

Abstract
Germline mutations in the succinate dehydrogenase complex subunit D gene are now known to be associated with hereditary paraganglioma–pheochromocytoma syndromes. Since the initial succinate dehydrogenase complex subunit D gene mutation was identified about a decade ago, more than 131 unique variants have been reported. We report the case of two siblings presenting with multiple paragangliomas and pheochromocytomas; they were both found to carry a mutation in the succinate dehydrogenase complex subunit D gene involving a substitution of thymine to guanine at nucleotide 236 in exon 3. This particular mutation of the succinate dehydrogenase complex subunit D gene has only been reported in one previous patient in Japan; this is, therefore, the first report of this pathogenic mutation in siblings and the first report of this mutation in North America. With continued screening of more individuals, we will be able to create a robust mutation database that can help us understand disease patterns associated with particular variants and may be a starting point in the development of new therapies for familial paraganglioma syndromes.
Keywords: Succinate dehydrogenase complex subunit D, germline mutation, pheochromocytoma, paraganglioma
Introduction
Paragangliomas (PGLs) are tumors arising from neuroendocrine tissues. Most parasympathetic PGLs are located in the head/neck and do not secrete hormones but can cause symptoms due to mass effect. Sympathetic PGLs, including pheochromocytomas (PCCs), are usually located in the lower mediastinum/abdomen/pelvis and cause symptoms related to hypersecretion of catecholamines.1
Hereditary syndromes associated with PCC include neurofibromatosis type 1 (NF1), multiple endocrine neoplasia type 2 (MEN2), and von Hippel-Lindau (VHL) disease; these syndromes are associated with germline mutations in the NF1, RET, and VHL genes, respectively.2 Advancements in molecular genetics have shown that most hereditary PGL/PCC syndromes are associated with mutations in the genes encoding one of the four subunits of succinate dehydrogenase (SDH), a crucial mitochondrial enzyme.3,4 The hereditary PGL/PCC syndromes demonstrate an autosomal-dominant inheritance pattern with incomplete penetrance; additionally, succinate dehydrogenase complex subunit D (SDHD) mutations tend to be pathologic only when the mutation is inherited from the father.5
Any patient with PCCs and/or PGLs should be screened for a hereditary PGL/PCC syndrome, especially those with multiple or recurrent tumors, onset before the age of 45 years, and positive family history.6 Since the mutations associated with hereditary PGL/PCC syndromes are mutually exclusive and particular mutations are associated with specific patterns of disease, a sequential method of genetic screening is recommended.7 For instance, SDHD mutations are particularly associated with head and neck PGLs, and patients presenting with such PGLs should be first tested for the SDH gene.8 Asymptomatic first-degree relatives of patients with a proven mutation should be screened, as early detection can alleviate risk of complications from catecholamine excess, mass effect, or malignant transformation.9
