

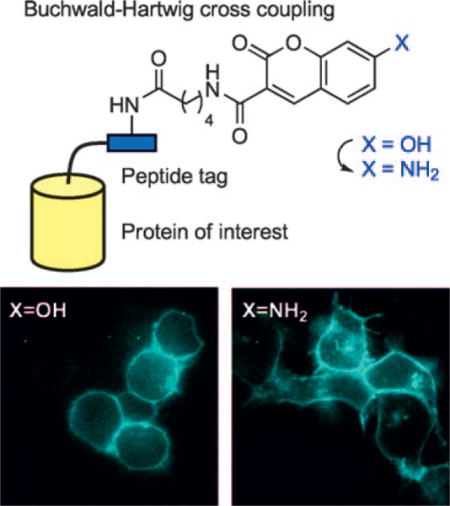
Graphical abstract
PRIME time: We report the synthesis of a pH-insensitive blue fluorophore, 7-aminocoumarin, by using palladium-catalyzed Buchwald–Hartwig cross coupling. 7-Aminocoumarin can be used to tag recombinant proteins on the cell surface and inside living cells through PRIME (probe incorporation mediated by enzymes), and unlike 7-hydroxycoumarin, can be visualized in acidic organelles such as endosomes.
Keywords: cell imaging, cross-coupling, enzyme engineering, fluorescent probes, palladium
To enable minimally invasive studies of proteins in their native context, it is desirable to tag proteins with small, bright reporter groups. Recently, our lab described PRIME (probe incorporation mediated by enzymes) technology for such tagging.[1–3] An engineered variant of Escherichia coli lipoic acid ligase (LplA) is used to covalently attach a fluorescent substrate, such as 7-hydroxycoumarin, onto a 13-residue peptide-recognition sequence (called LAP, for ligase acceptor peptide) that is genetically fused to a protein of interest (POI; Scheme 1A). The targeting specificity is derived from the extremely high natural sequence specificity of LplA.[4] PRIME was used to label and visualize various LAP-tagged cytoskeletal and adhesion proteins in living mammalian cells.
Scheme 1.
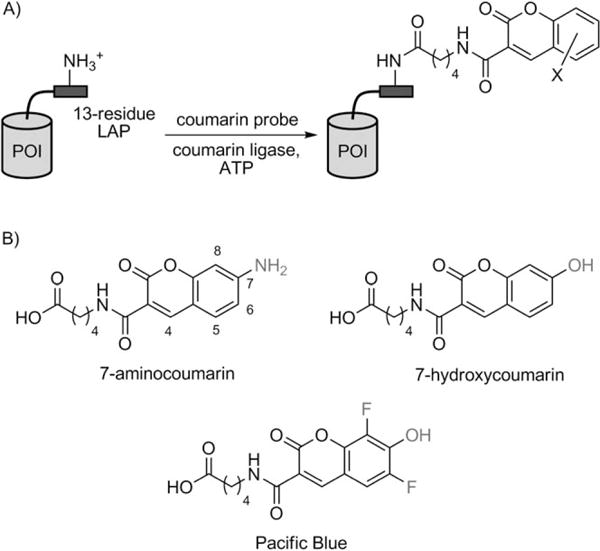
PRIME (probe incorporation mediated by enzymes) for site-specific labeling of proteins of interest (POIs) with coumarin fluorophores. A) Labeling scheme. Coumarin ligase is the W37V mutant of E. coli lipoic acid ligase (LplA).[1] LAP is a 13-residue recognition sequence for LplA.[16] B) Coumarin substrates for coumarin ligase. 7-Hydroxycoumarin and Pacific Blue have been previously described.[1] 7-Aminocoumarin was synthesized and characterized in this work.
One limitation of the 7-hydroxycoumarin probe used in our previous study is its pH-dependent fluorescence. The 7-OH substituent has a pKa of 7.5,[5] and the fluorophore is only emissive in its anionic form. Proteins labeled by PRIME with 7-hydroxycoumarin (on the extracellular or luminal side) therefore cannot be visualized in acidic compartments of the cell such as the endosome (pH 5.5–6.5[6]), where >90% of 7-hydroxycoumarin is expected to be neutral and therefore nonfluorescent. This problem prevents the use of 7-hydroxycoumarin for imaging receptor internalization and recycling, for example.
