.5, 0.75, 1, 1.5, 2, 4, 6, 8, 12 and 24 h after administration. Plasma samples were collected by centrifugation of collected blood immediately at 4000 rpm for 10 min at 4 °C and stored at −80 °C until analysis.
2.8.4. Excretion study
An excretion study was carried out in 6 fasted Wistar rats (3 female and 3 male). The rats were housed in separate metabolic cages after receiving a single oral dose of Rs-1 suspension at 70 mg/kg. Urine and feces samples were collected at 0–4, 4–8, 8–12, 12–24, 24–36, 36–48 and 48–60 h. The weight of feces and the volume of the urine samples were measured, after which all the samples were stored at –80 °C. Feces samples were individually homogenized in saline (1 g/20 mL) for 2 min to prepare the suspension for analysis.
2.9. Data analysis
The pharmacokinetic parameters, including the area under the plasma concentration–time curve from time zero to the last measurable plasma concentration point (AUC0−t), the area under the blood concentration–time curve from time zero to infinity (AUC0−∞), clearance (CL/F), volume of distribution (V/F), mean residence time (MRT), elimination half-life (t1/2), maximum concentration (Cmax) and time of reaching Cmax (Tmax) were evaluated by non-compartmental modeling using DAS Software (version 3.2.4, China State Drug Administration). The absolute oral bioavailability (F, %) in rats was calculated according to the following equation: F (%) = (AUCp.o.×Dosei.v.)/(AUCi.v.×Dosep.o.)×100.
3. Results and discussion
3.1. Optimization of LC–MS/MS conditions
During method development different mobile phase compositions (methanol–water and acetonitrile–water) with different buffers such as ammonium formate and formic acid were investigated to obtain chromatograms with good resolution and symmetric peak shapes. It was found that acetonitrile and water containing 0.1% formic acid could enhance the sensitivity of detection of both Rs-1 and IS. The elution gradient was also optimized to reduce a matrix effect and improve peak symmetry. As a result, a chromatogram of good peak shape and resolution was obtained on a shim-pack XR-ODS column (75 mm×3 mm, 2.2 μm, Shimadzu, Tokyo, Japan) using gradient mobile phase with acetonitrile (0.1% formic acid)–water (0.1% formic acid) in the gradient as described above, and the gradient mode was shown to be stable according to the method validation procedure.
To obtain better responses the MS conditions were optimized. The effect of positive and negative ionization modes (ESI) on the sensitivity of the analysis was investigated. The mass spectral response and signal stability of Rs-1 and IS (hesperetin) in negative mode was higher, and the MRM transitions were at m/z 453.2→411.2 for Rs-1 and m/z 301.1→163.9 for IS. The parameters of ionization DP, CE, EP, CXP and TEM were also optimized.
3.2. Sample preparation
Extraction methods with protein precipitation or LLE were investigated to remove interfering compounds from the samples. Organic solvents, such as methanol, ethanol, acetonitrile and acetone, were compared in developing the protein precipitation procedure along with the evaluations for matrix effect, recovery efficiency and reproducibility, but a serious ion suppression or matrix effect for analyte was observed in the LC–MS/MS analyses both for Rs-1 and IS, and could not meet the requirements of the pharmacokinetic study. Ethyl acetate, cyclohexane, n-hexane and diethyl ether were compared as LLE solvent, and ethyl acetate was found to be efficient in terms of low matrix effect, high recovery rate and good repeatability both for Rs-1 and IS except in feces samples, in which the optimal solvent was diethyl ether. Methanol and acetonitrile as the solvent to reconstitute biological samples after evaporation were also compared. It was found that tailing of peaks particularly of Rs-1 could occur when acetonitrile was used, and it was much better when methanol was used.