Whenever feasible, PCCs and PGLs are best treated with surgical resection.10 This should be preceded by hormonal blockade for secretory tumors. Prompt surgical resection of tumors in patients with succinate dehydrogenase complex subunit B (SDHB) mutations is especially critical due to elevated risk of malignant transformation and metastasis.11
Case report
A 42-year-old male presented for evaluation of refractory hypertension, with which he had struggled since the age of 15 years. He reported intermittent episodes of flushing, diaphoresis, anxiety, and headache. Blood pressure was 148/82 and heart rate was 94 beats per minute on a regimen of hydralazine and atenolol. On examination, there was notable fullness in the neck bilaterally, especially along the lateral aspects, although no discrete mass was palpable. Laboratory evaluation revealed markedly elevated 24-h urine normetanephrine and total vanillylmandelic acid. Levels of electrolytes, thyroid stimulating hormone, parathyroid hormone, and calcitonin were all within normal limits. Both computerized tomography (CT) and magnetic resonance imaging (MRI) of the neck showed bilateral carotid body tumors about 3 cm in size each, partially encasing both external carotid arteries (Figures 1 and 2). There was also evidence of a 1.2 × 1.1-cm2 PGL of the vagus nerve (i.e. glomus vagale). CT-abdomen showed three separate left adrenal masses and two right adrenal masses, measuring between 1.5 and 3.9 cm (Figure 3). A nuclear medicine I-123 meta-iodobenzylguanidine (MIBG) scan showed increased tracer uptake in both adrenal glands, consistent with bilateral PCCs (Figure 4). Within 1 month of diagnosis, the patient was started on phenoxybenzamine and subsequently underwent bilateral laparoscopic adrenalectomy. Pathology confirmed bilateral multinodular PCCs with focal vascular invasion, but lymph nodes were free of involvement. His blood pressure normalized, and he was discharged on hydrocortisone and fludrocortisone. Metanephrines normalized after adrenalectomy, and the patient no longer required any antihypertensives. Several months after adrenalectomy, the patient underwent resection of both PGLs, which were nonsecretory and pathologically confirmed to be PGLs. On recent follow-up, his blood pressure remains well controlled, and repeat metanephrine levels are within normal limits. Follow-up CT-chest/abdomen/pelvis shows no evidence of PCC recurrence. Repeat CT-neck shows no recurrence of bilateral carotid body PGLs, and the glomus vagale remains stable in size.
Figure 1.
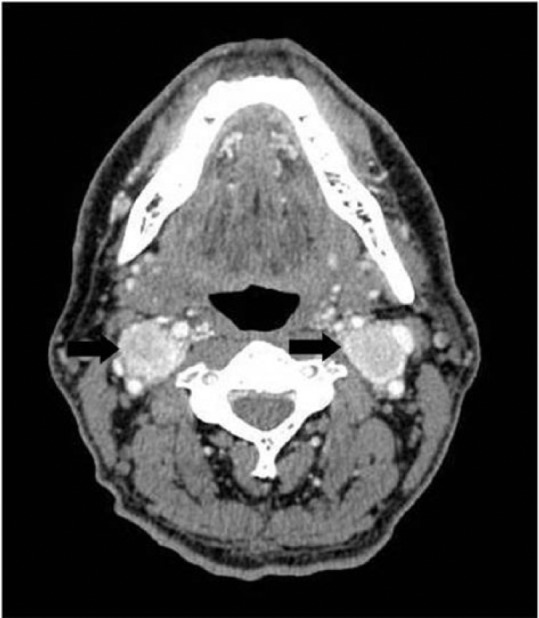
Contrast-enhanced CT of the neck at the level of the carotid bifurcation demonstrates intensely enhancing mass lesions at the level of the carotid bifurcations suggestive of paragangliomas (black arrows).
CT: computerized tomography.
Figure 2.

(a) Coronal T2-weighted and (b) axial T1-weighted MR sequences demonstrate mass lesions at the level of the carotid bifurcations (black arrows). There are numerous flow voids in the lesions reflecting the vascular nature of these tumors.
MR: magnetic resonance.
Figure 3.
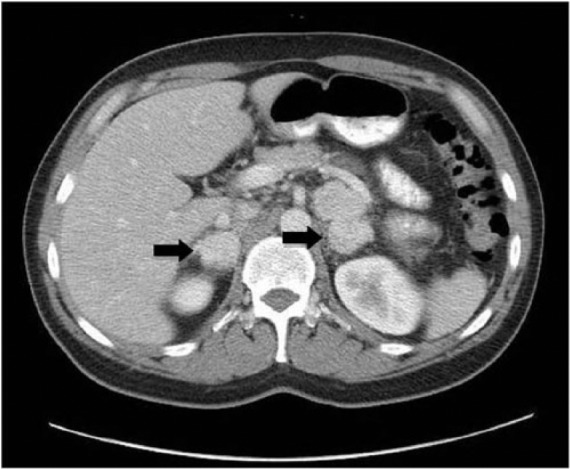
Contrast-enhanced CT imaging of the abdomen demonstrates multiple enhancing mass lesions in the adrenal glands bilaterally (black arrows).