A potential solution is to use 6,8-difluoro-7-hydroxycoumarin (Pacific Blue,[5] Scheme 1B), which has a reduced 7-OH pKa of 3.7. We found, however, that our engineered 7-hydroxycoumarin ligase, the W37V mutant of LplA, did not ligate an isosteric Pacific Blue substrate onto LAP efficiently.[1] It is likely that the ligase active site prefers neutral and hydrophobic substrates and therefore rejects Pacific Blue, which is predominantly anionic at physiological pH.
An alternative coumarin structure is 7-aminocoumarin (Scheme 1B). In contrast to 7-hydroxycoumarin and Pacific Blue, 7-aminocoumarin is expected to be both neutral and highly fluorescent over a wide range of pH values. We also predicted that it would be a substrate for W37VLplA, as it is sterically similar to 7-hydroxycoumarin and is uncharged at physiological pH.
The synthesis of the 7-aminocoumarin substrate 6 (Scheme 2) required a novel route, however. Previous synthetic routes to 7-aminocoumarin derivatives have used either Pechmann[7] or Perkin[8] condensations. The Pechmann reaction condenses aminoresorcinol with β-ketoesters and unavoidably produces 4-alkyl-substituted aminocoumarins. Based on our structure–activity studies, a substituent at the 4-position of coumarin is unlikely to be tolerated by LplA. The Perkin reaction condenses aminoresorcinoldehyde with malonic acid and requires N-alkylation to prevent spontaneous Schiff-base formation. An N-alkylated aminocoumarin would be considerably larger than 7-hydroxycoumarin and unlikely to be accepted by coumarin ligase.
Scheme 2.
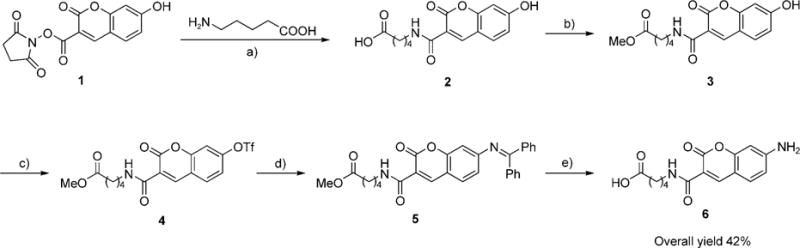
Synthesis of the 7-aminocoumarin substrate for coumarin ligase. a) Et3N, DMF, 98%; b) HCl, MeOH, 93%; c) Tf2O, pyridine, CH2Cl2, 87%; d) Pd(OAc)2, BINAP, Cs2CO3, benzophenone imine, THF, reflux, 70%; e) cat. HCl in THF/water 1:1, 76%.
To access the simple, minimally bulky 7-aminocoumarin structure 6, we devised a new synthetic route. The key feature is a palladium-catalyzed Buchwald–Hartwig cross coupling,[9,10] which converts the 7-OH group of 7-hydroxycoumarin into an unsubstituted primary aniline group. Our synthetic route (Scheme 2) begins with the 7-hydroxycoumarin substrate 2, which is protected as the methyl ester derivative 3. Triflic anhydride and pyridine were used to convert 3 to 7-triflylcoumarin 4 in 87% yield. The Buchwald–Hartwig cross coupling was then performed with benzophenone imine as a surrogate for ammonia.[11] We used a catalytic combination of Pd(OAc)2, 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP), and Cs2CO3, previously designed to give high coupling yields for electron-deficient aryl triflates and to reduce triflate hydrolysis.[12] The benzophenone imine-coumarin adduct 5 was obtained in 70% yield after gentle reflux with the catalyst system in THF. Benzophenone imine was then cleaved by acidic hydrolysis, which also hydrolyzed the methyl ester to give 6 in 76% yield. The overall yield for five synthetic steps was 42%.
We characterized the photophysical properties of 7-aminocoumarin 6 and compared them to those of the 7-hydroxycoumarin isostere 2. The excitation and emission maxima of 7-aminocoumarin are 380 nm/444 nm (Figure 1A), similar to those of 7-hydroxycoumarin (386 nm/448 nm[5]). The extinction coefficient of 7-aminocoumarin (18400m−1cm−1) is about half that of 7-hydroxycoumarin (36700m−1cm−1[5]). As expected, 7-aminocoumarin fluorescence is fairly constant across the pH range 3–10, whereas 7-hydroxycoumarin fluorescence drops sharply at pH values <6.5 (Figure 1B).
Figure 1.