3.3. Optimization of enzymatic hydrolysis conditions
As the reference substances of Rs-1 glucuronide and sulfate standards were not available, Cmax plasma and urine in the pretest of Rs-1 oral administration were used as substrates to optimize enzymatic hydrolysis conditions. The effect of different time and amount of the enzyme was optimized according to the response values of Rs-1. When 100 µL plasma or urine were used as substrate, it was difficult to reach equilibrium, with even 10,000 U β-glucuronidase or 100 U sulfatase being insufficient after 48 h. In order to reduce the amount of enzyme and plasma, 10 µL plasma or 20 µL urine were used to assay the Rs-1 glucuronide and sulfate conjugates. For β-glucuronidase, the amount of 1000, 2000, 3000, 5000 U and the incubation time of 1, 2 and 3 h were compared. It was found that after incubation at 37 °C for 2 h, Rs-1 glucuronide in 10 µL plasma or 20 µL urine with 2000 U β-glucuronidase can be totally hydrolyzed into Rs-1. For enzymolysis of Rs-1 sulfate, the amount of 5, 10, 20, 40 U and the incubation time of 2, 4, 12, 24, 36, 48 h were compared. It was found that 20 U sulfatase was sufficient. Accordingly, 36 and 12 h were the appropriate time to incubate for plasma and urine samples, respectively.
3.4. Method validation
The developed method was selective as no significant endogenous interference from plasma was observed at the retention times of Rs-1 and IS (Fig. 3) in chromatograms derived from processed blank samples from 6 different rats, as well as in urine (Fig. 4) and feces samples (Fig. 5).
Figure 3.

Representative MRM chromatograms of (A) blank rat plasma, (B) rat plasma spiked with Rs-1 at LLOQ, and (C) Rs-1 in plasma profiles 10 min after injection administration of Rs-1.
Figure 4.

Representative MRM chromatograms of (A) blank rat urine, (B) rat urine spiked with Rs-1 at LLOQ, and (C) Rs-1 in urine profiles 12–24 h after oral administration of Rs-1.
Figure 5.

Representative MRM chromatograms of (A) blank rat feces, (B) rat feces spiked with Rs-1 at LLOQ, and (C) Rs-1 in feces profiles 12–24 h after oral administration of Rs-1.
All calibration curves showed good linearity over the concentration ranges of 1.42–2172 ng/mL. The regression equations were as follows: plasma, Y=0.00239X+0.000922 (R=0.9991); urine, Y=0.00203X+0.00112 (R=0.9995); feces, Y=0.00305X+0.00155 (R=0.9994). The LLOQ of Rs-1 was 1.42 ng/mL for all matrices.
The intra-day and inter-day precision and accuracy data of Rs-1 in biological matrices are presented in Table 1. The intra-day and inter-day precision (RSD, %) values were both ≤10.9%, while the intra-day and inter-day accuracy (RE, %) were in the range of –3.7% to 2.3% and –2.2% to 1.5%, respectively. The results revealed that all values were in the acceptable ranges, indicating the method was reliable and reproducible for the determination of Rs-1 in plasma, urine and feces.
Table 1.
Intra- and inter-day precision and accuracy of Rs-1 in rat plasma, urine and feces (n=5).
| Matrix | Nominal concentration (ng/mL) | Intra-day |
Inter-day |
||
|---|---|---|---|---|---|
| Precision (RSD, %) | Accuracy (RE, %) | Precision (RSD, %) | Accuracy (RE, %) | ||
| Plasma | 1.42 | 5.0 | 1.3 | 3.9 | 1.4 |
| 3.56 | 5.4 | –1.3 | 3.5 | –1.1 | |
| 347.5 | 10.9 | –3.7 | 6.7 | 0.5 | |
| 1737 | 5.9 | –0.6 | 6.6 | 1.5 | |
| Urine | 1.42 | 8.1 | 2.0 | 6.4 | –0.1 |
| 3.56 | 1.2 | 0.4 | 4.1 | –1.7 | |
| 347.5 | 2.3 | –2.2 | 3.5 | 1.5 | |
| 1737 | 1.3 | –2.3 | 2.3 | –0.9 | |
| Feces | 1.42 | 3.7 | 2.3 | 5.4 | 0.8 |
| 3.56 | 3.1 | 2.1 | 3.3 | –0.4 | |
| 347.5 | 1.9 | 1.9 | 2.3 | 0.0 | |
| 1737 | 3.4 | –0.3 | 6.3 | –2.2 | |
The extraction recovery of Rs-1 and IS in plasma, urine and feces at 4 QC concentrations ranged 70.7%–90.4% and 83.1%–94.7%, and the matrix effect of Rs-1 and IS ranged 85.8%–100.4% and 78.1%–100.0%, respectively. Results are presented in Table 2 and indicate that the extraction and purification method in this study is suitable and reliable for Rs-1 and IS in plasma, urine and feces.
