

Abstract
Shank3 is a multi-domain, synaptic scaffolding protein that organizes proteins in the postsynaptic density of excitatory synapses. Clinical studies suggest that ~0.5% of autism spectrum disorder (ASD) cases may involve SHANK3 mutation/deletion. Patients with SHANK3 mutations exhibit deficits in cognition along with delayed/impaired speech/language and repetitive and obsessive/compulsive-like (OCD-like) behaviors. To examine how mutation/deletion of SHANK3 might alter brain function leading to ASD, we have independently created mice with deletion of Shank3 exons 4–9, a region implicated in ASD patients. We find that homozygous deletion of exons 4–9 (Shank3e4–9 KO) results in loss of the two highest molecular weight isoforms of Shank3 and a significant reduction in other isoforms. Behaviorally, both Shank3e4–9 heterozygous (HET) and Shank3e4–9 KO mice display increased repetitive grooming, deficits in novel and spatial object recognition learning and memory, and abnormal ultrasonic vocalizations. Shank3e4–9 KO mice also display abnormal social interaction when paired with one another. Analysis of synaptosome fractions from striata of Shank3e4–9 KO mice reveals decreased Homer1b/c, GluA2, and GluA3 expression. Both Shank3e4–9 HET and KO demonstrated a significant reduction in NMDA/AMPA ratio at excitatory synapses onto striatal medium spiny neurons. Furthermore, Shank3e4–9 KO mice displayed reduced hippocampal LTP despite normal baseline synaptic transmission. Collectively these behavioral, biochemical and physiological changes suggest Shank3 isoforms have region-specific roles in regulation of AMPAR subunit localization and NMDAR function in the Shank3e4–9 mutant mouse model of autism.
Keywords: autism spectrum disorder, Shank3, Phelan-McDermid syndrome, mouse model, grooming
Introduction
Autism spectrum disorder (ASD) is defined by the presence of social deficits, impaired communication, and stereotyped repetitive behaviors [APA, 2013]. Genetic studies implicate deletions and mutations of the SHANK3 gene as likely contributors to the behavioral and biological alterations associated with ASD and Phelan-McDermid (22q13 Deletion) Syndrome [Boccuto et al., 2013; Durand et al., 2007; Gauthier et al., 2009; Kolevzon et al., 2011; Moessner et al., 2007; Naisbitt et al., 1999; Tu et al., 1999; Turner 1999; Waga et al., 2011].
Shank3 is a member of the ProSAP/Shank family of postsynaptic scaffolding proteins [Boeckers, Bockmann, Kreutz, & Gundelfinger, 2002; Sheng & Kim, 2000] and is composed of five highly conserved protein domains that mediate a number of synaptic functions [Arons et al., 2012; Boeckers et al., 2001, 2005; Durand et al., 2007; Lim et al., 2001; Naisbitt et al., 1999; Peca et al., 2011; Sala et al., 2001; Sheng & Kim, 2000; Verpelli et al., 2011; Wang et al., 2011].
To date more than 14 point mutations, nucleotide insertions, microdeletions, microduplications, translocations, and chromosome deletions or rearrangements involving SHANK3 have been observed in genetic studies involving ASD patients [Bonaglia et al., 2001, 2006; Durand et al., 2007; Gauthier et al., 2009, 2010; Grabrucker, Schmeisser, Schoen, & Boeckers, 2011; Hamdan et al., 2011; Jiang & Ehlers, 2013; Kolevzon et al., 2011; Misceo et al., 2011; Moessner et al., 2007; Sykes et al., 2009]. Point mutations and deletions within Shank3 exons 4–9 have been shown to alter protein binding or to result in a complete loss of ankyrin repeat domain (ANK) function [Durand et al., 2007, 2012; Mameza et al., 2013].
In this study, we generate mice heterozygous (HET) or homozygous (KO) for Shank3 exon 4–9 deletion (Shank3e4–9) and examine functional consequences at multiple levels.
Materials and Methods
Generation of Shank3e4–9 Mutant Mice
A targeting construct was designed to delete exons 4–9 of Shank3 using Cre-loxP-mediated excision. The targeting vector was pBluescript II SK (+/−) (Agilent Technologies). The final construct had two homology arms, 5′ (2,194 bp) and 3′ (5,768 bp). A 4,581 bp DNA fragment containing exon 4–9 genomic DNA (2,575 bp) flanked by 2 loxP sequences and a Neo cassette (1,866 bp, positive selection) flanked by 2 frt sequences were inserted between 5′ and 3′ homology regions. For negative selection, a diphtheria toxin cassette was cloned adjacent to the 5′ end of the 5′ homology region. The construct, linearized by NotI, was electroporated into ES cells (129s6SvEvTac) and ES clones were selected for G418 resistance. ES clones with appropriately targeted recombination were identified by PCR using three primers (Forward: GAAACAGTGTGAGCGCCGTGTGATG; Reverse (1): GATGGATCTCTTGCCAACCATTCTC, Reverse (2): CAAATCCCTTCCCTGCATATAACTTCG); WT produced a 3,091 bp band while knock-in produced a 2,312 bp band. Accuracy of homologous recombination was confirmed by sequencing PCR products. Then genomic DNA from the ES cells was analyzed by Southern blotting to distinguish between targeted and wild-type Shank3 alleles. Positive ES clones were injected into blastocysts (C57BL6J strain) in our Transgenic Facility. Chimeras were bred with C57BL6J to confirm germ-line transmission, identified by PCR (primers: Forward: GTGGCCATTATTGCAGGGAACTTTGAG; Reverse: GTCTCAGAAGACCCTTCCTAGCACCTAATG); WT DNA produced a 329 bp band while knock-in DNA produced a 451 bp band. Knock-in mice were mated with mice expressing FLP1 recombinase to excise the Neo cassette generating conditional Shank3 mice. Conditional Shank3 mice were crossed with mice expressing Zp3-Cre to excise exon 4–9 in oocytes of female pups, and these were crossed with C57BL6J to excise exon 4–9 in all cells. Resulting progeny were genotyped using a combination of three primers as follows: Forward: GTGGAGGAATGAGACCAGAGTTGTTAGG, Reverse (1): GTGTCTAACCTGTCACCTAGCTTGCTCATCC, Reverse (2): GTCTCAGAAGACCCTTCCTAGCACCTAATG. WT DNA produced a 261 bp band; conditional knock-in produced a 301 bp band, and exon 4–9 deletion (Shank3e4–09) DNA produced a 491 bp band. Shank3e4–9 mice were further backcrossed with C57BL6J mice for at least five generations.
Western Blots
Synaptosomes and whole cell lysates were prepared as previously described in Kouser et al. [2013]. Western blotting was performed with antibodies previously described [Kouser et al., 2013].
Behavioral Overview
All mice tested were age- and sex-matched, littermate progeny of matings between heterozygous Shank3e4–9 mutants. Behavioral tests were performed by an experimenter blind to genotype of two separate cohorts; Cohort 1 consisted of 56 mice; n = 20 wildtype (WT, ten female and ten male), n = 16 heterozygous (HET, seven male and nine female), n = 20 homozygous (HOMO, ten female and ten male), that were 3–5 months of age at the start of testing. Cohort 1 consisted of 16 littermate triplets (WT/HET/HOMO) and 4 littermate pairs (WT/HOMO) typically from one litter for each. Cohort 1 underwent the following behaviors in order: elevated plus maze, dark/light, open field, locomotor, grooming, social interaction with caged adult, novel and spatial object recognition, rotarod, social interaction with free moving juvenile, olfactory tests, nesting behavior, marble burying, cued and contextual fear conditioning, Morris water maze, reversal of Morris water maze, visible water maze, pre-pulse inhibition of startle, and startle threshold. Cohort 2 consisted of 58 mice; n = 20 WT (ten female and ten male), n = 18 HET (eight female and ten male), n = 20 HOMO (ten female and ten male) with 18 triplets (WT/HET/HOMO) and 2 pairs (WT/HOMO). Cohort 2 underwent genotype/sex-matched social interaction test only. Analysis of behavioral data was conducted using StatPlus software (Version 2009, AnalystSoft, Alexandria, VA) using either two-way ANOVAs or three-way repeated measures ANOVA with genotype and sex as the main variables and trial, bouts, or time as the repeated measure where applicable. Post hoc planned comparisons were applied for significant effects and interactions. For detailed statistical results see Table 1.
Table 1.
Shank3e4–9 Results of Statistical Analyses
| Motor and Anxiety Tests (Fig. 4) | ||||
|
| ||||
| N = 20 WT, 16 HET, 20 KO | Locomotor Habituation | Beam Breaks Fig. 4A | Sex, Genotype, & Bin | 3-way rmANOVA; Main effect of Sex: F(1,1224)=22.12, P < 2.851E-06; Main effect of Genotype: F(2,1224)=6.09, P < 0.0023; Main effect of Bin: F(1,1223)=78.58, P < 0.000001; Main effect of Sex × Genotype Interaction: F(2,1224)=5.005, P < 0.006; Main effect of Sex × Bin Interaction: F(23,1224) = 0.20, P = 0.99; Main effect of Genotype × Bin Interaction: F(46,1224)=0.48, P = 0.99; Main effect of Sex × Genotype × Bin Interaction: F(46,1224) = 0.37, P = 0.99 |
| Post hoc Scheffe contrast of means (Sex: Female) | WT vs. HET: P = 0.747; WT vs. KO: P = 0.777; HET vs. KO: P = 0.998 | |||
| Post hoc Scheffe contrast of means (Sex: Male) | WT vs. HET: P = 0.999; WT vs. KO: P = 0.981; HET vs. KO: P = 0.987 | |||
| Post hoc Scheffe contrast of means (Sex: Female vs. Male) | WT: P = 0.734; HET: P = 0.327; KO: P = 0.392 | |||
| Post hoc Scheffe contrast of means (genotype) | WT vs. HET: P = 0.137; WT vs. KO: P < 0.00248; HET vs. KO: P = 0.411 | |||
| Ambulatory Movement | Total beam breaks | Sex and Genotype | 2-way ANOVA; Main effect of Sex: F(1,52)=11.93, P < 0.001; Main effect of Genotype: F(2,52)=0.58, P = 0.56; Main effect of Sex × Genotype Interaction: F(2,52) = 50.22, P = 0.79 | |
| Fine Movement | Total beam breaks | Sex and Genotype | 2-way ANOVA; Main effect of Sex: F(1,52)=1.03, P = 0.31; Main effect of Genotype: F(2,52)=0.16, P = 0.84; Main effect of Sex × Genotype Interaction: F(2,52)=1.04, P = 0.35 | |
| N = 20 WT, 16 HET, 20 KO | Accelerated Rotarod | Time to Fall off Fig. 4B | Sex, Genotype, and Trial | 3-way rmANOVA; Main effect of Sex: F(1,52)=40.81, P < 4.59E-10; Main effect of Genotype: F(2,52)=4.81, P < 0.008; Main effect of Trial: F(7,408)=16.46, P < 0.000001; Main effect of Sex × Genotype Interaction: F(2,52)=0.07, P = 0.93; Main effect of Sex × Trial Interaction: F(7,408)=1.21, P = 0.29; Main effect of Genotype × Trial Interaction: F(14,408)=50.25, P = 0.99, Main effect of Sex × Genotype × Trial Interaction: F(14,408)=0.45, P = 0.955 |
| Post hoc Scheffe contrast of means (genotype) | WT vs. HET: P < 0.017; WT vs. KO: P = 0.949; HET vs. KO: P < 0.039 | |||
| Post hoc Scheffe contrast of means (Sex: Female) | WT vs. HET: P = 0.064; WT vs. KO: P = 0.885; HET vs. KO: P = 0.056 | |||
| Post hoc Scheffe contrast of means (Sex: Male) | WT vs. HET: P = 0.111; WT vs. KO: P = 0.521; HET vs. KO: P = 0.399 | |||
| Post hoc Scheffe contrast of means (Sex: Female vs. Male) | WT: P < 0.0002; HET: P < 0.0003; KO: P < 0.011 | |||
| N = 20 WT, 16 HET, 20 KO | Open Field | Time in Center Fig. 4C | Sex and Genotype | 2-way ANOVA; Main effect of Sex: F(1,52)=0.854, P = 0.35; Main effect of Genotype: F(2,52)=0.21, P = 0.21; Main effect of Sex × Genotype Interaction: F(2,52)=0.28, P = 0.75 |
| Frequency in Center | Sex and Genotype | 2-way ANOVA; Main effect of Sex: F(1,52)=1.16, P = 0.28; Main effect of Genotype: F(2,52)=2.33, P = 0.10; Sex × Genotype Interaction: F(2,52)=0.736, P = 0.48 | ||
| Distance Travelled Fig. 4D | Sex and Genotype | 2-way ANOVA; No main effect of Sex: F(1,52)=1.16, P = 0.28; No main effect of Genotype: F(2,52)=2.33, P = 0.10; No main effect of Sex × Genotype Interaction: F(2,52)=0.73, P = 0.48 | ||