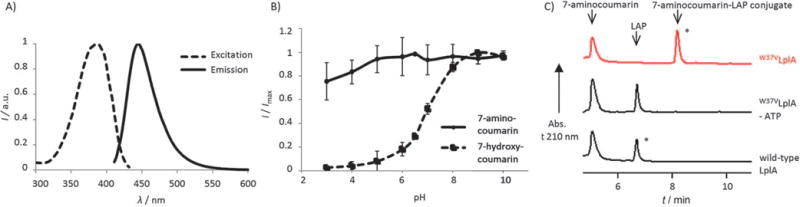
In vitro characterization of 7-aminocoumarin ligation. A) Fluorescence excitation and emission spectra for 7-aminocoumarin 6. B) pH profile for 7-amino and 7-hydroxycoumarins. Fluorescence intensity ratio (I at λmax divided by maximal fluorescence intensity Imax) is plotted against pH. Each measurement was performed in triplicate. Error bars: ±s.d. C) HPLC traces showing 7-aminocoumarin 6 ligation onto LAP peptide catalyzed by W37VLplA. The starred peaks were collected and analyzed by mass spectrometry (Figure S1). Negative controls (bottom two traces) are shown with ATP omitted or wild-type LplA in place of W37VLplA.
We next tested 7-aminocoumarin for ligation by LplA variants. Although W37VLplA is the best single mutant of LplA for 7-hydroxycoumarin ligation, we previously found that several other LplA single mutants also had coumarin ligation activity (W37I, G, A, S, and L[1]). We therefore tested these LplA variants along with W37VLplA for 7-aminocoumarin ligation onto LAP. As with 7-hydroxycoumarin, W37VLplA was still the best among these for ligation of 7-aminocoumarin (data not shown). Figure 1C shows an HPLC analysis of this ligation reaction. The starred peak in the HPLC trace was collected and analyzed by mass spectrometry to confirm its identity as the covalent adduct between 7-aminocoumarin and LAP (Figure S1 in the Supporting Information). Negative controls with ATP omitted, or W37VLplA replaced by wild-type LplA, gave no ligation product.
We compared the kinetics of 7-aminocoumarin and 7-hydroxycoumarin ligation by W37VLplA (Figure S2). With 500 μm of 7-aminocoumarin probe (likely saturating the ligase active site), 78% LAP was converted to product, compared to 46% conversion with 7-hydroxycoumarin, after a 55-minute reaction (Figure S2A). A twofold difference in reaction extent was also observed at lower probe concentration (100 μm) after 70 minutes (Figure S2B). At the reaction pH of 7.4, ~50% of 7-hydroxycoumarin is expected to be in the anionic form, whereas 7-aminocoumarin is neutral. The improved kinetics with 7-aminocoumarin likely reflects preferential binding of W37VLplA to neutral substrates.
7-Aminocoumarin 6 was then used for PRIME labeling in living mammalian cells. Neurexin-1β, a transmembrane neuronal synapse adhesion protein,[13] was fused to LAP at its extracellular N terminus, and labeled with 7-aminocoumarin and W37VLplA added to the growth medium. Figure 2A shows cell images after 20 min of 7-aminocoumarin labeling on human embryonic kidney (HEK) cells expressing LAP-neurexin-1β and a transfection marker (histone 2B fused to yellow fluorescent protein, H2B-YFP). A point mutation in the LAP sequence (Lys→Ala), or replacement of W37VLplA with wild-type LplA, eliminated 7-aminocoumarin labeling.
Figure 2.