Table 2.
Recovery and matrix effects of Rs-1 and IS in rat plasma, urine and feces (n=5).
| Compd. | Matrix | Nominal concentration (ng/mL) | Recovery |
Matrix effect |
||
|---|---|---|---|---|---|---|
| Mean (%) | RSD (%) | Mean (%) | RSD (%) | |||
| Rs-1 | Plasma | 1.42 | 90.4 | 12.3 | 92.9 | 13.4 |
| 3.56 | 86.4 | 5.2 | 90.1 | 8.3 | ||
| 347.5 | 86.4 | 1.9 | 95.9 | 4.6 | ||
| 1737 | 82.0 | 3.9 | 85.8 | 2.1 | ||
| Urine | 1.42 | 87.6 | 3.9 | 97.3 | 1.4 | |
| 3.56 | 80.0 | 6.7 | 96.9 | 1.7 | ||
| 347.5 | 77.1 | 5.5 | 98.4 | 2.2 | ||
| 1737 | 70.7 | 1.3 | 91.9 | 1.8 | ||
| Feces | 1.42 | 88.1 | 3.4 | 98.8 | 3.2 | |
| 3.56 | 89.7 | 0.7 | 100.4 | 0.8 | ||
| 347.5 | 82.6 | 2.7 | 97.4 | 1.6 | ||
| 1737 | 88.6 | 1.1 | 99.0 | 2.9 | ||
| IS | Plasma | 200 | 83.1 | 9.3 | 78.1 | 10.0 |
| Urine | 200 | 94.7 | 3.2 | 100.0 | 2.4 | |
| Feces | 200 | 85.5 | 1.1 | 84.5 | 1.7 | |
The stability of Rs-1 was investigated under a variety of conditions at 4 QC concentration levels and the results were summarized in Table 3. The QC samples were stable in all matrices at room temperature (25±3 °C) for 24 h and the RE% was –9.9% to 10.7%. Rs-1 was also considered to be stable after three freeze–thaw cycles (RE%, –6.7% to 3.3%). Long-term stability was stable for 20 days at −80 °C (RE%, –7.5% to 3.7%) and the ready-to-inject sample placed in autosampler at 25 °C for 24 h was also stable (RE%, –2.3% to 3.5%). All the results showed that Rs-1 was stable under those different conditions without any significant degradation.
Table 3.
Stability data of Rs-1 in plasma, urine and feces (n=5).
| Matrix | Nominal concentration (ng/mL) | Short-term (24 h) |
Freeze–thaw cycles |
Long-term (20 days) |
Auto-sampler (24 h) |
||||
|---|---|---|---|---|---|---|---|---|---|
| Accuracy (RE, %) | Precision (RSD, %) | Accuracy (RE, %) | Precision (RSD, %) | Accuracy (RE, %) | Precision (RSD, %) | Accuracy (RE, %) | Precision (RSD, %) | ||
| Plasma | 1.42 | –0.7 | 1.8 | 0.7 | 5.8 | 0.3 | 8.2 | –2.3 | 4.9 |
| 3.56 | –9.9 | 1.9 | –2.2 | 7.4 | –0.4 | 2.9 | –2.1 | 4.4 | |
| 347.5 | –3.9 | 7.0 | –1.0 | 5.9 | –0.4 | 5.6 | 1.5 | 4.1 | |
| 1737 | –0.9 | 5.9 | 3.3 | 5.2 | 0.4 | 0.9 | 0.1 | 6.0 | |
| Urine | 1.42 | 10.7 | 4.1 | –4.5 | 9.3 | –3.0 | 4.3 | –0.1 | 6.8 |
| 3.56 | 6.4 | 1.7 | 0.8 | 1.8 | 3.7 | 6.2 | –0.7 | 3.0 | |
| 347.5 | 4.5 | 3.1 | –6.5 | 2.9 | –4.0 | 2.1 | 3.5 | 1.3 | |
| 1737 | –0.6 | 0.9 | –6.7 | 1.5 | –7.5 | 1.4 | –1.4 | 1.8 | |
| Feces | 1.42 | 7.6 | 4.4 | –0.7 | 3.4 | 2.1 | 4.6 | 0.9 | 3.7 |
| 3.56 | –4.0 | 7.5 | 0.0 | 2.6 | –1.5 | 5.0 | 0.5 | 2.7 | |
| 347.5 | –0.3 | 1.7 | –0.8 | 2.7 | –0.2 | 1.8 | –0.6 | 4.2 | |
| 1737 | 0.0 | 9.5 | –2.1 | 6.0 | –1.0 | 2.4 | –0.4 | 8.5 | |
The precision and accuracy of the dilution experiments were evaluated as well and they all met the requirement. After incubation with β-glucuronidase or sulfatase, the determination of Rs-1 in plasma and urine was within the acceptable range as detailed in Table 4. The results proved that incubation with β-glucuronidase or sulfatase at 37 °C for the established time had no impact on the determination of Rs-1.