| N = 20 WT, 16 HET, 20 KO | Dark/Light | Latency to enter Light side Fig. 4E | Sex and Genotype | 2-way ANOVA; Main effect of Sex: F(1,52)=2.75, P = 0.137; Main effect of Genotype: F(2,52)=0.67, P = 0.51; Sex × Genotype Interaction: F(2,52)=0.61, P = 0.54 |
| Time in Light Side Fig. 4F | Sex and Genotype | 2-way ANOVA; Main effect of Sex: F(1,52)=0.58, P = 0.44; Main effect of Genotype: F(2,52)=0.35, P = 0.70; Sex × Genotype Interaction: F(1,52)=0.47, P = 0.62 | ||
| Time in Dark Side Fig. 4F | Sex and Genotype | 2-way ANOVA; Main effect of Sex: F(1,52)=0.58, P = 0.44; Main effect of Genotype: F(2,52)=0.35, P = 0.70; Sex × Genotype Interaction: F(2,52)=0.47, P = 0.62 | ||
| N = 20 WT, 16 HET, 20 KO | Elevated Plus Maze | Duration in Open Arms Fig. 4G | Sex and Genotype | 2-way ANOVA; Main effect of Sex: F(1,52)=0.32, P = 0.56; Main effect of Genotype: F(2,52)=51.58, P = 0.21; Sex × Genotype Interaction: F(2,52)=0.45, P = 0.63 |
| Duration in Closed Arms Fig. 4G | Sex and Genotype | 2-way ANOVA; Main effect of Sex: F(1,52)=3.24, P = 0.07; Main effect of Genotype: F(2,52)=2.09, P = 0.13; Sex × Genotype Interaction: F(2,52)=0.47, P = 0.63 | ||
| Total Distance travel Fig. 4H | Sex and Genotype | 2-way ANOVA; Main effect of Sex: F(1,52)=7.67, P < 0.007; Main effect of Genotype: F(2,52)=0.92, P = 0.40; Sex × Genotype Interaction: F(2,52)=0.1.21, P = 0.30 | ||
| Post hoc Bonferroni Test of means (Sex: Female vs. Male) | WT: P < 0.007, HET: P = 0.453; KO: P = 0.169 | |||
| Marble burying test | Marbles buried Fig. 4I | Sex and Genotype | 2-way ANOVA; Main effect of Sex: F(1,52)=2.846, P = 0.09; Main effect of Genotype: F(2,52)=1.124, P = 0.33; Sex × Genotype Interaction: F(2,52)=1.60, P = 0.21 | |
| Nest building test | Nest Width Fig. 4J | Sex, Genotype, Time | 3-way rmANOVA; Main effect of Sex: F(1,150)=8.13, P < 0.004; Main effect of Genotype: F(2,150)=1.19, P = 0.30; Main effect of Time: F(1,150)=7.01, P < 0.001; Main effect of Sex × Genotype Interaction: F(2,150)=0.411, P = 0.66; Main effect of Sex × Time: F(2,150)=0.110, P = 0.895; Main effect of Genotype × Time: F(4,150)=0.411, P = 0.663; Main effect of Sex × Genotype × Time: F(4,150)=0.18, P = 0.94 | |
| Post hoc Scheffe contrast of means (Sex: Female) | WT vs. HET: P = 0.814; WT vs. KO: P = 0.72; HET vs. KO: P = 0.597 | |||
| Post hoc Scheffe contrast of means (Sex: Male) | WT vs. HET: P = 0.674; WT vs. KO: P = 0.106; HET vs. KO: P = 0.288 | |||
| Post hoc Scheffe contrast of means (Sex: Female vs. Male) | WT: P < 0.010, HET: P = 0.150, KO: P = 0.339 | |||
| Nest Height Fig. 4K | Sex, Genotype, Time | 3-way rmANOVA; Main effect of Sex: F(1,150)=57.03, P < 0.0088; Main effect of Genotype: F(2,150)=2.87, P = 0.059; Main effect of Time: F(1,150)=6.49, P < 0.0019; Main effect of Sex × Genotype Interaction: F(2,150)=0.659, P = 0.518; Main effect of Sex × Time: F(2,150)=0.015, P = 0.984; Main effect of Genotype × Time: F(4,150)=0.47, P = 0.756; Main effect of Sex × Genotype × Time: F(4,150)=0.09, P = 0.98 | ||
| Post hoc Scheffe contrast of means (Sex: Female) | WT vs. HET: P = 0.596; WT vs. KO: P = 0.207; HET vs. KO: P = 0.557 | |||
| Post hoc Scheffe contrast of means (Sex: Male) | WT vs. HET: P = 0.222; WT vs. KO: P = 0.154; HET vs. KO: P < 0.019 | |||
| Post hoc Scheffe contrast of means (Sex: Female vs. Male) | WT: P = 0.146, HET: P < 0.011, KO: P = 0.380 | |||
|
| ||||
| Vocalization and Grooming (Fig. 5) | ||||
|
| ||||
| P4: N=8 WT, 20 HET, 6 KO; P6: N=21 WT, 10 HET, 14 KO; P8: N=26WT, 32 HET, 16 KO, P10: N=17 WT, 26 HET, 7 KO; P12: N=29 WT, 25 HET, 15 KO. | Ultrasonic vocalization Number of Calls Fig. 5A | Sex, Genotype, and Age | 3-way rmANOVA; Main effect of Sex: F(1,240)=0.519, P = 0.47; Main effect of Genotype: F(2,240)=8.23, P < 0.00035; Main effect of Day: F(4,240)=57.74, P < 0.000001; Main effect of Sex × Genotype Interaction: F(2,240)=2.68, P = 0.070; Main effect of Sex × Day Interaction: F(4,240)=0.156, P = 0.959; Main effect of Genotype × Day Interaction: F(8,240)=2.63, P < 0.0088; Main effect of Sex × Genotype × Day: F(8,240)=0.168, P = 0.994 | |
| Post hoc Scheffe contrast of means (Genotype) | WT vs. HET: P = 0.266, WT vs. KO: P < 0.00034, HET vs. KO: P < 0.0229 | |||
| Planned Comparison: Day 4 | ||||
| WT vs. HET | P < 0.029 | |||
| WT vs. KO | P < 0.003 | |||
| HET vs. KO | P = 0.098 | |||
| Day 6 | ||||
| WT vs. HET | P = 0.571 | |||
| WT vs. KO | P < 0.015 | |||
| HET vs. KO | P < 0.034 | |||
| Day 8 | ||||
| WT vs. HET | P = 0.893 | |||
| WT vs. KO | P = 0.882 | |||
| HET vs. KO | P = 0.810 | |||
| Day 10 | ||||
| WT vs. HET | P = 0.523 | |||
| WT vs. KO | P = 0.794 | |||
| HET vs. KO | P = 0.794 | |||
| Day 12 | ||||
| WT vs. HET | P < 0.043 | |||
| WT vs. KO | P = 0.928 | |||
| HET vs. KO | P = 0.135 | |||
| N = 20 WT, 16 HET, 20 KO | Grooming Time spent grooming Fig. 5B | Sex and Genotype | 2-way ANOVA; No Main effect of Sex: F(1,52)=2.85, P = 0.09; Main effect of Genotype: F(2,52)=6.48, P < 0.0031; No main effect of Sex × Genotype Interaction: F(2,52)=2.20, P = 0.12 | |
| Post hoc Scheffe contrast of means (Genotype) | WT vs. HET: P = 0.231; WT vs. KO: P < 0.011; HET vs. KO: P = 0.523 | |||
| Number of bouts Fig. 5C | Sex and Genotype | 2-way ANOVA; Main effect of Sex: F(1,52)=0.77, P = 0.38; Main effect of Genotype: F(2,52)=0.08, P = 0.91; Main effect of Sex × Genotype Interaction: F(2,52)=1.08, P = 0.34 | ||
| Time per bout (s) Fig. 5D | Sex and Genotype | 2-way ANOVA; Main effect of Sex: F(1,52)=2.85, P = 0.09; Main effect of Genotype: F(2,52)=6.48, P < 0.0031; Main effect of Sex × Genotype Interaction: F(2,52)=2.20, P = 0.12 | ||
| Post hoc Scheffe contrast of means (Genotype) | WT vs. HET: P = 0.080; WT vs. KO: P < 0.0038; HET vs. KO: P = 0.577 | |||
|
| ||||
| Social Interaction (Fig. 6) | ||||
|
| ||||
|
N = 10 WT, 8 HET, 10 KO pairs N = 20 WT, 16 HET, 20 KO |
Genotype and Sex matched social interaction Number of bouts Fig. 6A | Sex and Genotype | 2-way ANOVA; No Main effect of Sex: F(1,54)=0.77, P = 0.38; Main effect of Genotype: F(2,54)=3.93, P < 0.02; Sex × Genotype Interaction: F(1,54)=2.78, P = 0.07 | |
| Post hoc Scheffe contrast of means (Genotype) | WT vs. HET: P = 0.23; WT vs. KO: P < 0.003, HET vs. KO: P = 0.28 | |||
| Interaction time Fig. 6B | Sex and Genotype | 2-way ANOVA; No Main effect of Sex: F(1,54)=1.53, P = 0.22; Main effect of Genotype: F(2,54)=7.95, P < 0.001; Sex × Genotype Interaction: F(1,54)=0.52, P = 0.59 | ||
| Post hoc Scheffe contrast of means (Genotype) | WT vs. HET: P = 0.20; WT vs. KO: P < 0.0009, HET vs. KO: P = 0.15 | |||
| N = 20 WT, 16 HET, 20 KO | Total distance traveled | Sex and Genotype | 2-way ANOVA; Main effect of Sex: F(1,54)=2.57, P = 0.118; Main effect of Genotype: F(2,54)=0.471, P = 0.627; Sex × Genotype Interaction: F(1,54)=2.06, P = 0.143 | |
| Social Interaction with Juvenile Interaction Time Fig. 6C | Sex, Genotype, and Day | 3-way rmANOVA; Main effect of Sex: F(1,52)=4.36, P = 0.059; Main effect of Genotype: F(2,52)=1.47, P = 0.23; Main effect of Day: F(1, 52)=58.04, P < 0.000001; No main effect of Sex × Genotype Interaction: F(2,52)=2.12, P = 0.12; Main effect of Sex × Day Interaction: F(2,52)=0.87, P = 0.53; Main effect of Genotype × Day Interaction: F(2,52)=1.50, P = 0.20; Main effect of Sex × Genotype × Day: F(2,52)=1.03, P = 0.411 | ||
| Among Days: Sex & Genotype Day 1 | 2-way rmANOVA; Main effect of Sex: F(1,52)=18.56, P < 0.00008; Main effect of Genotype: F(2,52)=2.10, P = 0.13; Main effect of Sex and Genotype: F(1, 52)=0.427, P = 0.654; | |||
| Day 4 | 2-way rmANOVA; Main effect of Sex: F(1,52)=51.37, P < 3.27E-09; Main effect of Genotype: F(2,52)=2.10, P = 0.20; Main effect of Sex and Genotype: F(1, 52)=0.581, P = 0.562; | |||
| Trial (Initial Test vs. Recognition Test) within each Genotype Planned Comparison | WT: P < 0.000008, HET: P < 0.0005, KO: P < 0.00003 | |||
| Interaction bouts | Sex, Genotype, and Day | 3-way rmANOVA; Main effect of Sex: F(1,52)=1.78, P = 0.184; Main effect of Genotype: F(2,52)=0.251, P = 0.777; Main effect of Day: F(1,52)=61.49, P < 4.956E-12; No main effect of Sex × Genotype Interaction: F(2,52)=0.123, P = 0.884; Main effect of Sex × Day Interaction: F(1,52)=0.563, P = 0.454; Main effect of Genotype × Day Interaction: F(2,52)=1.94, P = 0.148; Main effect of Sex × Genotype × Day: F(2,52)=2.836, P = 0.063 | ||
| N = 20 WT, 16 HET, 20 KO | Social Interaction with a caged adult Interaction time Fig. 6D | Sex, Genotype and Target; | 3-way rmANOVA; Main effect of Sex: F(1,52)=2.49, P = 0.11; Main effect of Genotype: F(2,52)=0.76, P = 0.46; Main effect of Target: F(1,106)=3.19, P = 0.07; Main effect of Sex × Genotype Interaction: F(2,52)=2.31, P = 0.10; Main effect of Sex × Target: F(1,106)=1.50, P = 0.22; Main effect of Genotype × Target: F(2,106)=0.78, P = 0.45; Main effect of Sex × Genotype × Target: F(1,106)=1.20, P = 0.48 | |
| Latency to interact Fig. 6E | Sex & Genotype Trial 1 (empty) | 2-way rmANOVA; Main effect of Sex: F(1,52)=0.02, P = 0.86; Main effect of Genotype: F(2,52)=3.93, P < 0.026; Main effect of Sex × Genotype Interaction: F(2,52)=1.23, P = 0.298 | ||
| Post hoc Scheffe contrast of means (Genotype) | WT vs. HET: P = 0.15; WT vs. KO: P < 0.03, HET vs. KO: P = 0.81 | |||
| Sex & Genotype Trial 2 (caged mouse) | 2-way rmANOVA; Main effect of Sex: F(1,52)=1.29, P = 0.260; Main effect of Genotype: F(2,52)=1.11, P = 0.33; Main effect of Sex × Genotype Interaction: F(2,52)=0.95, P = 0.39 | |||
| Total distanced traveled | Sex, Genotype, and Target | 3-way rmANOVA; Main effect of Sex: F(1,52)=0.90, P = 0.33; Main effect of Genotype: F(2,52)=0.140, P = 0.869; Main effect of Target: F(2,103)=6.06, P < 1.048E-08; Main effect of Sex × Genotype Interaction: F(2,52)=1.07, P = 0.344; Main effect of Sex × Target Interaction: F(1,106)=0.931, P = 0.337; Main effect of Genotype × Target Interaction: F(2,103)=0.172, P = 0.841; Main effect of Sex × Genotype × Target Interaction: F(1,106)=0.045, P = 0.955 | ||
| Planned Comparison Inanimate | WT vs. HET: P = 0.10; WT vs. KO: P < 0.008 | |||
| Social | WT vs. HET: P = 0.55; WT vs. KO: P = 26 | |||
|
| ||||
| Spatial Learning (Fig. 7) | ||||
|
| ||||
| N = 20 WT, 16 HET, 20 KO | Object and Spatial Test Interaction Time(Baseline) Fig. 7B | Sex, Genotype, Object | 3-way rmANOVA; Main effect of Sex: F(1,52)=3.12, P = 0.07; Main effect of Genotype: F(2,52)=4.23, P < 0.01; Main effect of Object: F(2,52)=0.25, P = 0.77; Main effect of Sex × Genotype Interaction: F(2,52)=0.92, P = 0.39; Main effect of Sex × Object Interaction: F(2, 52)=1.81, P = 0.16; Main effect of Genotype × Object Interaction: F(4,143)=0.03, P = 0.99; Main effect of Sex × Genotype × Target Interaction: F(4,143)=0.34, P = 0.84 | |
| Post hoc Scheffe contrast of means (Genotype) | WT vs. HET: P = 0.871, WT vs. KO: P < 0.022, HET vs. KO: P = 0.146 | |||
| Planned Comparison: | ||||
| WT: Obj A vs. Obj B, Obj B vs. Obj C, Obj A vs. Obj C | P = 0.711, P = 0.748, P = 0.673 | |||
| HET Obj A vs. Obj B, Obj B vs. Obj C, Obj A vs. Obj C | P = 0.930, P = 0.748, P = 0.798 | |||
| KO Obj A vs. Obj B, Obj B vs. Obj C, Obj A vs. Obj C | P = 0.963, P = 0.569, P = 0.607 | |||