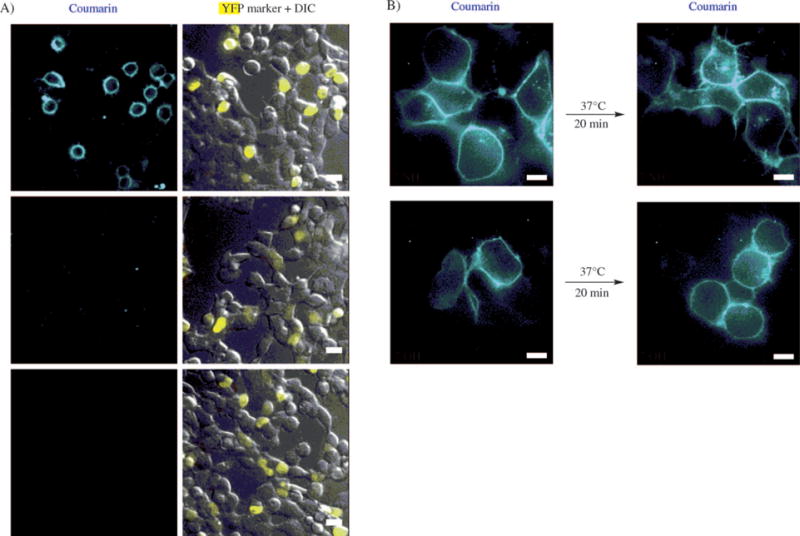
7-Aminocoumarin ligation to LAP-neurexin on the surface of living mammalian cells. A) HEK cells expressing LAP-neurexin-1β were labeled with 7-aminocoumarin and purified W37VLplA added to the culture medium. Negative controls are shown with a Lys→Ala mutation in LAP (second row), and with W37VLplA replaced by wild-type LplA (third row). H2B-YFP is the YFP transfection marker. Scale bars=20 μm. B) Visualization of internalized LAP-neurexin by using 7-aminocoumarin. Top row: HEK cells expressing LAP-neurexin were labeled as in (A), then incubated at 37°C for 20 min prior to imaging. The bottom row shows the same experiment but with 7-hydroxycoumarin instead of 7-aminocoumarin. Scale bars=10 μm.
To test the ability of 7-aminocoumarin to visualize neurexin in acidic endosomes, we incubated 7-aminocoumarin-labeled cells at 37°C for 20 min to allow endocytic internalization of surface pools of neurexin-1β. Figure 2B shows the appearance of internal 7-aminocoumarin puncta in cells after this 20-minute internalization period. In contrast, cells similarly labeled with 7-hydroxycoumarin and then incubated did not show substantial internal fluorescence due to quenching of the 7-hydroxycoumarin fluorescence in acidic compartments.
We also tested 7-aminocoumarin for intracellular protein labeling. To deliver the probe across the cell membrane, we derivatized the carboxylic acid of 7-aminocoumarin 6 as an acetoxymethyl (AM) ester (Figure 3A). Upon entering cells, the AM ester is cleaved by endogenous esterases,[14] releasing the parent 7-aminocoumarin probe 6. To perform intracellular protein labeling, HEK cells were transfected with expression plasmids for both the coumarin ligase, W37VLplA, and a LAP fusion protein. 7-Aminocoumarin-AM was incubated with cells for 10 min, then the medium was replaced over 60 min to allow endogenous anion transporters to clear excess unconjugated probe from the cytosol.[15] Figure 3B shows specific labeling in cells expressing LAP-tagged yellow fluorescent protein (LAP-YFP), but not in neighboring untransfected cells. An alanine mutation in the LAP sequence abolished 7-aminocoumarin labeling. To illustrate generality, we also labeled LAP-YFP targeted to the nucleus (LAP-YFP-NLS) and LAP fused to cytoskeletal protein β-actin.
Figure 3.
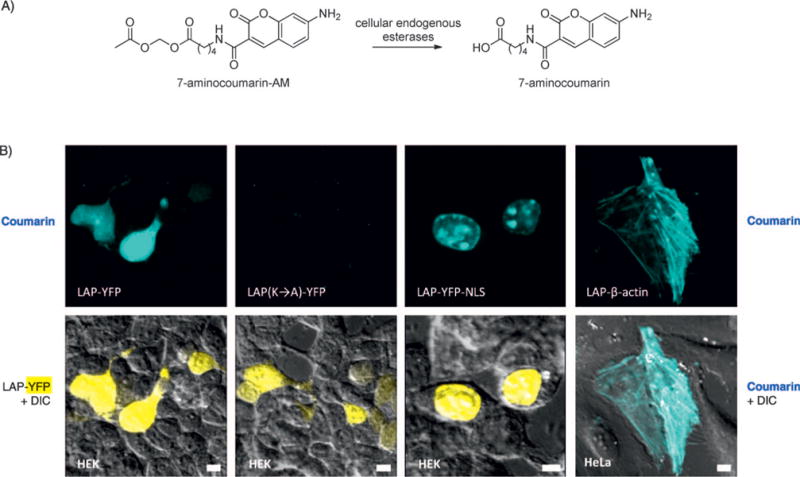
Site-specific protein labeling with 7-aminocoumarin inside living mammalian cells. A) Structure of membrane-permeant 7-aminocoumarin-acetoxymethyl (AM) ester, and the deprotection reaction catalyzed by endogenous esterases. B) Specific labeling of LAP in the cytosol, nucleus, and on a cytoskeletal protein (actin). Labeling was performed for 10 min with coexpressed W37VLplA. A negative control is shown with a Lys→Ala mutation in LAP (second column). NLS=nuclear localization sequence. Scale bars=10 μm.