Table 4.
Effect of β-glucuronidase and sulfatase to Rs-1 in plasma and urine (n=5).
| Matrix | Nominal concentration (ng/mL) |
β-Glucuronidase |
Sulfatase |
||
|---|---|---|---|---|---|
| Accuracy (RE, %) | Precision (RSD, %) | Accuracy (RE, %) | Precision (RSD, %) | ||
| Plasma | 1.42 | 4.1 | 3.7 | 2.0 | 3.5 |
| 3.56 | –6.5 | 3.9 | –3.0 | 5.1 | |
| 347.5 | –6.1 | 2.5 | –2.1 | 2.6 | |
| 1737 | 0.7 | 2.3 | 3.8 | 2.6 | |
| Urine | 1.42 | 4.8 | 3.7 | 5.6 | 3.1 |
| 3.56 | 1.6 | 7.0 | 3.0 | 4.1 | |
| 347.5 | –0.6 | 2.5 | –1.4 | 1.5 | |
| 1737 | –4.2 | 2.4 | –4.8 | 2.5 | |
3.5. Application of analytical method
3.5.1. Pharmacokinetics of Rs-1
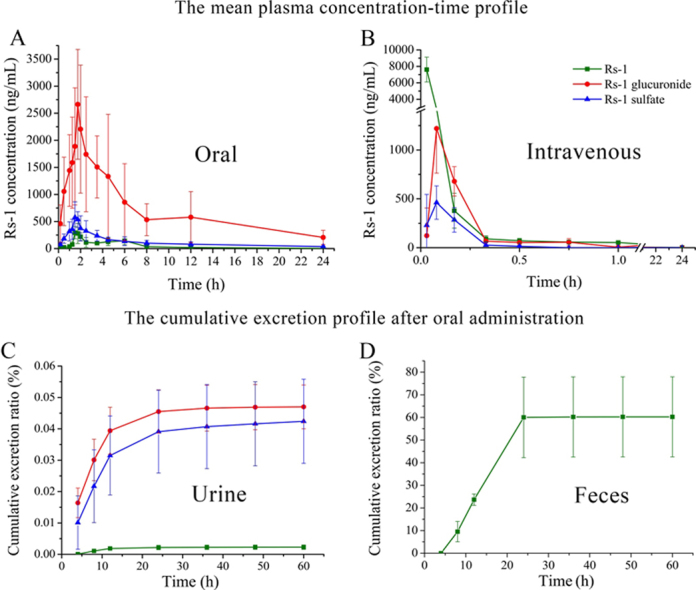
After a single oral administration of 70 mg/kg or a single intravenous injection of 1.37 mg/kg Rs-1 to rats (n=6), Rs-1 could be conjugated with glucuronic acid or sulfate. Rs-1 glucuronide and sulfate conjugates were characterized by the deprotonated molecular ion [M−H]− at m/z 629.1 and 533.2, which were 176 and 80 more than Rs-1 (m/z 453.2), respectively. The Rs-1 fragment was observed at m/z 453.2 in product ion mass spectra of Rs-1 glucuronide and sulfate conjugates (Fig. 6). The peak area of Rs-1 was enhanced after enzymolysis of the samples with β-glucuronidase or sulfatase, which indicated that it contains Rs-1 glucuronide and sulfate conjugates. The mean plasma concentration versus time profiles of Rs-1 and its conjugates (n=6 for each time point) are illustrated in Fig. 7, and the major pharmacokinetic parameters from non-compartment model analysis are presented in Table 5.
