

Abstract
Protein-protein interactions are essential for the basic biological machinery of the cell. This is important for processes like protein synthesis, enzyme kinetics, molecular assembly and signal transduction. A high number of macromolecular structural complexes are known due to recent advances in structure determination techniques. Therefore, it is of interest to develop an interactive tool to objectively analyze large protein complexes. Hence, we describe the development and utility of a web enabled application named ‘Protein-Protein Interaction in Macro-molecular Assembly’ (PIMA) for the analysis of large protein assemblies. The intricate details of physical interactions amongst protein subunits in a large complex are presented as simple user preferred interactive network diagrams
Background
Many biological processes in the cell are driven by protein subunit assemblies [1]. Therefore, it is important to understand inter-chain interactions and their contribution to the overall three-dimensional structure for macro-molecular stability. Earlier studies have provided insights on their interaction principles using known multi-meric protein structural complexes in the Protein Data Bank (PDB) [2]. The steric complementarity [3], inter atomic hydrogen bonds, salt bridges, hydrophobic effects and change in solvent accessible surface area (ASA) upon complex formation [4] has been noted as features of protein-protein interfaces in several complex molecular assemblies for cellular functions. Visual examination of various such aspects for each interaction in an assembly is often perplexing and frequently tedious to explain. Hence, there is need for a tool that can enable user defined interactive visualization of such parameters with corresponding quantitative values to illustrate several important binding features. Therefore, we describe a simple yet a powerful visualization interface (web driven) to objectively analyze and examine large protein assemblies.
Methodology
PPCheck application
PPCheck (a tool for quantifying protein-protein interactions) was used to identify interactions in macro-molecular assemblies. Residues within a Calpha - Calpha distance threshold of 10Å are initially identified and pseudo-energies are subsequently calculated depending on the nature of interaction [5]. A parallel processing version of PPCheck was developed for improved performance in this study as shown in (Figure 1)
Figure 1.

An illustration of the workflow for parallel processing of PPCheck in PIMA is shown.
Input options:
A formatted PDB file is uploaded using the web form to initiate PIMA analysis using default parameters as shown in (Figure 2a)
Figure 2.
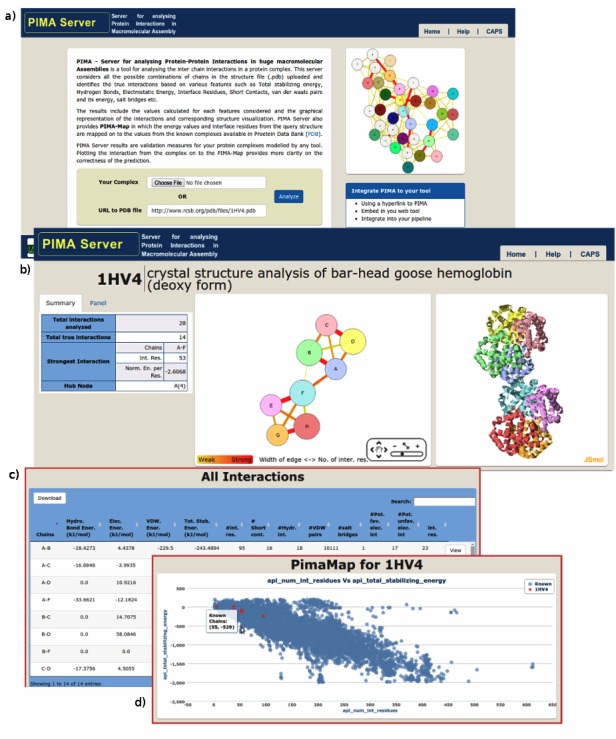
Input-Output interface of PIMA; (a) interface for user input and submission; (b) results with interaction network and 3D visualization; (c) tabular output of interactions; (d) PIMA map with energy values plotted along with known interactions.
Output options:
Interaction network and 3D visualization The inter-chain interactions are presented in the form of an interactive network graph along with the corresponding 3D molecular visualization according to the user instruction is shown in (Figure 2b). This component of the output page has two parts. The first part displays the chains (protein subunits) in the form of a graph-nodes (circles) and interactions in the form of graph-edges (lines). The second part displays the threedimensional representation of the molecule as shown in Figure 2b.