In summary, to extend PRIME technology to the imaging of proteins in acidic organelles while accommodating the steric and electronic constraints of our engineered coumarin ligase,[1] we have designed a new fluorescent ligase substrate. 7-Aminocoumarin was synthesized by a novel route, using palladium-catalyzed Buchwald–Hartwig cross coupling to efficiently convert the 7-OH substituent into a 7-NH2 substituent. We have demonstrated that 7-aminocoumarin can be site-specifically targeted to LAP fusion proteins by the coumarin ligase, both on the cell surface and inside living mammalian cells. PRIME tagging with this new probe represents one step in our ongoing effort to generalize PRIME for labeling any cellular protein with diverse fluorophore structures.
Experimental Section
Synthetic methods
All experiments were conducted with oven-dried glassware under N2 atmosphere and at ambient temperature (20–25°C) unless otherwise specified. All other chemicals were purchased from Alfa Aesar or Aldrich and used without further purification. 1H NMR, 13C NMR and 19F NMR spectra were recorded on a Varian Mercury spectrometer and referenced to the solvent. Chemical shifts are reported as δ values (ppm) referenced to the solvent residual signals: CD3OD, δH 3.31 ppm, δC 49.15 ppm; CD2Cl2, δH 5.32 ppm, δC 54.00 ppm; D2O, δH 4.80 ppm; CF3COOH for 19F NMR, δF −78.50 ppm. High-resolution mass spectra were obtained on a Bruker Daltonics APEXIV 4.7 Tesla Fourier transform mass spectrometer. Flash column chromatography was performed with 70–230 mesh silica gel.
Synthesis of 7-hydroxycoumarin 2
5-Aminovaleric acid (55 mg) and anhydrous triethylamine (0.1 mL) was added to a solution of 7-hydroxycoumarin-3-carboxylic acid succinimidyl ester 1 (50 mg, from AnaSpec) in anhydrous DMF (0.5 mL). The reaction proceeded for 4 h at 25°C in the dark. The mixture was diluted with ethyl acetate (10 mL) and HCl (10 mL, 1m). Layers were separated, and the aqueous layer was extracted with ethyl acetate (3×15 mL). The combined organic layer was washed with water and brine. The organic phase was dried over Na2SO4 and concentrated in vacuo. The residue was purified by preparatory thin-layer chromatography (silica gel, EtOAc/MeOH/acetic acid 90:5:5) to give 2 as yellow solid (48 mg, 98%). 1H NMR (400 MHz, CD3OD, 25°C): δ=8.75 (s, 1H), 7.66 (d, J=8.7 Hz, 1H), 6.87 (dd, J=2.1, 8.6 Hz, 1H), 6.76 (d, J=1.9, 1H), 3.54 (m, 2H; CH2), 2.31 (t, 2H; CH2), 1.68 (m, 4H; CH2); HR ESI-MS calcd: 306.0972 [M + H]+, obs.: 306.0983.
Synthesis of 7-hydroxycoumarin methyl ester 3
Aqueous HCl (0.1 mL, 1m) was added to a solution of 2 (5 mg) in MeOH (1 mL). The reaction proceeded for 24 h at 25°C. Purification by flash column chromatography (silica gel, 20:80 hexanes/EtOAc) afforded 3 (5 mg, 93%) as a yellow solid. 1H NMR (500 MHz, CD3OD, 25°C): δ=8.75 (s, 1H), 7.62 (d, J=8.6 Hz, 1H), 6.90 (d, J=8.6 Hz, 1H), 6.79 (s, 1H), 3.67 (s, 3H; CH3), 3.44 (m, 2H; CH2), 2.39 (t, 2H; CH2), 1.71 (m, 4H; CH2); 13C NMR (125 MHz, CD3OD, 25°C): δ=175.4, 165.3, 163.1, 157.9, 149.5, 132.5, 115.6, 114.1, 112.5, 103.1, 52.2, 40.7, 34.3, 29.7, 23.1; HR ESI-MS calcd: 320.1129 [M+H]+, obs.: 320.1139.