Figure 6.

Product ion spectra (MS/MS) of (A) Rs-1 glucuronide and (B) Rs-1 sulfate metabolites.
Figure 7.

The mean plasma concentration–time profile in rats after (A) oral administration of 70 mg/kg Rs-1 and (B) intravenous administration of 1.37 mg/kg Rs-1 (n=6).
Table 5.
Non-compartmental pharmacokinetics parameters of Rs-1, Rs-1 glucuronide and Rs-1 glucuronide after a single oral or intravenous administration in rat plasma.
| Parameter | Unit | Oral (70 mg/kg, n=6) |
Intravenous (1.37 mg/kg, n=6) |
||||
|---|---|---|---|---|---|---|---|
| Rs-1 | Rs-1 glucuronide | Rs-1 sulfate | Rs-1 | Rs-1 glucuronide | Rs-1 sulfate | ||
| AUC0–t | µg ⋅ h/L | 1,162.0±200.6 | 16,924.3±7944.0 | 2,822.8±967.9 | 1,004.7±209.9 | 299.005±64.9 | 96.2±34.1 |
| AUC(0–∞) | µg ⋅ h/L | 1,181.9±193.9 | 18,758.3±9293.0 | 3,303.2±992.6 | 1,017.6±206.3 | 299.006±64.9 | 96.2±34.1 |
| MRT(0–t) | h | 5.6±0.6 | 7.4±1.5 | 7.4±0.9 | 1.5±0.2 | 2.02±1.4 | 0.4±0.3 |
| MRT(0–∞) | h | 6.0±0.6 | 9.7±2.5 | 12.3±4.6 | 2.0±0.4 | 2.02±1.4 | 0.4±0.3 |
| t1/2z | h | 4.2±1.6 | 7.3±1.8 | 9.5±4.8 | 5.8±1.6 | 1.1±0.5 | 0.3±0.2 |
| Tmax | h | 1.7±0.2 | 2.0±0.7 | 1.7±0.2 | 0.03±0.00 | 0.10±0.04 | 0.09±0.05 |
| Vz/F | L/kg | 379.7±180.4 | 45.1±15.6 | 298.1±144.0 | 12.2±5.8 | 6.6±2.6 | 5.4±3.0 |
| CLz/F | L/h/kg | 60.6±10.1 | 4.4±1.8 | 23.8±11.0 | 1.4±0.3 | 4.7±1.1 | 16.5±8.6 |
| Cmax | µg/L | 399.0±97.8 | 2,984.7±745.7 | 714.8±176.1 | 7,611.7±1,510.5 | 1,258.3±389.4 | 488.8±172.0 |
| F (%) | – | 2.3 | – | – | – | – | – |
–Not applicable.
The profile revealed that after intravenous injection the elimination of Rs-1 and its two metabolites was very rapid, and the unmodified Rs-1 was the main compound. Rs-1 glucuronide and sulfate conjugates reached peak concentration at 5 min. At 10 min, Rs-1 glucuronide concentration (679.5±150.7 ng/mL) was higher than that of the parent compound (378.3±178.4 ng/mL) and Rs-1 sulfate (285.2±125.8 ng/mL). Rs-1 sulfate conjugate was cleared the fastest from the bloodstream since only trace amounts of Rs-1 sulfate (below the LLOQ) could be detected at 45 min, while at this time point the concentration of Rs-1 was 57.1±19.7 ng/mL and Rs-1 glucuronide was 55.7±36.1 ng/mL. Rs-1 glucuronide and Rs-1 were still present in plasma at 12 and 24 h, respectively.