| Interaction Time (Spatial test 7C) | Sex, Genotype, Object | 3-way rmANOVA; No Main effect of Sex: F(1,52)=1.49, P = 0.22; No Main effect of Genotype: F(2,52)=1.73, P = 0.18; Main effect of Object: F(2,143)=5.14, P < 0.007; Main effect of Sex × Genotype Interaction: F(2,52)=1.45, P = 0.23; Main effect of Genotype × Object Interaction: F(4,143)=1.01, P = 0.40; Main effect of Sex × Object Interaction: F(2, 143)=0.79, P = 0.45; Main effect of Sex × Genotype × Target Interaction: F(4,143)=0.18, P = 0.83 | ||
| Planned Comparison: | ||||
| WT: | ||||
| Obj B vs. Obj C | P = 0.39 | |||
| Obj B vs. Obj A | P < 0.018 | |||
| Obj C vs. Obj A | P < 0.004 | |||
| HET: | ||||
| Obj B vs. Obj C | P = 0.25 | |||
| Obj B vs. Obj A | P = 0.94 | |||
| Obj C vs. Obj A | P = 0.11 | |||
| KO: | ||||
| Obj B vs. Obj C | P = 0.96 | |||
| Obj B vs. Obj A | P = 0.32 | |||
| Obj C vs. Obj A | P = 0.42 | |||
| Interaction Time (Familiar vs. Novel)Fig. 7D | Sex, Genotype and Object | 3-way rmANOVA; Main effect of Sex: F(1,52)=4.78, P = 0.0505; Main effect of Genotype: F(2,52)=11.49, P < 0.00003; Main effect of Object: F(2,143)=5.09, P < 0.007; Main effect of Sex × Genotype Interaction: F(2,52)=0.27, P = 0.76; Main effect of Sex × Object Interaction: F(2,143)=0.704, P = 0.49; Main effect of Genotype × Object Interaction: F(4,143)=2.13, P = 0.08; Main effect of Sex × Genotype × Object Interaction: F(4,143)=0.69, P = 0.59 | ||
| Planned Comparison: | ||||
| WT: | ||||
| Obj B vs. Obj C | P < 0.0034 | |||
| Obj B vs. Obj B (Novel) | P = 0.773 | |||
| Obj C vs. Obj B (Novel) | P < 0.0085 | |||
| HET: | ||||
| Obj B vs. Obj C | P = 0.658 | |||
| Obj B vs. Obj B (Novel) | P = 0.250 | |||
| Obj C vs. Obj B (Novel) | P-0.140 | |||
| KO: | ||||
| Obj B vs. ObjC | P = 0.161 | |||
| Obj B vs. Obj B (Novel) | P < 0.048 | |||
| Obj C vs. Obj B (Novel) | P = 0.859 | |||
| Post hoc Scheffe contrast of means (Genotype) | WT vs. HET: P < 0.036, WT vs. KO: P < 0.00003, HET vs. KO: P = 0.25 | |||
| N = 20 WT, 16 HET, 20 KO | Morris Water Maze Latency to Reach Platform Fig. 7E (training) | Sex, Genotype & Day | 3-way rmANOVA; Main effect of Sex: F(1,52)=2.42, P = 0.12; Main effect of Genotype: F(2, 52)=0.25, P = 0.77; Main effect of Day: F(10,290)=25.33, P < 0.000001; Main effect of Sex × Genotype Interaction: F(2,52)=5.83, P < 0.0032; Main effect of Sex × Day Interaction: F(10,290)=2.85, P < 0.01; Main effect of Genotype × Day Interaction: F(10,290)=0.82, P = 0.62; Main effect of Sex × Genotype × Day Interaction: F(10,290)=0.23, P = 0.99 | |
| Latency to Reach Platform Fig. 7E (Reversal) | Sex, Genotype & Day | 3-way rmANOVA; Main effect of Sex: F(1,52)=0.435, P = 0.51;Main effect of Genotype: F(2, 52)=0.825, P = 0.439; Main effect of Day: F(10,290)=1.41, P = 0.238; Main effect of Sex × Genotype Interaction: F(2,52)=2.33, P = 0.099; Main effect of Sex × Day Interaction: F(10,290)=0.189, P = 0.903; Main effect of Genotype × Day Interaction: F(10,290)=0.161, P = 0.985; Main effect of Sex × Genotype × Day Interaction: F(10,290)=0.186, P = 0.980 | ||
| Distanced Traveled Fig. 7F (Training) | Sex, Genotype & Day | 3-way ANOVA; Main effect of Sex: F(1,290)=2.33, P = 0.127; Main effect of Genotype: F(2, 290)=0.233, P = 0.792; Main effect of Day: F(6,290)=28.42, P < 1.0 E-10; Main effect of Sex × Genotype Interaction: F(2,52)=3.10, P < 0.04; Main effect of Sex × Day Interaction: F(6,290)=3.86, P < 0.0009; Main effect of Genotype × Day Interaction: F(12, 290)=0.512, P = 0.906; Main effect of Sex × Genotype × Day Interaction: F(12, 290)=0.577, P =0.859 | ||
| Distanced Traveled Fig. 7F (Reversal) | Sex, Genotype & Day | 3-way rmANOVA; No Main effect of Sex: F(1,168)=1.43, P = 0.233 Main effect of Genotype: F(2,168)=0.325, P = 0.722; Main effect of Day: F(3,168)=0.70, P = 0.552; Main effect of Sex × Genotype Interaction: F(2,168)=2.01, P = 0.137; Main effect of Sex × Day Interaction: F(6,168)=2.71, P < 0.046; Genotype × Day Interaction: F(6,168)=0.209, P = 0.973; Sex × Genotype × Day Interaction: F(6, 168)=1.45, P = 0.196 | ||
| %Thigmotaxis Fig. 7G (Training) | Sex, Genotype & Day | 3-way rmANOVA; Main effect of Sex: F(1,52)=4.59, P < 0.032; Main effect of Genotype: F(1,52)=7.30, P < 0.00081; Main effect of Day: F(10,290)=68.76, P < 0.000001; Main effect of Sex × Genotype Interaction: F(2,290)=1.33, P = 0.26; Main effect of Sex × Day Interaction: F(10,290)=4.20, P < 0.00045; Main effect of Genotype × Day Interaction: F(10,290)=1.71, P = 0.06; No main effect of Sex × Genotype × Day Interaction: F(10,290)=0.58, P = 0.85 | ||
| Post hoc Scheffe contrast of means (Sex: Female) | WT vs. HET: P = 0.902; WT vs. KO: P = 0.114; HET vs. KO: P = 0.357 | |||
| Post hoc Scheffe contrast of means (Sex: Male) | WT vs. HET: P = 0.996; WT vs. KO: P = 0.375; HET vs. KO: P = 0.328 | |||
| Post hoc Scheffe contrast of means (Sex: Female vs. Male) | WT: P = 0.620; HET: P = 0.33; KO: P = 0.16 | |||
| Post hoc Scheffe contrast of means (Genotype) | WT vs. HET: P = 0.986, WT vs. KO: P < 0.00292, HET vs. KO: P < 0.0089 | |||
| %Thigmotaxis Fig. 7G (Reversal) | Sex, Genotype & Day | 3-way ANOVA; Main effect of Sex: F(1,52)=1.559, P = 0.213;Main effect of Genotype: F(2, 52)=0.164, P = 0.848; Main effect of Day: F(10,290)=17.50, P < 6.192E-10; Main effect of Sex × Genotype Interaction: F(2,52)=0.484, P = 0.616; Main effect of Sex × Day Interaction: F(10,290)=5.481, P < 0.0012; Main effect of Genotype × Day Interaction: F(10,290)=0.576, P = 0.74; Main effect of Sex × Genotype × Day Interaction: F(10,290)=1.481, P = 0.187 | ||
| Probe trial % Time in Quadrant Fig. 7H | Sex, Genotype & Quadrant | 3-way rmANOVA; Main effect of Sex: F(1,168)=4.25E-07, P = 0.99; Main effect of Genotype: F(2,168)=0.00001, P = 0.99; Main effect of Quadrant: F(3,168)=94.07, P < 0.00000001; Sex × Genotype Interaction: F(2,168)=0.00007, P = 0.99; Main effect of Sex × Quadrant Interaction: F(3,168)=0.192, P = 0.901; Main effect of Genotype × Quadrant Interaction: F(3,168)=2.43, P < 0.028; Main effect of Sex × Genotype × Quadrant Interaction: F(3,168)=0.44, P = 0.848 | ||
| Planned Comparisons (Quadrant) | WT: Target vs. Opposite P < 1.06E-09, Target vs. Right P < 3.05E-10, Target vs. Left P < 4.30E-05; HET: Target vs. Opposite P < 4.17E-06, Target vs. Right P < 4.13E-08, Target vs. Left P = 0.179; KO: Target vs. Opposite P < 2.633E-07, Target vs. Right P < 1.98E-07, Target vs. Left P = 0.055 | |||
| Reversal Probe trail % Time in Quadrant Fig. 7I | Sex, Genotype & Quadrant | 3-way rmANOVA; Main effect of Sex: F(1,168)=7.04E-07, P = 0.99; Main effect of Genotype: F(2,160)=3.84E-08, P = 0.99; Main effect of Quadrant: F(3,168)=42.51, P < 0.0000001; Sex × Genotype Interaction: F(2,168)=0.00001, P = 0.99; Sex × Quadrant Interaction: F(3,168)=0.579, P = 0.629; Main effect of Genotype × Quadrant Interaction: F(6,168)=2.53, P < 0.022; Sex × Genotype × Quadrant Interaction: F(6,168)=3.38, P = 0.0035 | ||
| Planned Comparisons (Quadrant) | WT: Target vs. Opposite P < 1.06E-09, Target vs. Right P < 3.05E-10, Target vs. Left P < 4.30E-05; HET: Target vs. Opposite P < 4.17E-06, Target vs. Right P < 4.13E-08, Target vs. Left P = 0.179; KO: Target vs. Opposite P < 2.633E-07, Target vs. Right P < 1.98E-07, Target vs. Left P = 0.055 | |||
| Visible Platform Latency to Platform (s) Fig. 7J | Sex, Genotype | 2-way rmANOVA; Main effect of Sex: F(1,168)=0.251, P = 0.618; Main effect of Genotype: F(2,160)=2.07, P = 0.13; Sex × Genotype Interaction: F(2,168)=1.312, P = 0.279 | ||
|
| ||||
| Acoustic Startle Threshold, Prepulse Inhibition, Fear Conditioning Fig. | ||||
|
| ||||
| N = 20 WT, 16 HET, 20 KO | Acoustic Startle Threshold Startle Response Fig. A | Sex, Genotype, Decibel | 3-way rmANOVA; Main effect of Sex: F(1,300)=0.05, P = 0.818; Main effect of Genotype: F(2,300)=0.351, P = 0.704; Main effect of Decibel: F(2,300)=32.48, P < 0.0000001; Main effect of Sex × Genotype Interaction: F(2,300)=1.00, P = 0.366; Main effect of Sex × Decibel Interaction: F(2, 300)=0.0181, P = 0.99; Main effect of Genotype × Decibel Interaction: F(10,300)=0.07, P = 0.99; Main effect of Sex × Genotype × Target Interaction: F(10,300)=0.256, P = 0.98 | |
| N = 20 WT, 16 HET, 20 KO | Prepulse Inhibition Initial Startle Response 1st 6 Fig. | Sex & Genotype | 2-way rmANOVA; Main effect of Sex: F(1,50)=0.06, P = 0.803; Main effect of Genotype: F(2,50)=1.14, P = 0.32; Sex × Genotype Interaction: F(2,50)=0.658, P = 0.521 | |
| Initial Startle Middle 12 Responses Fig. | Sex & Genotype | 2-way rmANOVA; Main effect of Sex: F(1,162)=0.07, P = 0.787; Main effect of Genotype: F(2,162)=3.56, P < 0.030; Sex × Genotype Interaction: F(2,162)=0.729, P = 0.483 | ||
| Post hoc Scheffe contrast of means (Genotype) | WT vs. HET: P = 0.201, WT vs. KO: P < 0.030, HET vs. KO: P = 0.811 | |||
| Initial Startle Middle 12 Responses Fig. | Sex & Genotype | 2-way rmANOVA; Main effect of Sex: F(1,50)=0.0067, P = 0.934; Main effect of Genotype: F(2,50)=1.02, P = 0.365; Sex × Genotype Interaction: F(2,50)=0.310, P = 0.734 | ||
| % Inhibition Fig. | Sex, Genotype & Day | 3-way rmANOVA; Main effect of Sex: F(1,52)=2.42, P = 0.12; Main effect of Genotype: F(2, 52)=0.25, P = 0.77; Main effect of Day: F(10,290)=25.33, P < 0.000001; Main effect of Sex × Genotype Interaction: F(2,52)=5.83, P < 0.0032; Main effect of Sex × Day Interaction: F(10,290)=2.85, P < 0.01; Main effect of Genotype × Day Interaction: F(10,290)=0.82, P = 0.62; Main effect of Sex × Genotype × Day Interaction: F(10,290)=50.23, P = 0.99 | ||
| N = 20 WT, 16 HET, 20 KO | Fear Conditioning Training: Pre-shock% Freezing | Sex & Genotype | Genotype Interaction: F(2,50)=0.479, P = 0.622 | |
| Post hoc Scheffe contrast of means (Genotype) | WT vs. HET: P < 0.014, WT vs. KO: P = 0.577, HET vs. KO: P = 0.131 | |||
| Training: Immediate post-shock% Freezing | Sex & Genotype | 2-way rmANOVA; Main effect of Sex: F(1,50)=17.87, P < 0.0001; Main effect of Genotype: F(2,50)=0.834, P = 0.440; Sex × Genotype Interaction: F(2,50)=0.685, P = 0.508 | ||
| of means (Sex: Female) | WT vs. HET: P = 0.557; WT vs. KO: P = 0.252; HET vs. KO: P = 0.852 | |||
| Post hoc Scheffe contrast of means (Sex: Male) | WT vs. HET: P = 0.951; WT vs. KO: P = 0.999; HET vs. KO: P = 0.938 | |||
| Post hoc Scheffe contrast of means (Sex: Female vs. Male) | WT: P = 0.160; HET: P < 0.009; KO: P < 0.002 | |||
| Test: Pre-cue % Freezing | Sex & Genotype | 2-way rmANOVA; Main effect of Sex: F(1,50)=3.91, P = 0.0533; Main effect of Genotype: F(2,50)=1.40, P = 0.255; Sex × Genotype Interaction: F(2,50)=1.181, P = 0.315 | ||
| Test: Cue % Freezing | Sex & Genotype | 2-way rmANOVA; Main effect of Sex: F(1,50)=1.262, P = 0.266; Main effect of Genotype: F(2,50)=2.246, P = 0.116; Sex × Genotype Interaction: F(2,50)=0.396, P = 0.674 | ||
Behavioral Tests
The elevated plus maze test was conducted as described previously [Etherton, Blaiss, Powell, & Sudhof, 2009]. Locomotor activity was measured as described previously [Etherton et al., 2009; Powell et al., 2004; Tabuchi et al., 2007]. The dark/light test was conducted as described previously [Blundell et al., 2009; Powell et al., 2004]. The rotarod test was conducted as described previously [Powell et al., 2004]. The open field test was conducted as described previously [Etherton et al., 2009].