Synthesis of 7-trifluoromethylsulfonylcoumarin methyl ester 4
Trifluoromethanesulfonic anhydride (30 μL, 0.18 mmol) was slowly added to a solution of 3 (38 mg, 0.12 mmol) in anhydrous dichloromethane (5 mL) and anhydrous pyridine (0.1 mL) at 0°C. The resulting mixture was stirred at room temperature for 2 h. The reaction was quenched with brine and diluted with ethyl acetate (10 mL). Layers were separated, and the aqueous layer was extracted with ethyl acetate (3×10 mL). The combined organic phase was dried over Na2SO4 and concentrated in vacuo to afford 4 (39 mg, 87%) as a brown solid. The product was used in the next reaction without further purification. 1H NMR (500 MHz, CD2Cl2, 25°C): δ=8.89 (s, 1H), 7.85 (d, J=8.7 Hz, 1H), 7.38 (d, J=2.1 Hz, 1H), 7.33 (dd, J=2.0, 8.7 Hz, 1H), 3.64 (s, 3H; CH3), 3.45 (m, 2H; CH2), 2.35 (t, 2H; CH2), 1.68 (m, 4H; CH2); 13C NMR (125 MHz, CD2Cl2, 25°C): δ=174.1, 161.1, 160.9, 155.3, 152.6, 147.25, 132.2, 119.2, 119.1, 117.9, 115.3, 110.7, 51.9, 39.9, 34.0, 29.4, 22.8; 19F NMR (300 MHz, CD2Cl2, 25°C): δ=−72.98; HR ESI-MS calcd: 452.0621 [M+H]+, obs.: 452.0611.
Synthesis of 7-diphenylmethyleneaminocoumarin methyl ester 5
An oven-dried flask was charged with (R)-(+)-BINAP (11 mg, 0.02 mmol), palladium(II) acetate (3 mg, 0.2 mmol), 4 (86 mg, 0.2 mmol), and cesium carbonate (164 mg, 0.5 mmol) and then purged with nitrogen. Benzophenone imine (46 mg, 0.025 mmol) and THF (5 mL) were added, and the mixture was stirred at reflux under nitrogen for 4 h. The mixture was cooled to room temperature, filtered, and concentrated. The yellow residue was purified by column chromatography (silica gel, hexanes/EtOAc 95:5→50:50) to give 5 (53 mg, 70%) as a yellow solid. 1H NMR (500 MHz, CD3OD, 25°C): δ=8.75 (s, 1H), 7.73 (d, J=8.7 Hz, 1H), 7.2–7.7 (m, 10H) 6.86 (dd, J=1.9, 8.6 Hz, 1H), 6.79 (s, 1H), 3.60 (s, 3H; CH3), 3.42 (m, 2H; CH2), 2.37 (t, 2H; CH2), 1.66 (m, 4H; CH2); 13C NMR (125 MHz, CD3OD, 25°C): δ=174.2, 170.1, 162.2, 158.0, 155.8, 148.3, 130.7, 130.5, 130.1, 129.8, 129.7, 128.8, 119.2, 116.6, 114.7, 108.1, 51.9, 39.7, 34.0, 30.2, 22.8; HR ESI-MS calcd: 483.1914 [M+H]+; obs.: 483.1932.
Synthesis of 7-aminocoumarin 6
HCl (0.5 mL, 1m) was added to a stirring solution of 5 (10 mg, 21 mmol) in THF/water (1:1, 10 mL). The mixture was stirred at 25°C for 48 h, then concentrated in vacuo. The yellow residue was purified by column chromatography (silica gel, EtOAc/MeOH/NH4OH 94:5:1) to afford 6 as a light yellow solid (5 mg, 76%). 1H NMR (500 MHz, D2O, 25°C): δ=8.30 (s, 1H), 7.36 (d, J=8.3 Hz, 1H), 6.66 (d, J=8.6 Hz, 1H), 6.40 (s, 1H), 3.36 (m, 2H; CH2), 2.29 (t, 2H; CH2), 1.66 (m, 4H; CH2); 13C NMR (125 MHz, CD3OD, 25°C): δ=181.8, 164.6, 163.3, 158.4, 148.9, 132.2, 113.5, 109.7, 109.5, 98.4, 39.8, 38.1, 29.8, 24.5; UV/Vis (phosphate buffer pH 7): λmax (ε)=380 nm (18400m−1cm−1); HR ESI-MS calcd: 303.0986 [M–H]−, obs.: 303.0973.