After oral administration of Rs-1 it was metabolized rapidly and Rs-1 glucuronide was the main compound, followed by Rs-1 sulfate. The mean AUC0–t of Rs-1, its glucuronide and sulfate conjugates were 1162.0±200.6, 16,924.3±7944.0, and 2822.8±967.9 µg⋅h/L, respectively. The time-to-peak concentration (Tmax) of Rs-1 and its sulfate conjugate were observed at about 1.7 h, and Rs-1 glucuronide had a slight delay at about 2.0 h. The apparent volume of distribution (V) value suggested that Rs-1, Rs-1 glucuronide and sulfate could extensively distribute into organs and tissues. Comparing the clearance and t1/2 values of Rs-1, Rs-1 glucuronide and sulfate, Rs-1 could be most quickly eliminated from the circulatory system. The absolute oral bioavailability of Rs-1 was 2.3%, and the total absorption rose to 31.5% in addition to its glucuronide and sulfate metabolites. It could be observed from the concentration-time curve that there were double peaks (approximately at 4.5–6 h) of Rs-1 (Fig. 7), and that was the same as obtained previously with resveratrol. The most popular explanation for the double peaks was enterohepatic circulation. The resveratrol glucuronide could be deconjugated in the gut to the active compound, resveratrol, by bacterial glucuronidases and then reabsorbed14, 24. Those results revealed that Rs-1 was poorly bioavailable, which might be in part due to the low absorption and extensive metabolism.
3.5.2. Excretion of Rs-1
Urine and feces were collected at different exposure times after oral administration of 70 mg/kg Rs-1. Rs-1 glucuronide and sulfate conjugates were also detected in the urine samples by MS/MS, and the peak area of Rs-1 was also enhanced after the enzymolysis of the samples with β-glucuronidase or sulfatase. While in feces, only unchanged Rs-1 was detected, and the peak area of Rs-1 showed no enhancement after the enzymolysis with β-glucuronidase or sulfatase. As shown in Fig. 8, 60.3% of the gavaged dose of Rs-1 was excreted via the feces in unchanged form over 60 h after oral gavage. The major duration of excretion of Rs-1 in feces was 8–24 h and 60% of the gavage dose was detected in this period. With the decreasing Rs-1 concentration in the rat, the excretion ratio was reduced and accounted for only 0.3% during the 24–60 h period; 0.09% was excreted through the urine 60 h after oral gavage. The major forms of Rs-1 in urine were Rs-1 glucuronide and sulfate, which were 0.047% and 0.042% of the gavaged dose, and Rs-1 was slightly less than 0.003%. 8–24 h was also the main period of excretion in urine and decreased with time. The data showed that Rs-1 was mainly excreted via the feces in unchanged form after oral gavage, and most appeared to be unabsorbed according to the drug concentration in plasma.
Figure 8.

(A) Urinary and (B) fecal cumulative excretion profile of Rs-1 in rats after single oral dose of 70 mg/kg Rs-1 (n=6).
Viniferins (resveratrol oligomers) are found in wine and some popular herbal medicines, and have been linked to many health-promoting properties in humans. Rs-1 is an important kind of viniferin, and its pharmacological activities have been widely studied. Its pharmacokinetics show poor water solubility and high metabolism, especially after oral administration. As with resveratrol, in order to improve the poor oral bioavailability, increase the solubility, and protect it from being metabolized, structural modification to the phenolic hydroxyl, such as methylation25, 26 and acetylation27, should be considered in future studies. Moreover, this formulation research is also needed to increase the absorption.
4. Conclusions
In summary, a rapid and sensitive LC–MS/MS method for the determination of Rs-1 in rat plasma, urine and feces was developed and validated, and the method was successfully applied to determine the bioavailability, metabolism and excretion of Rs-1 in rats. To the best of our knowledge, this is the first paper to report the metabolism and excretion of Rs-1. Moreover, the method of Rs-1 measurement in plasma in this study had a shorter sample turnover rate of 4 min per sample and higher sensitivity with LLOQ of 1.42 ng/mL compared with a previous study21. The absolute oral bioavailability of Rs-1 was 2.3%, and the total absorption rose to 31.5% with addition of its glucuronide and sulfate metabolites. 60.3% of the gavaged dose of Rs-1 was excreted via the feces in unchanged form after oral gavage. The results indicate that both poor absorption and extensive metabolism are important factors leading to the poor bioavailability of Rs-1, which can provide a basis for further studies on structural modification and dosage form design.