Recordings of pup ultrasonic vocalizations were conducted as described previously [Bader et al., 2011]. On postnatal days 4–12 individual pups were separated from the dam and placed in a weigh boat inside a sound-attenuated box. Vocalizations were recorded from a microphone ~9 inches above the mouse for 5 min and analyzed using Avisoft SASLab Pro (Glienicke, Germany).
Novel and spatial object recognition tests were performed essentially as described [Lee, Hunsaker, & Kesner, 2005; Save, Poucet, Foreman, & Buhot, 1992]. Specifically, mice were habituated for 4 days to a square, open arena (44 × 44 × 44 cm, ~ 7 lux) with spatial cues affixed to the arena walls. Each mouse was subjected to 1 trial lasting 5 min per day during 4 days of habituation. On the 5th day, “testing day,” all mice received 7, 5-min trials each with 6 or 45 min between each trial (see Fig. 8A). Three, 50 mL conical tubes were filled with water and arranged accordingly for the first 5 trails. Each object was ~12.5 cm from the closest wall. Prior to trial 6 (spatial test), object A was moved to the opposite corner. Prior to trial 7 (object test) object B was changed to a stationary ping-pong ball. Mice were recorded using CleverSys ObjectScan, (Reston VA).
Figure 8.
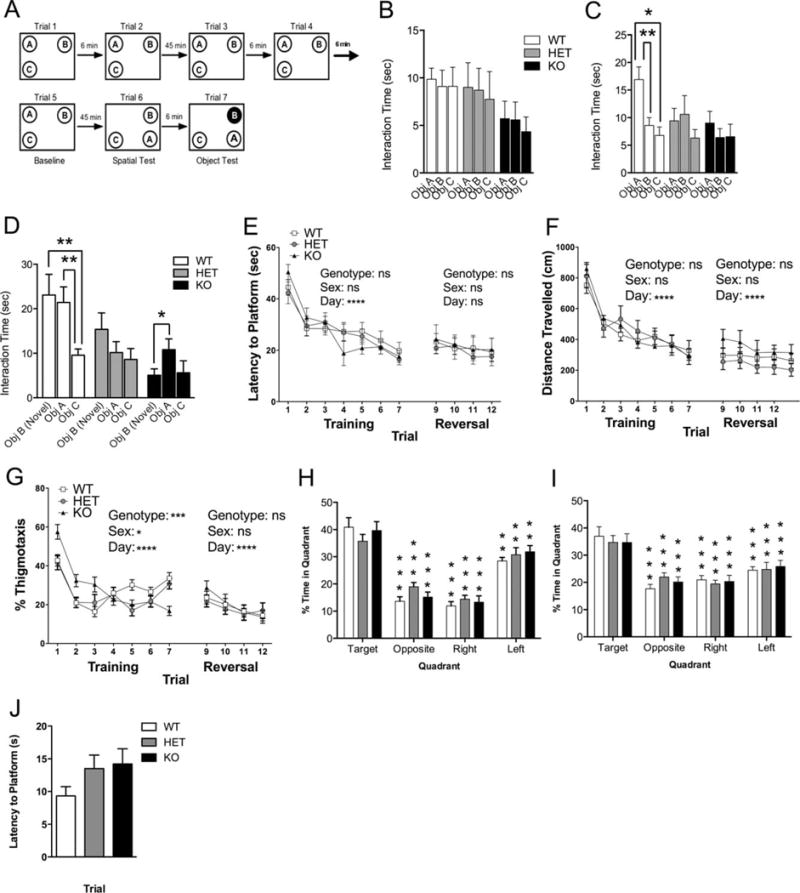
Shank3e4–9 mutant mice exhibit significantly impaired spatial and novel object recognition. (A) Schematic representation of the spatial learning and object recognition test. For four consecutive days mice were habituated to the arena (44 × 44 × 44 cm) for 5 min (not shown in schematic). Following habituation all mice received 7 trials each with inter-trial interval as depicted in schematic and described in Methods. (B) The mean time spent interacting with objects A, B, and C (baseline; trial 5). (C) Mean interaction time during trial 6 (spatial test) with objects A, B, and C after A has been moved to a novel location. (D) The mean interaction time with novel object B and familiar objects A and C (trial 7, novel object recognition test). Following 7 days of Morris water maze training we analyzed (E) latency to platform, (F) total distance traveled to reach platform, and (G) % thigmotaxis (*P < 0.05; **P < 0.01, ***P < 0.001, n = 20 WT, 16 HET, 20 KO). Probe trials conducted one day after training on day 8 (H) and one day following reversal training, day 13 (I) showed no difference in spatial preference among groups. (J) Latency to reach the platform in the visible platform version of the water maze conducted at the end of all testing.
Social interaction tests
Social interaction with a novel juvenile target mouse was performed essentially as described [Blundell et al., 2009; Kwon et al., 2006; Tabuchi et al., 2007]. Social interaction with a caged adult was performed as described [Blundell, Blaiss et al., 2010]. Social interaction with genotype- and sex-matched pairs was performed by pairing mice with a sex- and genotype-matched partner within the same cohort. Matched pairs were derived from separate cages and never previously housed together. Mouse pairs were placed into an open field (44 × 44 × 44 cm) at separate ends and allowed to interact for 5 min under dim lighting (~7 lux).
Mice were habituated to a novel cage for 10 min, followed by a 10 min test period in which total time spent grooming was measured. Time spent grooming the face, head, body, or tail was considered grooming.
The Morris water maze task was conducted as previously described [Powell et al., 2004].
Both prepulse inhibition and startle response were conducted as previously described [Blundell, Kaeser, Sudhof, & Powell, 2010]. Marble burying task was conducted as described previously [Blundell, Kaeser, Sudhof, & Powell, 2010]. Nesting behavior was conducted as previously described [Etherton et al., 2009]. Cued and contextual fear conditioning was performed essentially as described previously [Powell et al., 2004].
Electrophysiology
All recordings were performed at 33 ± 0.5°C, and all data were collected using Clampex (pClamp software suite version 10.2; Molecular Devices, Sunnyvale, CA). Experiments were conducted as previously described in detail [Kouser et al., 2013]. For striatal recordings, the stimulating electrode was placed just inside the striatum below the corpus callosum (Fig. 10A) ~150–200 μm from the recorded MSN. The distance between the recording electrode and the stimulating electrode was kept constant within these bounds. Whole-cell patch clamp recordings in hippocampus and striatum were carried out in the presence of 100 μM picrotoxin to block fast inhibitory transmission, and began 5–10 min (NMDA/AMPA ratio) or 10–15 min (miniature excitatory postsynaptic currents [mEPSCs]) following successful break-in.
Figure 10.
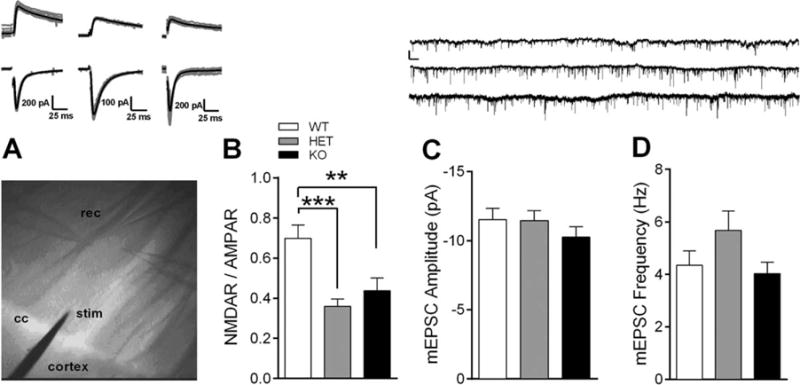
Striatal excitatory transmission is impaired following Shank3 exon 4–9 deletion. (A) Image capture of electrode placement in dorsal striatum using IR-DIC microscopy at 10× resolution. Stimulating electrode (stim) was placed just inside of the corpus collosum (cc) and patch clamp electrodes (rec) were used to record whole-cell EPSCs from MSNs 150–200 μm away. Inset: 10 consecutive traces (gray) and average trace (black) from WT (left), HET (middle), and KO mice (right) at −70 mV (bottom) and +40 mV (top). Scale bar: 200 pA (WT), 400 pA (HET), 170 pA (KO), 25 ms. (B) NMDA/AMPA ratio is decreased in Shank3e4–9 HET and KO mice (n = 18 WT, 17 HET, and 18 KO cells). (C) Mean mEPSC amplitude (Inset: 1-min raw traces from WT (top), HET (middle), KO (bottom) mice. Scale bar: 15 pA; 1.5 s) and (D) mean frequency of events are unaffected in HET and KO mice (n = 25 WT, 29 HET, and 20 KO cells). **P < 0.01, ***P <0.001.
Acute coronal slices containing the hippocampus or thalamocortical slices containing dorsal striatum (350–400 μm thick) were made using a vibrating microtome (Vibratome, Bannockburn, IL) as previously described [Kouser et al., 2013]. Extracellular field recordings were performed on male mice 3–4 weeks of age. Whole cell recordings were performed in the hippocampus of male mice at 14–17 days of age and at 3–4 weeks of age in striatum.
Octahydro-12-(hydroxymethyl)-2-imino-5,9:7,10a-dimethano-10aH[1,3]dioxocino[6,5-d]pyrimidine-4,7,10,11,12-pentolTetrodotoxin (TTX, Tetrodotoxin), picrotoxin, N-(2,6-Dimethylphenylcarbamoylmethyl)triethylammonium chloride (QX314), and (RS)-3,5-Dihydroxyphenylglycine (DHPG) were obtained from Tocris Bioscience (Minneapolis, MN). CsMethanesulfonate and CsCl were obtained from Sigma-Aldrich (St. Louis, MO). All other reagents were obtained from Fisher Scientific (Waltham, MA).
Results
Genetic Targeting of Exons 4–9 of the Shank3 Gene
Shank3e4–9 mice were generated as described above (Fig. 1A). Southern blot confirmed correct incorporation of targeting construct (Fig. 1B).
Figure 1.
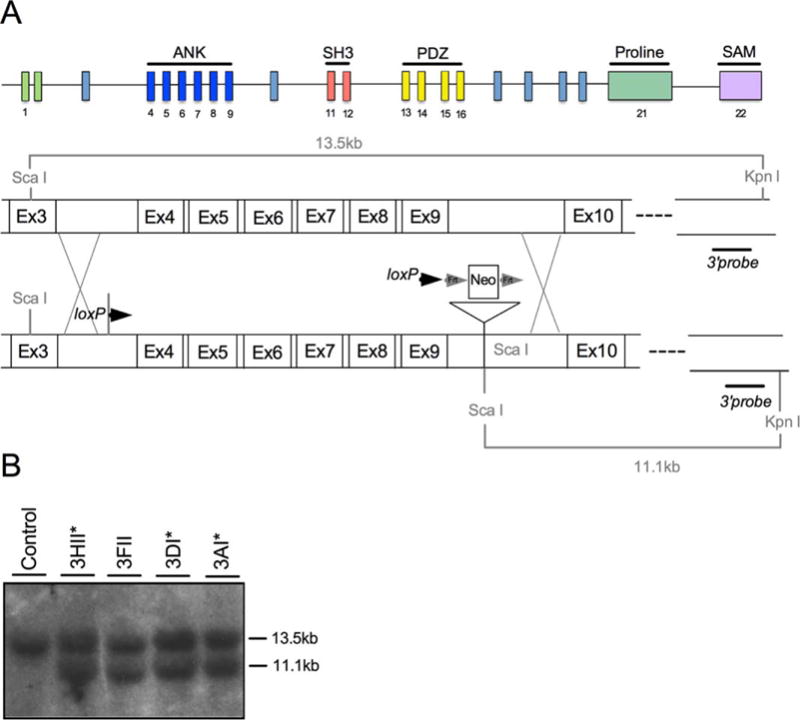
Genetic deletion of Shank3 exons 4–9. (A) Schematic of the Shank3 gene displaying exons and their respective domains (ANK—Ankyrin repeat domain; SH3—Src Homology 3 domain; PDZ—PSD95/Dlg1/Zo1 domain; SAM—Sterile alpha motif) (top). Schematic of the targeted portion exons 4–9 of Shank3 (middle) and the insertion of the targeting construct following recombination (bottom). (B) Southern blot of ScaI and KpnI-digested DNA from control (lane 1) and neo-resistant ES cells (lanes 2–5) reveals 13.5 kb and 11.1 kb ScaI and KpnI fragments reflecting proper targeting in clones that were selected for blastocyst injections (asterisks).
Altered Shank3 Expression in Striatal Lysates from Shank3e4–9 Mutants
To characterize Shank3 isoform expression, Shank3 antibodies against the C- or N-terminals (supplied by Paul Worley) were blotted on whole striatal lysates from 3–6 month old mice. The focus on dorsal striatum was due to the predominant expression of Shank3 vs. Shank1 and Shank2 in this region [Peca et al., 2011]. Expanding on previously published, incomplete biochemical characterizations of Shank3 in similar models [Bozdagi et al., 2010; Peca et al., 2011; Wang et al., 2011], Western blots revealed loss of the two highest molecular weight bands of Shank3 detected by the C-terminal (C-1 and C-2, P < 0.0001 for each) and N-terminal (N-1 and N-2, P < 0.0001 for each) Shank3 antibodies in homozygotes (Fig. 2A) and a decrease in these bands in the Shank3e4–9 HET (Fig. 2A C-1: P = 0.0351; C-2: P = 0.0111; N-1: P < 0.0001; N-2: P < 0.0001). We also saw a decrease in both HET and KO mice of C-3 (HET: P = 0.0477; KO: P = 0.0020) and C-7 (HET: P = 0.0126; KO: P = 0.0001) bands as well as the N-3 band (HET: P = 0.0170; KO: P = 0.0003) (Fig. 2A). Thus, deletion of Shank3 exons 4–9 results in a more complex alteration of Shank3 isoforms than previously reported [Bozdagi et al., 2010; Peca et al., 2011; Wang et al., 2011].