CT: computerized tomography.
Figure 4.
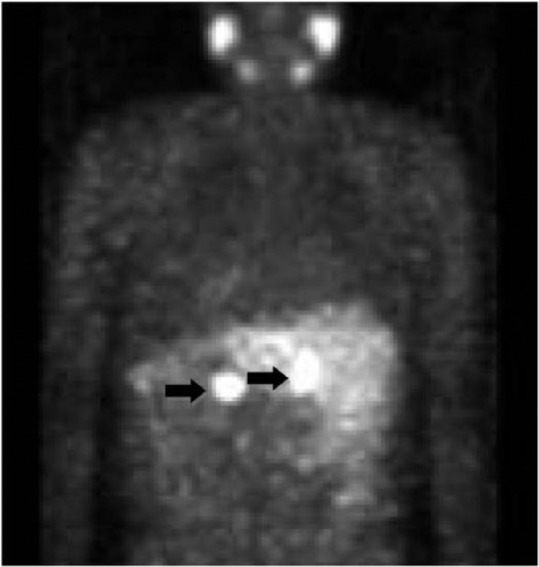
I-123 MIBG imaging demonstrates radiotracer uptake in the bilateral adrenal glands consistent with pheochromocytomas (black arrows).
MIBG: meta-iodobenzylguanidine.
The patient underwent genetic counseling and testing. He tested negative for the MEN2a/b, VHL, and NF1 mutations but further testing revealed a novel mutation of the SDHD gene involving a substitution of thymine to guanine at nucleotide 236 in exon. Among hundreds of observed SDHD mutations, this particular mutation was identified only in one previous patient in Japan who was affected with the PGL/PCC syndrome, although details about that patient’s presentation are not available.
The patient’s first-degree family members were evaluated; his father had passed away and was not available for gene testing but his brother tested positive for the same SDHD gene mutation. He also had multiple PGLs including a right adrenal PCC, left peri-adrenal PGL, left peri-aortic PGL, and bilateral carotid body PGLs. The PCC and abdominal PGLs were first resected followed by the right carotid body PGL; the left carotid body PGL was left intact due to its sub-centimeter size and nonfunctioning status.
The University of Pittsburgh Medical Center does not require ethics approval for reporting individual cases. The patients provided written informed consent for their information and images to be included in this article and for publication in an international medical journal.
Molecular genetic analysis
After appropriate informed consent, a sample of the patients’ blood was sent for mutation analysis of the patient’s SDHD gene available as a clinical assay at the University of Pittsburgh Medical Center. In brief, genomic DNA was extracted from 1 mL of whole blood on the MagNA Pure LC (Roche Diagnostics) using manufacturer’s instructions into a total elution volume of 200 µL. From an aliquot of this elution, polymerase chain reaction (PCR) amplification of exons 1 through 4 of the SDHD gene and flanking intron-exon boundaries was performed on an ABI 9700 Thermocycler (Applied Biosystems Incorporated). Following PCR amplification, 2 µL of the PCR product were prepared using the same primers in forward and reverse directions using the ABI BigDye Terminator Kit (Applied Biosystems Incorporated) according to manufacturer’s instructions and sequenced on an ABI 3130xl sequencer (Applied Biosystems Incorporated; 50 cm capillary, POP6 polymer) using ABI Sequencing Analysis Software 5.3.1 and Mutation Surveyor 4.06 (Softgenetics) for analysis of sequencing data using NG_012337 reference sequence for the SDHD gene. Sequencing for the SDHB and succinate dehydrogenase complex subunit C (SDHC) genes was performed using the same method with primers specific for all exons and intron-exon boundaries of those genes.