Synthesis of 7-aminocoumarin-AM
Silver(I) oxide (6 mg, 30 μmol) followed by acetoxymethyl bromide (1.5 μL, 15 μmol) was added to a stirring solution of 7-aminocoumarin 6 (3 mg, 9 μmol) in anhydrous acetonitrile (1 mL). The mixture was stirred at 25°C for 12 h, then concentrated in vacuo. The yellow residue was purified by column chromatography (silica gel, EtOAc/hexane 8:1) to afford 7-aminocoumarin-AM as a light yellow solid (3 mg, 81% yield). 1H NMR (300 MHz, CDCl3, 25°C): δ=8.39 (s, 1H), 7.28 (d, J=8.7 Hz, 1H), 6.70 (dd, J=8.6, 2.4 Hz, 1H), 6.45 (d, J=2.4 Hz, 1H), 5.72 (s, 2H), 3.34 (m, 2H; CH2), 2.31 (t, 2H; CH2), 2.09 (s, 3H; CH3), 1.70 (m, 4H; CH2); HR ESI-MS calcd: 377.1343 [M+H]+, obs.: 377.1348.
7-Aminocoumarin and 7-hydroxycoumarin pH profiles (Figure 1B)
Fluorescence emission was recorded for 150 μm solutions by using a TECAN Safire Microplate Reader and a plastic transparent-bottomed 384-well plate (Greiner). pH 3–6 buffers were prepared by mixing different ratios of acetic acid (0.1m) and sodium acetate-trihydrate (0.1m) solutions. pH 7–10 buffers were prepared by mixing different ratios of Na2HPO4 (0.1m) and either HCl (0.1m; for pH 7–9 buffers) or NaOH (0.1m; for pH 10 buffer). Final pH adjustments in all buffer solutions were made by adding small amounts of HCl (1m) or NaOH (1m).
In vitro 7-aminocoumarin ligation reactions (Figures 1C and S2)
For Figure 1C, reactions were assembled as follows: LplA enzyme (2 μm), LAP2 synthetic peptide (150 μm, sequence: GFEIDKVWYDLDA[16]), 7-aminocoumarin 6 probe (500 μm), ATP (5 mm), and Mg(OAc)2 (5 mm) in Na2HPO4 (25 mm, pH 7.2). The reaction mixture was incubated at 30°C for 2 h and quenched with EDTA (final concentration 100 mm). The mixture was analyzed on a Varian Prostar HPLC by using a reversed-phase C18 Microsorb–MV 100 column (250×4.6 mm). Chromatograms were recorded at 210 nm. We used a 10-min gradient of 30–60% acetonitrile in water with 0.1% trifluoroacetic acid at a flow rate of 1 mLmin−1. LAP2 had a retention time of 7 min; after ligation to 7-aminocoumarin, tR increased to 9 min.
For Figure S2, W37VLplA (2 μm) and coumarin probe (500 μm) were used in one case. Aliquots from the reaction were collected and quenched with EDTA over 55 min. For the other case, W37VLplA (1 μm) and coumarin probe (100 μm) were used, and aliquots were collected and quenched over 70 min. After HPLC analysis, percent product conversions were calculated by dividing the product peak area by the sum of (product+starting material) peak areas.
Mass spectrometric analysis of peptides (Figure S1)
Starred peaks from Figure 1C were manually collected and injected into an Applied Biosystems 200 QTRAP mass spectrometer. The flow rate was 3 μLmin−1, and mass spectra were recorded under the positive-enhanced multicharge mode.
Mammalian cell culture
Human embryonic kidney (HEK) cells were cultured in Dulbecco’s modified Eagle medium (DMEM; Cell-gro, Manassa, VA, USA) supplemented with 10% v/v fetal bovine serum (PAA Laboratories, Etobicoke, Ontario, Canada). For imaging, cells were plated as a monolayer on glass coverslips. Adherence of HEK cells was promoted by precoating the coverslip with fibronectin (50 μgmL−1, Millipore). All cells were maintained at 37°C under 5% CO2.
PRIME cell-surface labeling (Figure 2)
HEK cells were transfected at ~70% confluency with expression plasmids for LAP4.2[16]-neurexin-1β (400 ng for a 0.95 cm2 dish) and H2B-YFP (100 ng) using Lipofectamine 2000 (Invitrogen). 18 h after transfection, cells were treated with W37VLplA enzyme (10 μm), coumarin probe (200 μm), ATP (1 mm), and Mg(OAc)2 (5 mm) in cell growth medium for 20 min at room temperature. After excess labeling reagents had been removed by replacing the medium 2–3 times, cells were immediately imaged or incubated at 37°C for 20 min to allow cell-surface protein turnover.