Figure 2.
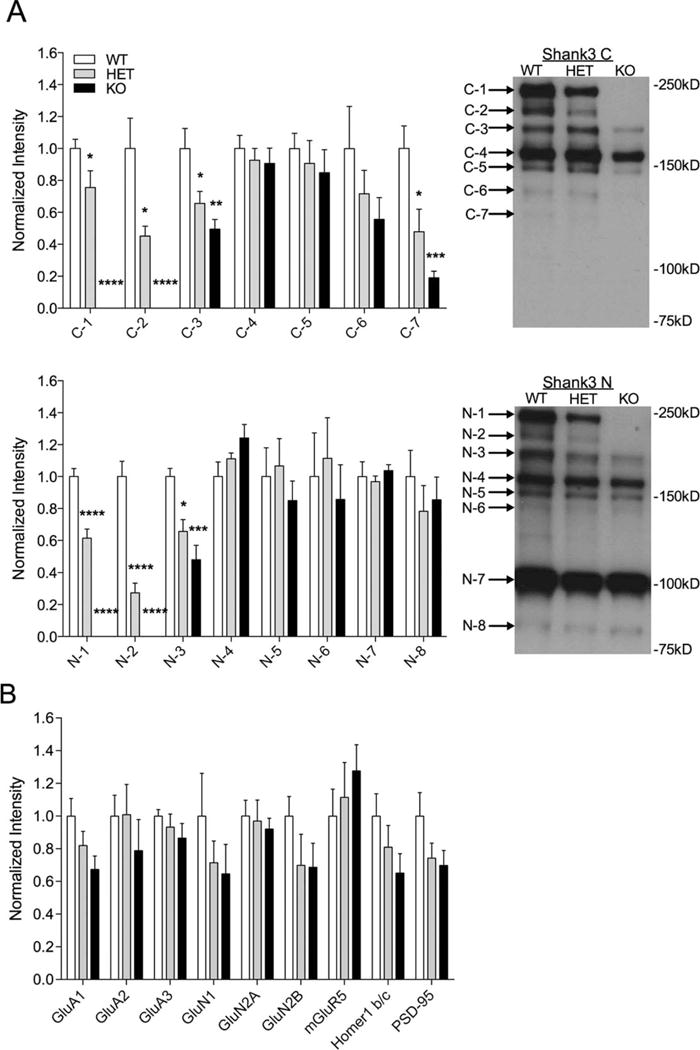
Altered Shank3 isoform expression from whole striatum lysates in Shank3e4–9 mutant mice. (A) Quantification and representative western blots of striatum whole tissue lysates with C-terminal Shank3 antibody (top) and N-terminal Shank3 antibody (bottom) showing decrease (HET) or complete loss (KO) of the C-1, C-2, N-1, and N-2 bands of Shank3 using Shank3 C and N antibodies in Shank3e4–9 mutants compared to WT. Additionally, there was a significant decrease in C-3, C-7, and N-3 bands in both HET and KO mice. (B) Quantification of other synaptic proteins from striatal lysates shows no significant differences. For each analysis, data were normalized to β-actin levels and then to the average of WT (*P < 0.05, **P < 0.01, ***P < 0.001, n = 8 WT, 7 HET, 8 KO).
Normal Synaptic Protein Expression in Striatal Lysates of Shank3e4–9 Mutants
We probed whole striatal lysates for synaptic proteins known to bind directly or indirectly to Shank3. Other than Shank3, we did not observe significant changes in protein expression in the whole tissue lysates of the striatum (Fig. 2B).
Altered Shank3 and Synaptic Protein Localization in Shank3e4–9 Striatal Synaptosomes
Although levels of Shank3 binding partners were not altered in whole lysates, loss of Shank3 might alter synaptic localization of its binding partners. To determine the effect of deletion of Shank3 exons 4–9 on subcellular localization, we isolated striatal synaptosome fractions and compared synaptic protein levels in HET and KO mice to WT littermates. Similar to whole striatal lysates, Shank3e4–9 KO mice showed loss of the largest two bands of Shank3 using both C-terminal (C-1 HET: P = 0.0076; C-2 HET: P < 0.0001; C-1 and C-2 KO: P < 0.0001) and N-terminal (HET: N-1: P < 0.0001; N-2 P = 004; KO: N-1, P < 0.0001 and N-2, P = 0.0014) antibodies (Fig. 3A). We also saw significant decreases in C-3 (P = 0.0013), C-5 (P = 0.0002), C-6 (P < 0.0001), and C-7 (P < 0.0001) bands of Shank3 in the Shank3e4–9 KO mice in striatal synaptosomes (Fig. 3A), as well as N-3 (P < 0.001), N-4 (P = 0.0225), and N-5 (P = 0.0044) bands using the N-terminal antibody. Thus, synaptic alterations in Shank3 isoforms largely parallel whole lysates with additional abnormal synaptic localization of some isoforms.
Figure 3.
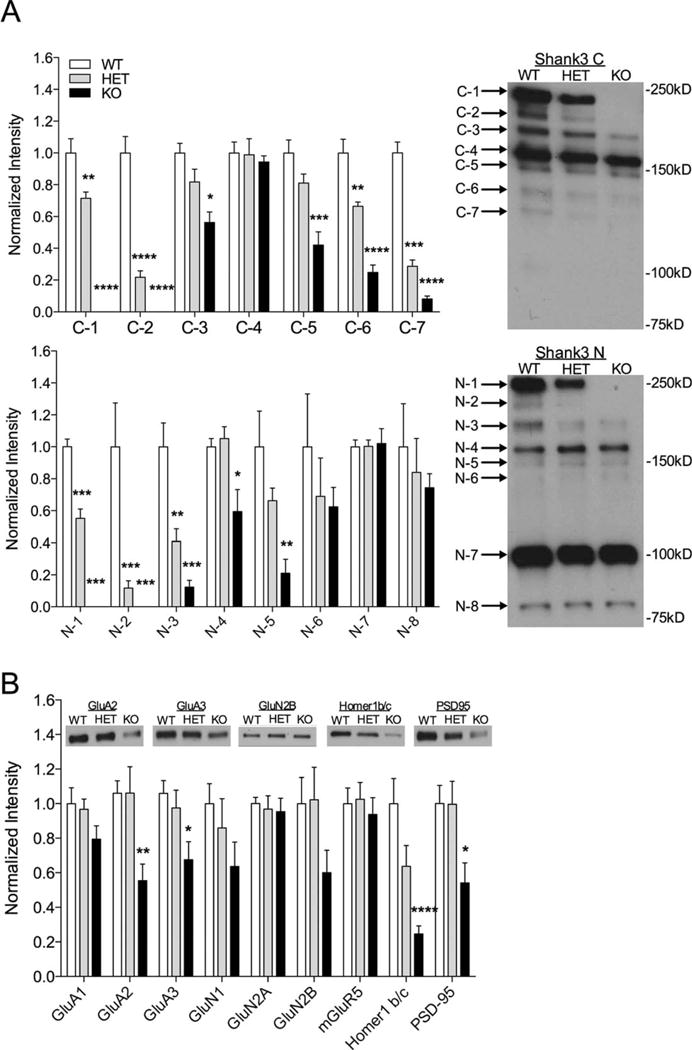
Striatal synaptosome analysis in Shank3e4–9 mutant mice. (A) Quantification and representative Western blots of striatal synaptosomes with C-terminal Shank3 antibody (top) and N-terminal Shank3 antibody (bottom). There is complete loss of the C-1, C-2, N-1, and N-2 bands of Shank3 using Shank3 C and N antibodies in Shank3e4–9 KO mice and significant decrease in same bands in HET mice compared to WT. Significant decreases are also observed in HET and KO mice for C-5, C-6, C-7, and N-3 and in KO mice only for C-3, C-5, N-4, and N-5 (*P < 0.05, **P < 0.01, ***P < 0.001 as indicated, n = 6) (B) Quantification of other synaptic proteins from striatal synaptosomes shows significant decreases in GluA2, GluA3, Homer1 b/c, and PSD95 in Shank3e4–9 KO mice. For each analysis, data were normalized to β-actin levels and then to the average of WT. Representative blots are shown inset for proteins showing significant differences. (*P < 0.05, **P < 0.01, ***P < 0.001, n = 11 WT, 11 HET, 12 KO).
In addition to Shank3, we found significant decreases in synaptosome levels of GluA2 (P = 0.0077), GluA3 (P = 0.0191), Homer1b/c (P < 0.0001), and PSD-95 (P = 0.0263) (Fig. 3B) in Shank3e4–9 KOs. We found no differences in other glutamate receptor subunits in Shank3e4–9 KO mice, nor did we see significant differences in synaptic proteins in the Shank3e4–9 HET mice, though a strong trend toward decreased Homer1b/c was evident (Fig. 3B). We interpret these data as an alteration in synaptic localization of these proteins. Of course, altered synaptosome localization of receptor subunits could mean decreased localization within the synaptic membrane surface or decreased localization to internal membrane stores. Functional consequences of such changes or lack thereof are better concluded from synaptic electrophysiology experiments.
Shank3e4–9 Mutants Display Several Normal Behaviors
Our initial behavioral tests determined whether Shank3e4–9 mutants displayed motor or motor learning deficits. Shank3e4–9 mutant mice exhibited normal locomotor activity and locomotor habituation (Fig. 4A). We further analyzed both ambulatory movement (breaking two different beams consecutively) and fine movement (breaking of the same beam two or more times consecutively) and also found no differences (not shown). Both motor coordination and learning were unchanged on the accelerating rotarod (Fig. 4B).
Figure 4.
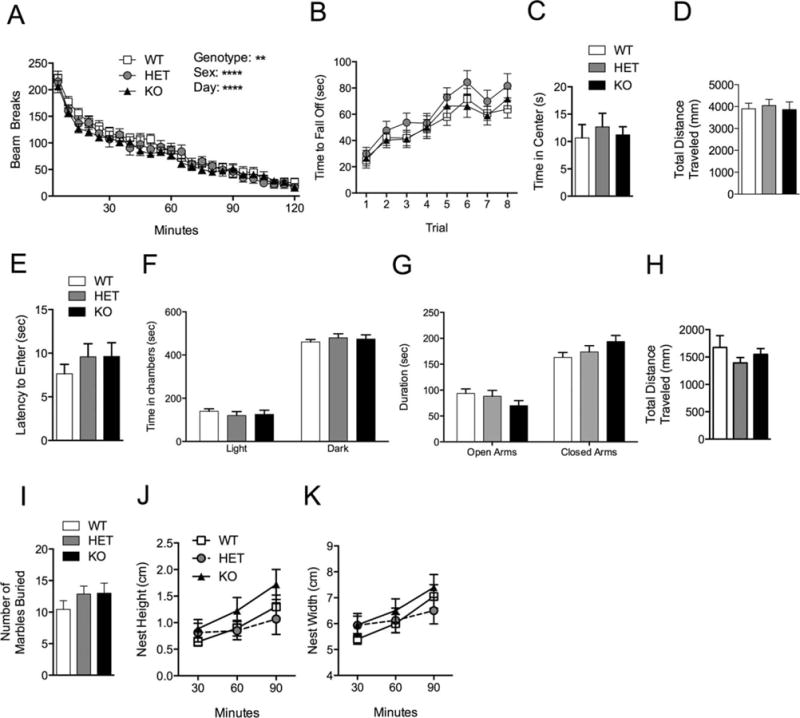
Motor and anxiety tests in Shank3e4–9 mutant mice. (A) All genotypes showed similar locomotor habituation over 2 hr in the locomotor box. (B) In the rotarod test all genotypes showed similar motor learning and coordination over 2 days and 8 trials. In the open field test all genotypes spent a similar amount of time in the center (C) and traveled similar distances (D). (E) In the dark/light test all three genotypes showed similar latencies to enter the light chamber from the dark chamber; additionally all genotypes spent equivalent times in either the dark or light chamber (F). In the elevated plus maze all three genotypes spent a similar percentage of time in open arms and closed arms respectively (G) and traveled similar distances in this task (H). All genotypes performed similarly in the marble burying test (I) and nest building test (J–K); (n = 20 WT, 16 HET, 20 KO).
Using tests of anxiety-like behavior including open field, dark/light, and elevated plus maze tasks, Shank3e4–9 mutants displayed no behavioral differences. In the open field all three genotypes displayed similar time in the center (Fig. 4C genotype, P = 0.21) and traveled similar distances (Fig. 4D genotype, P = 0.10). In the dark/light test, Shank3e4–9 mutants and controls displayed equivalent latencies to enter the light chamber (Fig. 4E genotype, P = 0.51) and spent equivalent time in either chamber (Fig. 4F genotype, P = 0.70). In the elevated plus maze Shank3e4–9 KO mice displayed similar time in the closed arms (genotype, P = 0.13) and open arms (genotype, P = 0.21) compared to WT (Fig. 4G) and traveled similar distances (Fig. 4H genotype, P = 0.40). All three genotypes performed similarly on the marble burying (Fig. 4I) and nest building tests (Fig. 4J–K).
We tested the acoustic startle reflex and prepulse inhibition in Shank3e4–9 mutant mice, and found that they performed comparably to WT control mice (Fig. 5A,B). Shank3e4–9 mutant mice also performed similar to WT mice in both cued and contextual fear conditioning (Fig. 5C,D). These data suggest an absence of generalized behavioral dysfunction in Shank3e4–9 mutants.
Figure 5.