Review of the sequencing data by molecular genetic pathologists revealed a heterozygous substitution of thymine to guanine at nucleotide 236 (c.236T > G) in exon 3 of the SDHD gene present in both forward and reverse sequences (Figure 5). No significant change from wild-type sequences was found in any of the exons of SDHB or SDHC for this patient. The observed change in SDHD alters the second nucleotide of codon 79, resulting in a missense amino acid variant p.Leu79Arg. This p.Leu79Arg variant is listed in the Leiden Open Variation Database for SDHD (http://chromium.liacs.nl/LOVD2/SDH; accessed 22 May 2013) as having been identified in only one previous patient in Japan and having a Sorting Intolerant from Tolerant (SIFT) score of 0.00 (<0.05 is interpreted as most likely damaging or pathogenic). Evolutionary conservation analysis via Basic Local Alignment Search Tool (BLAST) showed chimpanzees, birds, and amphibians conserve the leucine residue at this codon, while most ungulates, rodents, insects, and nematodes substitute the chemically very similar isoleucine. Arginine has chemical properties quite different from leucine, or isoleucine, and could be expected to have a more significant effect on the SDHD protein and function, consistent with the SIFT analysis reported at the Leiden Open Variation Database. Since the p.Leu79Arg variant had been identified in only one other patient, this result was interpreted as an American College of Medical Genetics and Genomics (ACMG) category 3 variant (“may or may not be responsible for disease”12), although SIFT and evolutionary conservation analysis suggested a slight propensity to pathogenicity.
Figure 5.
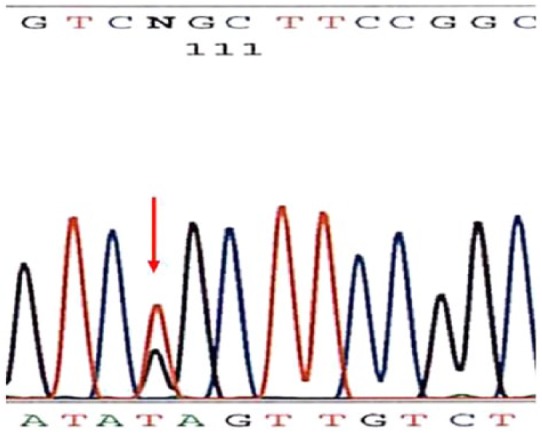
Sequence analysis showing the heterozygous substitution of thymine to guanine at nucleotide 236 (c.236T > G) in exon 3 of the SDHD gene present in both forward and reverse sequences.
SDHD: succinate dehydrogenase complex subunit D.
Discussion
The mechanism by which SDH complex mutations lead to development of PGLs and PCCs is unclear. Based on the knowledge that the SDH complex is critical in the tricarboxylic acid (TCA) cycle and the aerobic respiratory chains of mitochondria, it has been postulated that SDH mutations lead to cellular hypoxia and induction of angiogenic factors.13 Several studies have supported this hypothesis; one study has shown high expression of angiogenic factors in human PGLs, and another study of two adults with nonsense SDHD mutations showed high expression of genes encoding for hypoxia-inducible angiogenic factors.14 This hypothesis is further supported by the fact that PGLs are found more commonly in those living at high altitudes.15–17 Advancements in molecular genetics have allowed us to detect new mutations in the SDH complex, and this knowledge may lead to further understanding of the mechanism of tumorogenesis associated with such mutations.
To the best of the authors’ knowledge, this is the first report in North America of a mutation in the SDHD gene involving a substitution of thymine to guanine at nucleotide 236 in exon 3. This variant was previously described in one Japanese patient and is the first report of this mutation being associated with a hereditary PGL/PCC syndrome in two symptomatic brothers.
At the time of this case report, there are 131 unique SDHD gene mutations listed in the TCA cycle gene mutation database. As we continue to evaluate the entire coding region of the SDHD gene, we are likely to discover further variants. As we add to the database, we will be able to delineate patterns of disease associated with particular variants. This will allow us to provide effective genetic counseling and perhaps be the impetus for research and development of targeted therapies.
Conclusion
Patients with PGL/PCC should undergo sequential genetic testing for mutations based on the pattern of disease.