PRIME intracellular labeling (Figure 3)
HEK or HeLa cells were transfected with expression plasmids for W37VLplA (20 ng) and LAP substrate (LAP2-YFP, LAP2-YFP-NLS, or LAP2-β-actin; 400 ng) by using Lipofectamine 2000. 18 h after transfection, cells were treated with 7-aminocoumarin-AM (20 μm) in serum-free DMEM for 10 min at 37°C. Excess coumarin probe was removed by washing the cells with cell-growth medium (4×15 min). Cells were then imaged live.
Fluorescence imaging
Cells were imaged in Dulbecco’s phosphate-buffered saline (DPBS) in confocal mode. We used a Zeiss Axiovert 200M inverted microscope with a 40× oil-immersion objective. The microscope was equipped with a Yokogawa spinning-disk confocal head, a Quad-band notch dichroic mirror (405/488/568/647), and 405 (diode), 491 (DPSS), and 561 nm (DPSS) lasers (all 50 mW). 7-Aminocoumarin (405 laser excitation, 445/40 emission), YFP (491 laser excitation, 528/38 emission), and DIC images were collected by using Slidebook software. Fluorescence images in each experiment were normalized to the same intensity ranges. Acquisition times ranged from 10–1000 ms.
Supplementary Material
Acknowledgments
Funding was provided by the NIH (R01 GM072670) and MIT. X.J. was supported by John Reed (MIT Class of 1961) and Paul E. Gray (MIT Class of 1954) Funds, administered by the MIT Undergraduate Research Opportunity Program. We thank Jennifer Yao for LplA enzymes, Daniel S. Liu and Katharine A. White for plasmids, David Surry (MIT) for synthetic advice, and Peng Zou for helpful feedback on the manuscript.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cbic.201000414.
References
- 1.Uttamapinant C, White KA, Baruah H, Thompson S, Fernandez-Suarez M, Puthenveetil S, Ting AY. Proc Natl Acad Sci USA. 2010;107:10914–10919. doi: 10.1073/pnas.0914067107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baruah H, Puthenveetil S, Choi YA, Shah S, Ting AY. Angew Chem Int Ed. 2008;47:7018–7021. doi: 10.1002/anie.200802088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fernandez-Suarez M, Baruah H, Martinez-Hernandez L, Xie KT, Baskin JM, Bertozzi CR, Ting AY. Nat Biotechnol. 2007;25:1483–1487. doi: 10.1038/nbt1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cronan JE, Zhao X, Jiang YF. Adv Microb Physiol. 2005;50:103–146. doi: 10.1016/S0065-2911(05)50003-1. [DOI] [PubMed] [Google Scholar]
- 5.Sun WC, Gee KR, Haugland RP. Bioorg Med Chem Lett. 1998;8:3107–3110. doi: 10.1016/s0960-894x(98)00578-2. [DOI] [PubMed] [Google Scholar]
- 6.Demaurex N. News Physiol Sci. 2002;17:1–5. doi: 10.1152/physiologyonline.2002.17.1.1. [DOI] [PubMed] [Google Scholar]
- 7.Rosen T. Comp Org Syn. 1991;2:395–408. [Google Scholar]
- 8.Kappe T, Mayer C. Synthesis. 1981:524–526. [Google Scholar]
- 9.Guram AS, Buchwald SL. J Am Chem Soc. 1994;116:7901–7902. [Google Scholar]
- 10.Paul F, Patt J, Hartwig JF. J Am Chem Soc. 1994;116:5969–5970. [Google Scholar]
- 11.Wolfe JP, Ahman J, Sadighi JP, Singer RA, Buchwald SL. Tetrahedron Lett. 1997;38:6367–6370. [Google Scholar]
- 12.Ahman J, Buchwald SL. Tetrahedron Lett. 1997;38:6363–6366. [Google Scholar]
- 13.Craig AM, Kang Y. Curr Opin Neurobiol. 2007;17:43–52. doi: 10.1016/j.conb.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsien RY. Annu Rev Neurosci. 1989;12:227–253. doi: 10.1146/annurev.ne.12.030189.001303. [DOI] [PubMed] [Google Scholar]
- 15.Oh YK, Straubinger RM. Pharm Res. 1997;14:1203–1209. doi: 10.1023/a:1012158924547. [DOI] [PubMed] [Google Scholar]
- 16.Puthenveetil S, Liu DS, White KA, Thompson S, Ting AY. J Am Chem Soc. 2009;131:16430–16438. doi: 10.1021/ja904596f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.