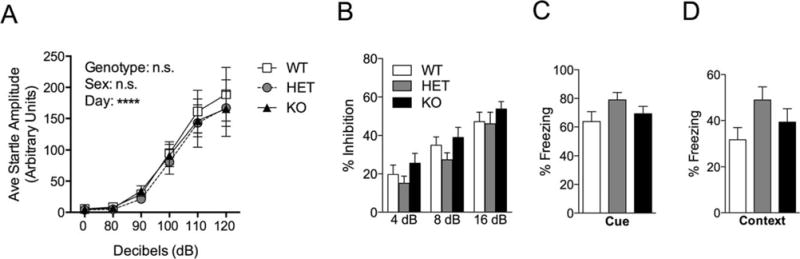
Startle, Prepulse inhibition and Fear Conditioning in Shank3e4–9 mutant mice. (A) All three genotypes displayed similar startle amplitude following a range of dB stimuli. (B) Prepulse inhibition of acoustic startle is unchanged among the genotypes. All three genotypes were tested in a one trial cue-dependent (C) and context-dependent (D) fear conditioning paradigm. There was no significant difference among genotypes in in level of freezing (n = 20 WT, 16 HET, 20 KO).
Altered Repetitive Behavior and Ultrasonic Vocalizations in Shank3e4–9 Mutants
A core diagnostic criterion for ASD is stereotyped repetitive behavior [Bodfish, Symons, Parker, & Lewis, 2000; Turner, 1999]; therefore we monitored grooming behavior for 10 min in a novel home-cage following a 10-min habituation period. Shank3e4–9 KO mice displayed almost twice as much time grooming compared to WT controls with HET mice trending toward increased grooming (Fig. 6A WT vs. HET: P = 0.08, WT vs. KO: P < 0.0024). This difference was not due to increased number of grooming bouts (Fig. 6B P = 0.91), but rather to increased time grooming per bout in both HET and KO groups (Fig. 6C WT vs. HET: P < 0.01; WT vs. KO: P < 0.0006).
Figure 6.
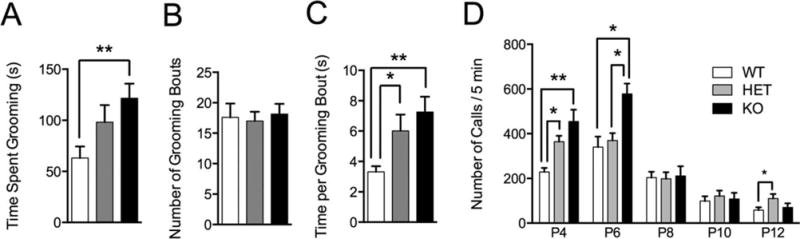
Vocalization and grooming abnormalities in Shank3e4–9 mutant mice. (A) Shank3e4–9 KO mice displayed increased time spent grooming during the observation period. (B) All genotypes display similar number of grooming bouts during the 10-min observation period. (C) Both KO and HET mice spend more time grooming per bout than WT mice. (*P < 0.05; **P < 0.01, n = 20 WT, 16 HET, 20 KO). (D) Both HET and KO mice display abnormalities in the number of ultrasonic calls following separation from their mother early in life. At age P4 and P6 KO mice display an increase in number of calls compared to WT mice, while HET mice displayed increased calls at ages P4 and P12 (*P < 0.05; **P < 0.01, P4: n = 8 WT, 20 HET, 6 KO; P6: n = 21 WT, 10 HET, 14 KO; P8: n = 26 WT, 32 HET, 16 KO, P10: n = 17 WT, 26 HET, 7 KO; P12: n = 29 WT, 25 HET, 15 KO).
Ultrasonic vocalization (USV) in pups is a developmentally regulated behavior in mice and can be altered in mouse models of ASD [Bader et al., 2011; Penagarikano et al., 2011; Wohr, Roullet, Hung, Sheng, & Crawley, 2011]. Therefore, we recorded USVs from pups after temporary isolation from their Shank3e4–9 HET mothers for 5 min at ages P4–P12. Shank3e4–9 HET and KO mice displayed significantly more calls than WT controls early in development (Fig. 6D, P4, WT vs. KO: P < 0.003; WT vs. HET: P < 0.029), and Shank3e4–9 KO displayed more calls at P6 (WT vs. KO: P < 0.0015). Additionally, Shank3e4–9 HET mice displayed more calls than either WT or KO mice later in development (P12, WT vs. HET: P < 0.04).
Social Deficits in Shank3e4–9 KO Mice
Because social deficits are a core characteristic of ASD, we tested Shank3e4–9 mutant mice in three social interaction tests. In the most direct test of reciprocal, adult, social interaction, we paired our Shank3e4–9 mutant mice according to sex and genotype and tracked the social interaction of these pairs using CleverSys Social-Scan software. Shank3e4–9 HET mutants and WT littermate controls displayed similar numbers of physical interaction bouts (Fig. 7A, P = 0.23) and time interacting (Fig. 7B, P = 0.20). Shank3e4–9 KO mice, however, interacted less times (Fig. 6A, P < 0.003) and spent less time physically interacting with their sex and genotype-matched counterpart than WTs (Fig. 7B, P < 0.0009). Total distance traveled by all groups was equivalent (not shown).
Figure 7.
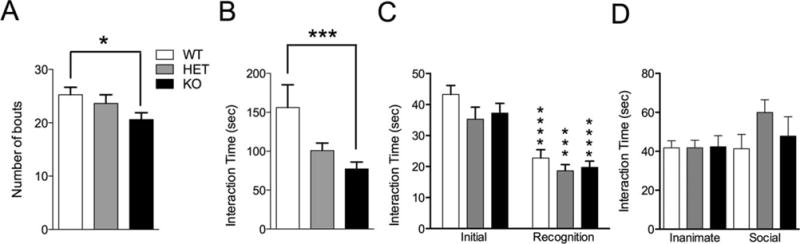
Social interaction in Shank3e4–9 mutant mice. Direct social interaction between age/sex-matched adult pairs of mice of the same genotype scored as (A) number of interaction bouts and (B) time spent interacting. (C) Interaction with a juvenile target mouse. All genotypes displayed similar time interacting during the initial and recognition periods of the juvenile social interaction test. (D) Time spent interacting with an empty cage in an open arena (inanimate) followed by time spent interacting with a social target in that cage. No difference was observed in time spent interacting among genotypes. (*P < 0.05, ***P < 0.001, ****P < 0.0001; n = 20 WT, 16 HET, 20 KO).
Using other social interaction tests, we did not identify differences. When pairing experimental mice with a novel juvenile, both Shank3e4–9 mutants and WTs display similar time interacting (Fig. 7C, P = 0.13). When presented with the same juvenile mouse 4 days later, both Shank3e4–9 mutant mice and controls display decreased interaction time with the now familiar mouse, and social interaction time was not different (Fig. 7C, P = 0.20). Shank3e4–9 mutant mice were next tested in the presence of an empty cage followed by a novel caged adult mouse. Shank3e4–9 mutants and WTs display similar time interacting with the caged adult mouse (Fig. 7D, P = 0.46). Total distance traveled was equivalent (not shown).
Object Recognition Deficits in Shank3e4–9 Mutant Mice
Many studies report intellectual disability in ASD patients [Steele, Minshew, Luna, & Sweeney, 2007; Williams, Goldstein, Carpenter, & Minshew, 2005]. We used a spatial and object recognition task to test Shank3e4–9 mutants’ ability to recognize a familiar object moved to an unfamiliar location and a novel object placed in a familiar location. Mice were habituated to an empty chamber for 5 min/day for 4 days prior to being familiarized with three identical objects arranged in specific locations relative to cues on the walls of the box (Fig. 8A). No group showed a preference for a specific object during habituation (Fig. 8B). Forty-five minutes after five such training sessions lasting 6 min each, the mice were tested for spatial novelty recognition in which object A was moved to a previously unoccupied location (Fig. 8A “Trial 6”). During the spatial test both Shank3e4–9 KO and HET mice showed no preference for object A in the new location while WTs showed a significant preference for object A in the new location (Fig. 8C WT: Obj A vs. Obj B, P < 0.018; Obj A vs. Obj C, P < 0.004). In the novel object recognition test, object A remained in its new location and object B was replaced with a stationary ping-pong ball (Fig. 8A; Trial 7, filled circle B). WT mice showed a significant preference for the new object B in trial 7 vs. the familiar control object C (Fig. 8D WT: Obj B vs. Obj C, P < 0.0034), while Shank3 mutants showed no preference for novel object B over control object C (Fig. 8D HET: Obj B vs. Obj C, P = 0.658; KO: Obj B vs. Obj C, P = 0.161).
To test longer-term spatial memory in Shank3e4–9 mutant mice we used the Morris water maze. All genotypes displayed similar latencies to swim to the hidden platform (Fig. 8E) and similar distances traveled (Fig. 8F), however, Shank3e4–9 KO mice displayed an increase in % thigmotaxis (Fig. 8G; thigmotaxis = percentage of time within 9 cm of the wall) across training days (WT vs. KO, P < 0.002; Het vs. KO, P < 0.008). A probe trial showed no difference in spatial preference among groups (Fig. 8H). Upon reversal training, Shank3e4–9 mutants and WT controls displayed similar latencies and distances to reach the hidden platform (Fig. 8E,F, days 9–12), and subsequent probe trial showed spatial preference in all groups (Fig. 8I). In a visible platform test, there was no significant difference among groups in their latency to the platform (Fig. 8J).
Altered Hippocampal Synaptic Plasticity in Shank3e4–9 Mutants
Extracellular “field” and whole-cell patch clamp electrophysiology were used to determine the effect of Shank3e4–9 deletion on synaptic function and plasticity in the CA1 region of the hippocampus. Following a single 100 Hz train for 1 s we observed significantly decreased magnitude of long-term potentiation (LTP) in Shank3e4–9 KO vs. WT (Fig. 9A). There was a main effect of genotype on the magnitude of LTP 50–60 min following LTP induction (Fig. 9A,B One-Way ANOVA: F2,20 = 4.06, P = 0.03; n = 8 (WT), 8 (HET), and 7 (KO) slices), and post hoc analysis identified a significant decrease in LTP from KO mice compared to WT (Fig. 9A,B WT: 137.50 ± 10.15, KO: 106.0 ± 5.40; Dunnet’s multiple comparisons, P < 0.05).
Figure 9.
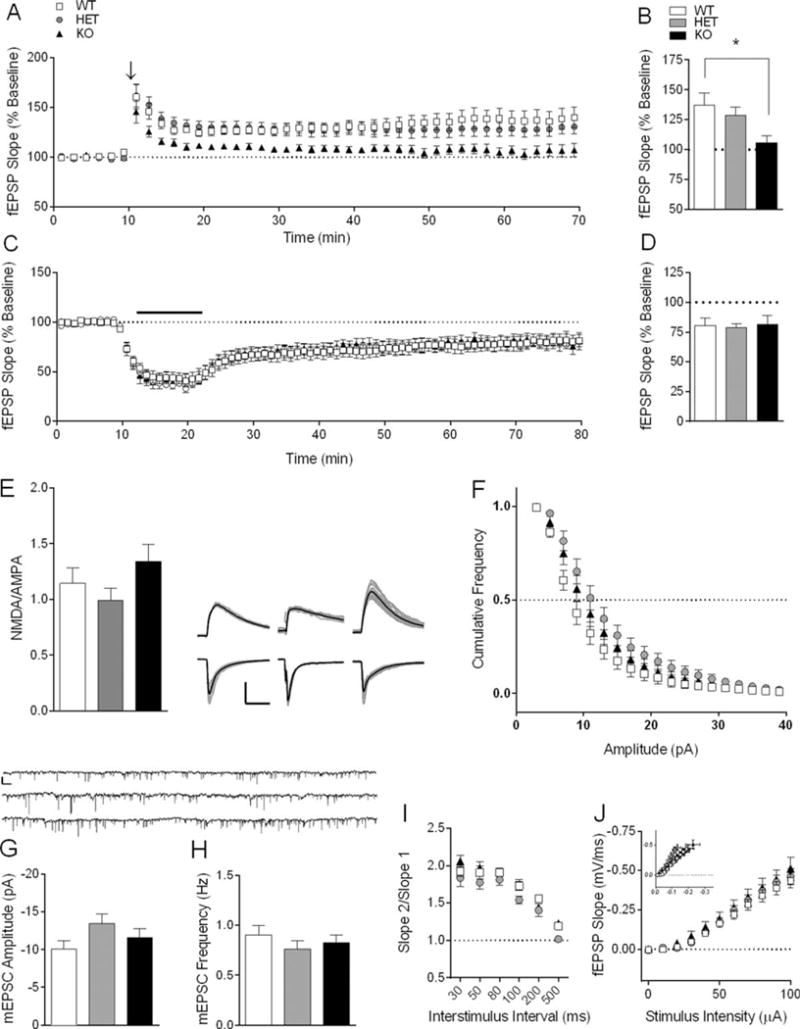
Synaptic plasticity and basal synaptic transmission at hippocampal CA3-CA1 synapses in Shank3e4–9 HET and KO mice. (A) LTP is decreased in Shank3e4–9 KO mice, but not in Shank3e4–9 HET mice. Arrow indicates 100 Hz conditioning stimulus. Inset: Average of 10 consecutive traces immediately preceding 100 Hz stimulation for 1 s (black) and at 60 min post-tetanus (gray) in WT (left), HET (middle), and KO (right) mice. Scale bar: 0.2 mV; 5 ms. (B) Summary data of mean fEPSP slope for final 10 min of recording normalized to pre-tetanus baseline (n = 8 WT, 8 HET, and 7 KO slices). (C) mGluR-LTD is normal in Shank3e4–9 HET and KO mice. Bar indicates 10 min bath application of 100 μM DHPG. Inset: Average of ten consecutive traces immediately preceding DHPG wash-in (black) and at 60 min after the start of DHPG washout (gray) in WT (left), HET (middle), and KO (right) mice. Scale bar: 0.2 mV; 5 ms. (D) Summary data of mean fEPSP slope for final 10 min of recording normalized to pre-DHPG baseline (n = 12 WT, 9 HET, 8 KO slices). *P < 0.05. (E) NMDA/AMPA ratio is unchanged in Shank3e4–9 HET and KO mice. Inset: ten consecutive traces (gray) and average trace (black) from WT (left), HET (middle), and KO mice (right) at −70 mV (bottom) and +40 mV (top) (n = 30 WT, 35 HET, and 24 KO cells). Scale bar: 200 pA, 50 ms. (F) Cumulative frequency plot of mEPSC amplitude, (G) mean mEPSC amplitude, and (H) mean frequency of events are unaffected in Shank3e4–9 HET and KO mice. Inset: 1 min raw traces from WT (top), HET (middle), KO (bottom) mice. Scale bar: 15 pA; 1.5 s (n = 21 WT, 19 HET, 19 KO cells). (I) Paired-pulse ratio is not affected in Shank3e4–9 HET or KO mice at interstimulus intervals 30–500 ms (n = 8 WT, 9 HET, 10 KO slices). (J) Input-output relationship of stimulus intensity to fEPSP slope is unchanged in HET and KO mice compared to WT controls. Inset: fEPSP slope plotted against fiber volley amplitude (n = 15 WT, 10 HET, 7 KO slices).