Acknowledgments
This study was supported by Fundamental Research Funds for the Central Universities–China (2014CX16). We thank Chunsuo Yao, from Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, for providing the analyte Rs-1.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
References
- 1.Baur J.A., Sinclair D.A. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006;5:493–506. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- 2.Pervaiz S. Resveratrol: from grapevines to mammalian biology. FASEB J. 2003;17:1975–1985. doi: 10.1096/fj.03-0168rev. [DOI] [PubMed] [Google Scholar]
- 3.Saiko P., Szakmary A., Jaeger W., Szekeres T. Resveratrol and its analogs: defense against cancer, coronary disease and neurodegenerative maladies or just a fad? Mutat Res. 2008;658:68–94. doi: 10.1016/j.mrrev.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 4.Kopp P. Resveratrol, a phytoestrogen found in red wine. A possible explanation for the conundrum of the ‘French paradox’? Eur J Endocrinol. 1998;138:619–620. doi: 10.1530/eje.0.1380619. [DOI] [PubMed] [Google Scholar]
- 5.Breuil A.C., Adrian M., Pirio N., Meunier P., Bessis R., Jeandet P. Metabolism of stilbene phytoalexins by Botrytis cinerea: 1. Characterization of a resveratrol dehydrodimer. Tetrahedron Lett. 1998;39:537–540. [Google Scholar]
- 6.Cichewicz R., Kouzi S.M. Dimerization of resveratrol by the grapevine pathogen Botrytis cinerea. J Nat Prod. 2000;63:29–33. doi: 10.1021/np990266n. [DOI] [PubMed] [Google Scholar]
- 7.Nicotra S., Cramarossa M.R., Mucci A., Pagnoni U.M., Riva S., Forti L. Biotransformation of resveratrol: synthesis of trans-dehydrodimers catalyzed by laccases from Myceliophtora thermophyla and from Trametes pubescens. Tetrahedron. 2004;60:595–600. [Google Scholar]
- 8.Yu B.B., Han X.Z., Lou H.X. Oligomers of resveratrol and ferulic acid prepared by peroxidase-catalyzed oxidation and their protective effects on cardiac injury. J Agric Food Chem. 2007;55:7753–7757. doi: 10.1021/jf0711486. [DOI] [PubMed] [Google Scholar]
- 9.Wilkens A., Paulsen J., Wray V., Winterhalter P. Structures of two novel trimeric stilbenes obtained by horseradish peroxidase catalyzed biotransformation of trans-resveratrol and (–)-epsilon-viniferin. J Agric Food Chem. 2010;58:6754–6761. doi: 10.1021/jf100606p. [DOI] [PubMed] [Google Scholar]
- 10.Cichewicz R.H., Kouzi S.A. Resveratrol oligomers: structure, chemistry, and biological activity. Stud Nat Prod Chem. 2002;26:507–579. [Google Scholar]
- 11.Ohyama M., Tanaka T., Ito T., Iinuma M., Bastow K.F., Lee K.H. Antitumor agents 200. Cytotoxicity of naturally occurring resveratrol oligomers and their acetate derivatives. Bioorg Med Chem Lett. 1999;9:3057–3060. doi: 10.1016/s0960-894x(99)00520-x. [DOI] [PubMed] [Google Scholar]
- 12.Zhong C., Liu X.H., Chang J., Yu J.M., Sun X. Inhibitory effect of resveratrol dimerized derivatives on nitric oxide production in lipopolysaccharide-induced RAW 264.7 cells. Bioorg Med Chem Lett. 2013;23:4413–4418. doi: 10.1016/j.bmcl.2013.05.058. [DOI] [PubMed] [Google Scholar]
- 13.Ito T., Akao Y., Yi H., Ohguchi K., Matsumoto K., Tanaka T. Antitumor effect of resveratrol oligomers against human cancer cell lines and the molecular mechanism of apoptosis induced by vaticanol C. Carcinogenesis. 2003;24:1489–1497. doi: 10.1093/carcin/bgg105. [DOI] [PubMed] [Google Scholar]