Even asymptomatic first-degree relatives of patients found to have a mutation should be screened, given the autosomal-dominant inheritance pattern and incomplete penetrance.
The initial SDH complex mutation was identified slightly over a decade ago, and we are likely to continue to identify new mutational variants.
Novel variants should be reported to the mutation database; this will ultimately assist us in providing genetic counseling, identifying patterns of disease associated with particular mutations, and developing new treatment modalities.
Footnotes
Declaration of conflicting interests: The authors declare that there is no conflict of interest.
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
References
- 1. Kirmani S, Young WF. Hereditary paraganglioma-pheochromocytoma syndromes. In: Pagon RA, Adam MP, Bird TD, et al. (eds) Gene reviews (Internet). Seattle, WA: University of Washington, 1993. [PubMed] [Google Scholar]
- 2. Pacak K, Linehan WM, Eisenhofer G, et al. Recent advances in genetics, diagnosis, localization, and treatment of pheochromocytoma. Ann Intern Med 2001; 134: 315–329. [DOI] [PubMed] [Google Scholar]
- 3. Benn DE, Gimenez-Roqueplo AP, Reilly JR, et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab 2006; 91: 827–836. [DOI] [PubMed] [Google Scholar]
- 4. Baysal BE, Ferrell RE, Willett-Brozick JE, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000; 287: 848–851. [DOI] [PubMed] [Google Scholar]
- 5. Baysal BE. Genomic imprinting and environment in hereditary paraganglioma. Am J Med Genet C Semin Med Genet 2004; 129: 85–90. [DOI] [PubMed] [Google Scholar]
- 6. Young WF., Jr. Endocrine hypertension, vol. 12 Philadelphia, PA: Saunders Elsevier, Inc, 2011, pp. 545–580. [Google Scholar]
- 7. Welander J, Soderkvist P, Gimm O. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr Relat Cancer 2011; 18: R253–R276. [DOI] [PubMed] [Google Scholar]
- 8. Neumann HP, Pawlu C, Peczkowska M, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 2004; 292: 943–951. [DOI] [PubMed] [Google Scholar]
- 9. Gujrathi CS, Donald PJ. Current trends in the diagnosis and management of head and neck paragangliomas. Curr Opin Otolaryngol Head Neck Surg 2005; 13: 339–342. [DOI] [PubMed] [Google Scholar]
- 10. Young WF., Jr. Paragangliomas: clinical overview. Ann N Y Acad Sci 2006; 1073: 21–29. [DOI] [PubMed] [Google Scholar]
- 11. Pasini B, Stratakis CA. SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma-paraganglioma syndromes. J Intern Med 2009; 266: 19–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. American College of Medical Genetics and Genomics (ACMG) Laboratory Practice Committee Working Group. ACMG recommendations for standards for interpretation of sequence variations. Genet Med 2000; 2(5): 302–303. [Google Scholar]
- 13. Saraste M. Oxidative phosphorylation at the fin de siècle. Science 1999; 283: 1488–1493. [DOI] [PubMed] [Google Scholar]
- 14. Gimenez-Roqueplo AP, Favier J, Rustin P, et al. The R22X mutation of the SDHD gene in hereditary paraganglioma abolishes the enzymatic activity of complex II in the mitochondrial respiratory chain and activates the hypoxia pathway. Am J Hum Genet 2001; 69: 1186–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sobol SM, Dailey JC. Familial multiple cervical paragangliomas: report of a kindred and review of the literature. Otolaryngol Head Neck Surg 1990; 102: 382–390. [DOI] [PubMed] [Google Scholar]
- 16. McCaffrey TV, Meyer FB, Michels VV, et al. Familial paragangliomas of the head and neck. Arch Otolaryngol Head Neck Surg 1994; 120: 1211–1216. [DOI] [PubMed] [Google Scholar]
- 17. Shedd DP, Arias JD, Glunk RP. Familial occurrence of carotid body tumors. Head Neck 1990; 12: 496–499. [DOI] [PubMed] [Google Scholar]