Since Shank3 interacts indirectly with Group 1 mGluRs at the synapse, we induced mGluR-dependent long-term depression (LTD) at CA3-CA1 synapses by bath application of 100 μM DHPG for 10 min. Magnitude of mGluR-LTD at 50–60 min is not altered in Shank3e4–9 KO mice (Fig. 9C,D One-Way ANOVA: F2,26 = 0.04, P = 0.96; n = 12 (WT), 9 (HET), and 8 (KO) slices).
Synaptic Transmission at Hippocampal Synapses is Preserved in Shank3e4–9 Mutants
Because Shank3 interacts indirectly with both NMDA and AMPA receptors, we investigated changes in the relative contribution of NMDA and AMPA receptor-mediated currents to excitatory postsynaptic currents (EPSCs). NMDA/AMPA ratio in the hippocampus is not affected by deletion of Shank3 exons 4–9 (Fig. 9E, One-Way ANOVA: F(2,86) = 1.78, P = 0.17; n = 30 (WT), 35 (HET), and 24 (KO) cells). The cumulative frequency of mEPSC amplitude (Fig. 9F, Kolmagorov–Smirnov two-sample test P > 0.1) and mean mEPSC amplitude (Fig. 9G, One-Way ANOVA: F2,56 = 1.92, P = 0.16) in hippocampal area CA1 are unaffected by deletion of Shank3 exons 4–9. Furthermore, we do not observe any change in mEPSC frequency in Shank3e4–9 mutants (Fig. 9H, One-Way ANOVA: F2,56 = 0.55, P = 0.58; n = 21 (WT), 19 (HET), 19 (KO) cells). Also, Shank3e4–9 deletion had no effect on paired pulse ratio (Fig. 9I, RM Two-Way ANOVA: Genotype F2,24 = 3.15, P = 0.06, Interval F5,120 = 101.2, P < 0.0001, Genotype × Interval F10,120 = 0.88, P = 0.56; n = 8 WT, 9 HET, and 10 KO slices). We found no difference in the input/output (I/O) relationship of stimulus intensity to fEPSP slope in Shank3e4–9 KO compared to WT (Fig. 9J, RM Two-Way ANOVA: Genotype F2,29 = 0.52, P = 0.60, Intensity F10,290 = 182.5, P < 0.0001, Genotype × Intensity F20,290 = 0.32, P = 0.99; n = 15 WT, 10 HE, and 7 KO slices).
Striatal Excitatory Transmission is Impaired in Shank3e4–9 Mutant Mice
Because Shank3 is the predominant Shank isoform in striatum and Shank3e4–9 KO and HET mice demonstrate increased self-grooming, we next examined striatal synaptic function (Fig. 10A). Increased self-grooming is an OCD-like behavior previously attributed to altered striatal synaptic transmission in mouse models [Blundell, Blaiss et al., 2010; Peca et al., 2011; Wan et al., 2013; Wan, Feng, & Calakos, 2011; Welch et al., 2007]. NMDA/AMPA ratio is significantly decreased at glutamatergic synapses onto medium spiny neurons from Shank3e4–9 HET and KO mice compared to WT (Fig. 10B, One-Way ANOVA: F2,50 = 9.43, P < 0.001; n = 18 (WT), 17 (HET), 18 (KO) cells). Because we found no difference in mEPSC amplitude (Fig. 10C, One-Way ANOVA: F2,71 = 0.75, P = 0.48; n = 25 WT, 29 HET, 20 KO cells), the decrease in NMDA/AMPA ratio is likely due to decreased NMDA receptor function. We also observed no difference in mEPSC frequency in striatum (Fig. 10D, One-Way ANOVA: F2,71 = 1.99, P = 0.14; n = 25 WT, 29 HET, 20 KO cells).
Discussion
We generated and extensively characterized a mutant mouse model of relevance to autism caused by SHANK3 deletion/mutation. We demonstrate effects of this deletion on multiple Shank3 isoforms. We also demonstrate novel alterations in striatal synaptic biochemistry and function not previously examined in related Shank3 models. Additionally, decreased synaptic plasticity in the hippocampus and impaired striatal excitatory transmission in Shank3e4–9 mutant mice correlate with deficits in spatial object recognition and increased repetitive grooming, behaviors known to be modulated by hippocampal and striatal manipulations respectively.
Previous studies [Bozdagi et al., 2010; Han et al., 2013; Peca et al., 2011; Wang et al., 2011] analyzing Shank3 expression in mutant models were limited to only one or a few higher molecular weight isoforms. Studies have shown the possibility of more than 20 Shank3 isoforms derived from six promoters and a number of alternatively spliced exons [Jiang & Ehlers, 2013; Wang et al., 2011; Wang, Xu, Bey, Lee, & Jiang, 2014]. Our expanded analysis indicates a reduction in at least three additional lower molecular weight isoforms in Shank3e4–9 mice.
Examination of Shank3 protein bands in striatal synaptosomes reveals decreased synaptic localization of additional Shank3 immunoreactive bands. Striatal synaptosomes also demonstrate decreases in Homer1b/c, PSD-95, and GluA2 and GluA3 subunits. Interestingly, there were no changes in any of the synaptic proteins measured in hippocampal synaptosomes (not shown). These data suggest striatum-specific synaptic alterations in synaptic biochemistry.
Striatal synaptic function has not been reported in any Shank3 exon 4–9 deletion mouse model to date in spite of Shank3 being the predominant Shank family member in striatum [Peca et al., 2011]. Our data demonstrate a decrease in NMDA/AMPA ratio in both HET and KO Shank3e4–9 mice at striatal synapses with no change in mEPSC amplitude, suggesting decreased NMDAR-mediated synaptic responses. NMDAR subunits are known to interact indirectly with Shank3 through PSD-95, and PSD-95 levels are reduced in striatal synaptosomes. It will be of great interest to understand the mechanism by which loss of Shank3 leads to reduced NMDAR-mediated synaptic currents. Our striatal synaptosome data suggest that decreased synaptic NMDAR subunits may not be involved, though future experiments will explore the many possible mechanisms for decreased striatal NMDAR function in this model. We do not consider our observed striatal synaptosome preparation changes to be inconsistent with our striatal electrophysiology findings because synaptosome preparations are crude, do not represent synaptic surface receptor subunits exclusively, and multiple mechanisms for alterations in NMDAR function are possible outside of altered NMDAR subunit number.
The one finding consistent among all Shank3 mutant models published to date is a decrease in NMDAR-mediated hippocampal LTP [Bozdagi et al., 2010; Kouser et al., 2013; Wang et al., 2011; Yang et al., 2012]. Our data support a decrease in hippocampal LTP in the Shank3 exon 4–9 mice as previously [Bozdagi et al., 2010; Yang et al., 2012] and replicated [Wang et al., 2011], making this the most replicated phenotype among Shank3 mutants.
We find no alteration in hippocampal NMDA/AMPA ratio, mEPSC amplitude, mEPSC frequency, baseline synaptic transmission, or paired pulse ratio (PPR) at multiple interstimulus intervals. These findings directly replicate those of Wang et al. demonstrating no alterations in input/output curves, PPR, or mEPSC frequency or amplitude in their Shank3 exon 4–9 deletion model [Wang et al., 2011]. This is important as both our findings and those of Wang et al. directly contradict the findings of Buxbaum’s group reporting a decrease in AMPAR-mediated input/output curves, decreased mEPSC amplitude, increased mEPSC frequency, and decrease PPR at a single interstimulus interval in area CA1 of hippocampus in their Shank3 exon 4–9 deletion model [Bozdagi et al., 2010; Yang et al., 2012]. The reason for this discrepancy is not clear.
While speculation on differences among laboratories using different methodologies and even different mice is not always fruitful, we offer some potential explanations for the electrophysiological discrepancies in hippocampus between Buxbaum’s group and both our findings and the same findings by the Jiang group. One major difference between the Jiang/Powell Labs and Buxbaum lab is the lack of a difference in hippocampal input/output (I/O) curves. I/O curves are notorious for their variability which depends on stimulating electrode type, placement, age, and many other factors. This variability is best accounted for by interleaving mutant and control animals in a coordinated fashion. It is also accounted for by increasing the number of animals and slices used in each experiment. In Bozdagi et al.’s initial publication on the heterozygous mutants, they used only four mice per genotype with 2–3 slices per mouse. In their followup publication, they replicated this difference with an N = 9 slices per genotype (again using 2–3 slices per mouse, making a maximum of five mice or as few as three mice per group). Their experiments also appear to have been done in the presence of low Mg2+. Our studies were performed with higher n’s and with physiologic levels of Mg2+ in the bath. Similarly, when recording mEPSCs, they used an n = 7–8 for WT and heterozygous mice, while our studies used n’s of 19–21 cells to determine mEPSC frequency and amplitude. Thus, it is possible that our results more closely reflect the population due to the greater power of our experiments. The Jiang publication also lists n = 23 slices from eight mice for their I/O curves and N = 14 for mEPSC experiments. We performed extracellular recordings (I/O curves, LTP, and PPR) on mice age 3–4 weeks; the Jiang lab tested mice at 2–4 months of age; the Buxbaum lab tested mice at 4–6 weeks of age. It is possible that we have collectively identified a transient, developmental window of synaptic dysfunction at age 4–6 weeks. Our whole cell recordings were performed in hippocampus at 14–17 days of age. The age of whole cell recordings in the Buxbaum group is not clear from their paper. It appears that whole cell recordings in the Jiang group were from mice 2–4 months of age. It is also possible that there are subtle differences in the constructs used to target exons 4–9 among the different models. This could in theory affect alternative splicing and lead to differences in some of the lower molecular weight isoforms of Shank3 that we have examined using N-terminal antibodies in this manuscript. This would be readily tested through the use of these same antibodies in all exon 4–9 models. That said, the reason for such discrepancies in I/O curves, mEPSCs, and Paired Pulse Ratio remain to be determined.
Importantly, our behavioral results are largely consistent with thorough behavioral characterization of a similar Shank3 mouse model performed in the Crawley lab with Shank3 exon 4–9 mice [Yang et al., 2012]. Both of us describe increased repetitive grooming and decreased novel object recognition as robust phenotypes. Independent replication of behavioral studies across laboratories is of critical importance to autism research.
Increased grooming is a relatively robust behavioral phenotype in this model that now replicates across three different laboratories [Wang et al., 2011; Yang et al., 2012].
Deficiencies in object recognition in Shank3e4–9 KO mice were also observed by Jiang et al. [Wang et al., 2011] and in Shank3e4–9 HET mice by Crawley et al. [Yang et al., 2012], again providing robust behavioral phenotypes in this model. Previously, our laboratory characterized a novel Shank3 mutant mouse (Shank3ΔC/ΔC, exon 21 deletion) that displayed neophobia to novel objects and novel environments [Kouser et al., 2013]. We do not find similar effects in Shank3e4–9 mutants.
In our hands, Shank3e4–9 mutant pups also displayed significantly more USVs than WT. Vocalization abnormalities have been observed in other ASD mouse models [Scattoni, Gandhy, Ricceri, & Crawley, 2008], as well as in adult Shank3e4–9 KO mice from the Jiang lab [Wang et al., 2011]. However, the Crawley lab study did not observe vocalization abnormalities in both Shank3e4–9 HET and KO mice even at P4 [Yang et al., 2012]. The different outcomes for pup USV data could possibly be accounted for by differences in lengths of recording and separation of pups from their mother or by other subtle differences in behavioral environments or handling across the two laboratories. Alternatively, this may be a less robust phenotype.
We also observed deficits in reciprocal social interaction in Shank3e4–9 KO mice interacting with another KO mouse. Curiously, altered reciprocal social interaction was only observed when KO mice were interacting with each other, but not when KO mice were interacting with a WT juvenile target. The Crawley lab demonstrated normal sociability on the three-chambered social approach task in Shank3 exon 4–9 deletion mice [Yang et al., 2012], while the Jiang lab demonstrated significantly decreased sociability in these mice using this task [Wang et al., 2011]. The Crawley lab demonstrated mildly altered juvenile play in Shank3 mutants in some cohorts as well [Yang et al., 2012]. Thus, social phenotypes in this model are subtle, and it remains unclear which social phenotypes are most robust and reproducible in this model.
Overall, deletion of autism-relevant Shank3 exons 4–9 results in multiple abnormalities in synaptic function and behavior. Our findings provide evidence that Shank3e4–9 mutant mice represent a valid model of relevance to autism and Phelan-McDermid Syndrome and provide evidence that NMDARs may be a viable therapeutic target in this model.
Acknowledgments
We thank Dr. Paul Worley for Shank3 antibody. Supported by NIH (R01HD069560, R21HD065290, & R01HD069560-S1 to CMP), Autism Speaks (CMP), Autism Speaks and Autism Science Foundation Postdoctoral Fellowships (HES), The Hartwell Foundation (CMP), Ed and Sue Rose Distinguished Professorship in Neurology (CMP), and gifts from Drs. Clay Heighten and Debra Caudy and BRAINS for Autism (CMP).
Footnotes
Authors contributions: T.C.J., H.E.S., J.M.R., and C.M.P. designed research; T.C.J., H.E.S., Z.X., J.M.R., and S.L. performed research; T.C.J., H.E.S., and J.M.R. analyzed data; T.C.J., H.E.S., J.M.R., and C.M.P. wrote the paper.
Contributor Information
Thomas C. Jaramillo, Department of Neurology and Neurotherapeutics, University of Texas Southwestern Medical Center, Dallas, Texas
Haley E. Speed, Department of Neurology and Neurotherapeutics, University of Texas Southwestern Medical Center, Dallas, Texas
Zhong Xuan, Department of Neurology and Neurotherapeutics, University of Texas Southwestern Medical Center, Dallas, Texas.
Jeremy M. Reimers, Department of Neurology and Neurotherapeutics, University of Texas Southwestern Medical Center, Dallas, Texas
Shunan Liu, Department of Neurology and Neurotherapeutics, University of Texas Southwestern Medical Center, Dallas, Texas.