- 14.Walle T., Hsieh F., DeLegge M.H., Oatis J.E., Jr, Walle U.K. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab Dispos. 2004;32:1377–1382. doi: 10.1124/dmd.104.000885. [DOI] [PubMed] [Google Scholar]
- 15.Zhang R., Mao P., Zhang T., Ma C., Jin B., Li T. Pharmacokinetic analysis and tissue distribution of Vam3 in the rat by a validated LC–MS/MS method. Acta Pharm Sin B. 2015;5:254–263. doi: 10.1016/j.apsb.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Langcake P., Pryce R.J. A new class of phytoalexins from grapevines. Experientia. 1977;33:151–152. doi: 10.1007/BF02124034. [DOI] [PubMed] [Google Scholar]
- 17.Waffo-Teguo P., Lee D., Cuendet M., Mérillon J.M., Pezzuto J.M., Kinghorn A.D. Two new stilbene dimer glucosides from grape (Vitis vinifera) cell cultures. J Nat Prod. 2001;64:136–138. doi: 10.1021/np000426r. [DOI] [PubMed] [Google Scholar]
- 18.Langcake P., Pryce R.J. Oxidative dimerisation of 4-hydroxystilbenes in vitro–production of a grapevine phytoalexin mimic. J Chem Soc Chem Commun. 1977;7:208–210. [Google Scholar]
- 19.Pezet R., Gindro K., Viret O., Spring J.L. Glycosylation and oxidative dimerization of resveratrol are respectively associated to sensitivity and resistance of grapevine cultivars to downy mildew. Physiol Mol Plant Pathol. 2004;65:297–303. [Google Scholar]
- 20.Kong LL, Wang X, Ma L, Wang XB, inventors. (7R, 8R)-Trans-δ-viniferin, its preparation method and application for reducing blood sugar. China patent CN ZL200810243995.9. 2009 May 20
- 21.Liu Q., Liao X., Xu J., Zhao J., Luo J., Kong L. Development and validation of a sensitive and selective LC–MS/MS method for the determination of trans δ-veniferin, a resveratrol dehydrodimer, in rat plasma and its application to pharmacokinetics and bioavailability studies. J Chromatogr B. 2014;958:124–129. doi: 10.1016/j.jchromb.2014.03.026. [DOI] [PubMed] [Google Scholar]
- 22.US Food and Drug Administration. Guidance for industry: bioanalytical method validation. 2001. Available from: 〈http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf〉.
- 23.European Medicines Agency. Guideline on bioanalytical method validation. 2012. Available from: 〈http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf〉. [DOI] [PubMed]
- 24.Marier J.F., Vachon P., Gritsas A., Zhang J., Moreau J.P., Ducharme M.P. Metabolism and disposition of resveratrol in rats: extent of absorption, glucuronidation, and enterohepatic recirculation evidenced by a linked–rat model. J Pharmacol Exp Ther. 2002;302:369–373. doi: 10.1124/jpet.102.033340. [DOI] [PubMed] [Google Scholar]
- 25.Lin H.S., Ho P.C. A rapid HPLC method for the quantification of 3,5,4′-trimethoxy-trans-stilbene (TMS) in rat plasma and its application in pharmacokinetic study. J Pharm Biomed Anal. 2009;49:387–392. doi: 10.1016/j.jpba.2008.10.042. [DOI] [PubMed] [Google Scholar]
- 26.Kapetanovic I.M., Muzzio M., Huang Z., Thompson T.N., McCormick D.L. Pharmacokinetics, oral bioavailability, and metabolic profile of resveratrol and its dimethylether analog, pterostilbene, in rats. Cancer Chemoth Pharm. 2011;68:593–601. doi: 10.1007/s00280-010-1525-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang L., Liu X., Wang Q., Cheng S., Zhang S., Zhang M. Pharmacokinetics, tissue distribution and excretion study of resveratrol and its prodrug 3,5,4′-tri-O-acetylresveratrol in rats. Phytomedicine. 2013;20:558–563. doi: 10.1016/j.phymed.2012.12.012. [DOI] [PubMed] [Google Scholar]