Each node represents a single chain in the complex. It uses the color-coding from the standard JSMol coding (color) library that enables easy visualization of the studied molecule for establishing an imaginary yet a qualitative rational relationship. This is in-addition to the labels used to describe chain (protein subunit) names in the PDB file. The size of the node represents the size (the number of atoms) of the chain. An edge represents a true interaction between two chains and the width represents the number of interface residues (usually referred as interface size). The strength of the interactions (PPCheck energies) is represented using the color gradient from red (strong) to yellow (weak). The features added to each component in PIMA are highly interactive with each other that enable the user to reasonably probe into the structure and nature of the interactions at the interfaces. PIMA analysis also displays data such as the number of atoms and the protein sequence for the molecule under study.
Search and sort interactions
Interactions identified in the protein complex using PIMA analysis are displayed as a table with relevant parameter values for subsequent interpretation (Figure 2c). The interactive table enables the user to sort the rows with essential parameter values in the columns. This helps to search and find a specific interaction where required. Interface residue pairs for the interacting chains (subunits) along with the type of interactions are also provided for each pair of protein subunits in the assembly complex.
Energy plot
This plot displays the energy values of each interaction for the input complex along with other known physical interactions described and or derived using data provided in the PDB file (Figure 2d). The interactions from the input complexes are marked with a distinguishable color from other known interactions. It is also possible to zoom (in and out) the plot to examine patterns within a specific energy range in addition to the complete energy profile for interaction.
Tool description
PIMA Server is a tool to analyze and visualize inter-chain interactions in a huge protein-protein assembly complex. This helps to study the stability of the complex for functional inferences.
Implementation
The PIMA server back end is implemented using Python, Bio- Python [6],[7] and Python-CGI technologies. The user-friendly web interface is implemented using HTML5, CSS, JavaScript and JQuery technologies. The chain interaction map is implemented using Cytoscape web [8] and the 3D molecular visualization of the protein molecule is implemented using JSMOL [9]. Parallel processing is achieved using the multiprocessing module in Python.
Performance
PIMA produces results in 17 seconds for a protein assembly complex (PDB ID: 1HV4) with 8 chains and 9288 non-hydrogen atoms. It should be noted that the performance of the system vary according to the number of chains in the complex, the size of the chains and the load on the server.
Conclusion
PIMA is a powerful interactive graphical resource to abstract and objectively conceptualize the inter-chain relationships in huge protein assemblies. The results are presented as simple two-dimensional networks without compensating on the details of interactions.
Acknowledgments
OKM would like to thank Mr. Anshul Sukhwal for providing the PPCheck Algorithm. He also thanks the Vice Chancellor of SASTRA University, Tamilnadu, India. The work is supported by Centre for Excellence Grant (BT/01/COE/09/01) funded by Department of Biotechnology, India. The authors also thank NCBS (TIFR) for infrastructural support in this study.
Footnotes
Citation:Mathew & Sowdhaminiet al. Bioinformation 12(1): 9-11 (2016)
References
- 1.King RW, et al. Science. 1996;274:1652. doi: 10.1126/science.274.5293.1652. [DOI] [PubMed] [Google Scholar]
- 2.Jones S, Thornton JM. Proc Natl Acad Sci U S A. 1996;93:13. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsai CJ, et al. Protein Science. 1997;6:1793. doi: 10.1002/pro.5560060901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chakravarty D, et al. Protein Sci. 2013;22:1453. doi: 10.1002/pro.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sukhwal A, Sowdhamini R. Molecular Biosystems. 2013;9:1652. doi: 10.1039/c3mb25484d. [DOI] [PubMed] [Google Scholar]
- 6.Chapman B, Chang J. ACM SIGBIO Newsletter. 2000;20:15. [Google Scholar]
- 7.Cock PJA, et al. Bioinformatics. 2009;25:1422. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lopes CT, et al. Bioinformatics. 2010;26:2347. doi: 10.1093/bioinformatics/btq430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanson RM, et al. Israel Journal of Chemistry. 2013;53:207. [Google Scholar]