Craig M. Powell, Department of Neurology and Neurotherapeutics, University of Texas Southwestern Medical Center, Dallas, Texas Department of Psychiatry and Neuroscience Graduate Program, University of Texas Southwestern Medical Center, Dallas, Texas.
References
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders, fifth edition (DSM-5) 5th. Arlington, VA: American Psychiatric Publishing; 2013. p. 991. [Google Scholar]
- Arons MH, Thynne CJ, Grabrucker AM, Li D, Schoen M, Cheyne JE, Garner CC. Autism-associated mutations in ProSAP2/Shank3 impair synaptic transmission and neurexin-neuroligin-mediated transsynaptic signaling. Journal of Neuroscience. 2012;32(43):14966–14978. doi: 10.1523/JNEUROSCI.2215-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader PL, Faizi M, Kim LH, Owen SF, Tadross MR, Alfa RW, Shamloo M. Mouse model of Timothy syndrome recapitulates triad of autistic traits. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(37):15432–15437. doi: 10.1073/pnas.1112667108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell J, Blaiss CA, Etherton MR, Espinosa F, Tabuchi K, Walz C, Powell CM. Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. Journal of Neuroscience. 2010;30(6):2115–2129. doi: 10.1523/JNEUROSCI.4517-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell J, Kaeser PS, Sudhof TC, Powell CM. RIM1alpha and interacting proteins involved in presynaptic plasticity mediate prepulse inhibition and additional behaviors linked to schizophrenia. Journal of Neuroscience. 2010;30(15):5326–5333. doi: 10.1523/JNEUROSCI.0328-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell J, Tabuchi K, Bolliger MF, Blaiss CA, Brose N, Liu X, Powell CM. Increased anxiety-like behavior in mice lacking the inhibitory synapse cell adhesion molecule neuroligin 2. Genes, Brain and Behavior. 2009;8(1):114–126. doi: 10.1111/j.1601-183X.2008.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccuto L, Lauri M, Sarasua SM, Skinner CD, Buccella D, Dwivedi A, Schwartz CE. Prevalence of SHANK3 variants in patients with different subtypes of autism spectrum disorders. European Journal of Human Genetics. 2013;21(3):310–316. doi: 10.1038/ejhg.2012.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodfish JW, Symons FJ, Parker DE, Lewis MH. Varieties of repetitive behavior in autism: Comparisons to mental retardation. Journal of Autism and Developmental Disorders. 2000;30(3):237–243. doi: 10.1023/a:1005596502855. [DOI] [PubMed] [Google Scholar]
- Boeckers TM, Bockmann J, Kreutz MR, Gundelfinger ED. ProSAP/Shank proteins—A family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. Journal of Neurochemistry. 2002;81(5):903–910. doi: 10.1046/j.1471-4159.2002.00931.x. [DOI] [PubMed] [Google Scholar]
- Boeckers TM, Kreutz MR, Winter C, Zuschratter W, Smalla KH, Sanmarti-Vila L, Gundelfinger ED. Proline-rich synapse-associated protein-1/cortactin binding protein 1 (ProSAP1/CortBP1) is a PDZ-domain protein highly enriched in the postsynaptic density. Annals of Anatomy. 2001;183(2):101. doi: 10.1016/S0940-9602(01)80024-8. [DOI] [PubMed] [Google Scholar]
- Boeckers TM, Liedtke T, Spilker C, Dresbach T, Bockmann J, Kreutz MR, Gundelfinger ED. C-terminal synaptic targeting elements for postsynaptic density proteins ProSAP1/Shank2 and ProSAP2/Shank3. Journal of Neurochemistry. 2005;92(3):519–524. doi: 10.1111/j.1471-4159.2004.02910.x. [DOI] [PubMed] [Google Scholar]
- Bonaglia MC, Giorda R, Borgatti R, Felisari G, Gagliardi C, Selicorni A, Zuffardi O. Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. American Journal of Human Genetics. 2001;69(2):261–268. doi: 10.1086/321293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaglia MC, Giorda R, Mani E, Aceti G, Anderlid BM, Baroncini A, Zuffardi O. Identification of a recurrent breakpoint within the SHANK3 gene in the 22q13.3 deletion syndrome. Journal of Medical Genetics. 2006;43(10):822–828. doi: 10.1136/jmg.2005.038604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdagi O, Sakurai T, Papapetrou D, Wang X, Dickstein DL, Takahashi N, Buxbaum JD. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Molecular Autism. 2010;1(1):15. doi: 10.1186/2040-2392-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Bourgeron T. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nature Genetics. 2007;39(1):25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand CM, Perroy J, Loll F, Perrais D, Fagni L, Bourgeron T, Sans N. SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Molecular Psychiatry. 2012;17(1):71–84. doi: 10.1038/mp.2011.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etherton MR, Blaiss CA, Powell CM, Sudhof TC. Mouse neurexin-1alpha deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t] Proceedings of the National Academy of Sciences of the United States of America. 2009;106(42):17998–18003. doi: 10.1073/pnas.0910297106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier J, Champagne N, Lafreniere RG, Xiong L, Spiegelman D, Brustein E, S2D Team De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(17):7863–7868. doi: 10.1073/pnas.0906232107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier J, Spiegelman D, Piton A, Lafreniere RG, Laurent S, St-Onge J, Rouleau GA. Novel de novo SHANK3 mutation in autistic patients. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2009;150B(3):421–424. doi: 10.1002/ajmg.b.30822. [DOI] [PubMed] [Google Scholar]
- Grabrucker AM, Schmeisser MJ, Schoen M, Boeckers TM. Postsynaptic ProSAP/Shank scaffolds in the cross-hair of synaptopathies. Trends in Cell Biology. 2011;21(10):594–603. doi: 10.1016/j.tcb.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Hamdan FF, Gauthier J, Araki Y, Lin DT, Yoshizawa Y, Higashi K, Michaud JL. Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. American Journal of Human Genetics. 2011;88(3):306–316. doi: 10.1016/j.ajhg.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K, Holder JL, Jr, Schaaf CP, Lu H, Chen H, Kang H, Zoghbi HY. SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature. 2013;503(7474):72–77. doi: 10.1038/nature12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YH, Ehlers MD. Modeling autism by SHANK gene mutations in mice. Neuron. 2013;78(1):8–27. doi: 10.1016/j.neuron.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolevzon A, Cai G, Soorya L, Takahashi N, Grodberg D, Kajiwara Y, Buxbaum JD. Analysis of a purported SHANK3 mutation in a boy with autism: Clinical impact of rare variant research in neurodevelopmental disabilities. Brain Research. 2011;1380:98–105. doi: 10.1016/j.brainres.2010.11.005. [DOI] [PubMed] [Google Scholar]
- Kouser M, Speed HE, Dewey CM, Reimers JM, Widman AJ, Gupta N, Powell CM. Loss of predominant shank3 isoforms results in hippocampus-dependent impairments in behavior and synaptic transmission. Journal of Neuroscience. 2013;33(47):18448–18468. doi: 10.1523/JNEUROSCI.3017-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Parada LF. Pten regulates neuronal arborization and social interaction in mice. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t] Neuron. 2006;50(3):377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Hunsaker MR, Kesner RP. The role of hippocampal subregions in detecting spatial novelty. Behavioral Neuroscience. 2005;119(1):145–153. doi: 10.1037/0735-7044.119.1.145. [DOI] [PubMed] [Google Scholar]
- Lim S, Sala C, Yoon J, Park S, Kuroda S, Sheng M, Kim E. Sharpin, a novel postsynaptic density protein that directly interacts with the shank family of proteins. Molecular and Cellular Neuroscience. 2001;17(2):385–397. doi: 10.1006/mcne.2000.0940. [DOI] [PubMed] [Google Scholar]
- Mameza MG, Dvoretskova E, Bamann M, Honck HH, Guler T, Boeckers TM, Kreienkamp HJ. SHANK3 gene mutations associated with autism facilitate ligand binding to the Shank3 ankyrin repeat region. Journal of Biological Chemistry. 2013;288(37):26697–26708. doi: 10.1074/jbc.M112.424747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misceo D, Rodningen OK, Baroy T, Sorte H, Mellembakken JR, Stromme P, Frengen E. A translocation between Xq21.33 and 22q13.33 causes an intragenic SHANK3 deletion in a woman with Phelan-McDermid syndrome and hypergonadotropic hypogonadism. American Journal of Medical Genetics Part A. 2011;155A(2):403–408. doi: 10.1002/ajmg.a.33798. [DOI] [PubMed] [Google Scholar]
- Moessner R, Marshall CR, Sutcliffe JS, Skaug J, Pinto D, Vincent J, Scherer SW. Contribution of SHANK3 mutations to autism spectrum disorder. American Journal of Human Genetics. 2007;81(6):1289–1297. doi: 10.1086/522590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Sheng M. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron. 1999;23(3):569–582. doi: 10.1016/s0896-6273(00)80809-0. [DOI] [PubMed] [Google Scholar]
- Peca J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Feng G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011;472(7344):437–442. doi: 10.1038/nature09965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, Geschwind DH. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. 2011;147(1):235–246. doi: 10.1016/j.cell.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell CM, Schoch S, Monteggia L, Barrot M, Matos MF, Feldmann N, Nestler EJ. The presynaptic active zone protein RIM1alpha is critical for normal learning and memory. [Comparative Study Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.] Neuron. 2004;42(1):143–153. doi: 10.1016/s0896-6273(04)00146-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C, Piech V, Wilson NR, Passafaro M, Liu G, Sheng M. Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron. 2001;31(1):115–130. doi: 10.1016/s0896-6273(01)00339-7. [DOI] [PubMed] [Google Scholar]
- Save E, Poucet B, Foreman N, Buhot MC. Object exploration and reactions to spatial and nonspatial changes in hooded rats following damage to parietal cortex or hippo-campal formation. Behavioral Neuroscience. 1992;106(3):447–456. [PubMed] [Google Scholar]
- Scattoni ML, Gandhy SU, Ricceri L, Crawley JN. Unusual repertoire of vocalizations in the BTBR T+tf/J mouse model of autism. PLoS ONE. 2008;3(8):e3067. doi: 10.1371/journal.pone.0003067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Kim E. The Shank family of scaffold proteins. Journal of Cell Science. 2000;113(Pt 11):1851–1856. doi: 10.1242/jcs.113.11.1851. [DOI] [PubMed] [Google Scholar]
- Steele SD, Minshew NJ, Luna B, Sweeney JA. Spatial working memory deficits in autism. Journal of Autism and Developmental Disorders. 2007;37(4):605–612. doi: 10.1007/s10803-006-0202-2. [DOI] [PubMed] [Google Scholar]
- Sykes NH, Toma C, Wilson N, Volpi EV, Sousa I, Pagnamenta AT, International Molecular Genetic Study of Autism Consortium Copy number variation and association analysis of SHANK3 as a candidate gene for autism in the IMGSAC collection. European Journal of Human Genetics. 2009;17(10):1347–1353. doi: 10.1038/ejhg.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM, Südhof TC. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t] Science. 2007;318(5847):71–76. doi: 10.1126/science.1146221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, Worley PF. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of post-synaptic density proteins. Neuron. 1999;23(3):583–592. doi: 10.1016/s0896-6273(00)80810-7. [DOI] [PubMed] [Google Scholar]
- Turner M. Annotation: Repetitive behaviour in autism: A review of psychological research. Journal of Child Psychology and Psychiatry. 1999;40(6):839–849. [PubMed] [Google Scholar]
- Verpelli C, Dvoretskova E, Vicidomini C, Rossi F, Chiappalone M, Schoen M, Sala C. Importance of Shank3 protein in regulating metabotropic glutamate receptor 5 (mGluR5) expression and signaling at synapses. Journal of Biological Chemistry. 2011;286(40):34839–34850. doi: 10.1074/jbc.M111.258384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waga C, Okamoto N, Ondo Y, Fukumura-Kato R, Goto Y, Kohsaka S, Uchino S. Novel variants of the SHANK3 gene in Japanese autistic patients with severe delayed speech development. Psychiatric Genetics. 2011;21(4):208–211. doi: 10.1097/YPG.0b013e328341e069. [DOI] [PubMed] [Google Scholar]
- Wan Y, Ade KK, Caffall Z, Ilcim Ozlu M, Eroglu C, Feng G, Calakos N. Circuit-selective striatal synaptic dysfunction in the Sapap3 knockout mouse model of obsessive-compulsive disorder. Biological Psychiatry. 2013;75(8):623–630. doi: 10.1016/j.biopsych.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Feng G, Calakos N. Sapap3 deletion causes mGluR5-dependent silencing of AMPAR synapses. Journal of Neuroscience. 2011;31(46):16685–16691. doi: 10.1523/JNEUROSCI.2533-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, McCoy PA, Rodriguiz RM, Pan Y, Je HS, Roberts AC, Jiang YH. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Human Molecular Genetics. 2011;20(15):3093–3108. doi: 10.1093/hmg/ddr212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Xu Q, Bey AL, Lee Y, Jiang YH. Transcriptional and functional complexity of Shank3 provides a molecular framework to understand the phenotypic heterogeneity of SHANK3 causing autism and Shank3 mutant mice. Molecular Autism. 2014;5:30. doi: 10.1186/2040-2392-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JM, Lu J, Rodriguiz RM, Trotta NC, Peca J, Ding JD, Feng G. Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature. 2007;448(7156):894–900. doi: 10.1038/nature06104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DL, Goldstein G, Carpenter PA, Minshew NJ. Verbal and spatial working memory in autism. Journal of Autism and Developmental Disorders. 2005;35(6):747–756. doi: 10.1007/s10803-005-0021-x. [DOI] [PubMed] [Google Scholar]
- Wohr M, Roullet FI, Hung AY, Sheng M, Crawley JN. Communication impairments in mice lacking Shank1: Reduced levels of ultrasonic vocalizations and scent marking behavior. PLoS One. 2011;6(6):e20631. doi: 10.1371/journal.pone.0020631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Bozdagi O, Scattoni ML, Wohr M, Roullet FI, Katz AM, Crawley JN. Reduced excitatory neurotransmission and mild autism-relevant phenotypes in adolescent Shank3 null mutant mice. Journal of Neuroscience. 2012;32(19):6525–6541. doi: 10.1523/JNEUROSCI.6107-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]