

Abstract
Since the discovery of Xin repeat-containing proteins in 1996, the importance of Xin proteins in muscle development, function, regeneration, and disease has been continuously implicated. Most Xin proteins are localized to myotendinous junctions of the skeletal muscle and also to intercalated discs (ICDs) of the heart. The Xin gene is only found in vertebrates, which are characterized by a true chambered heart. This suggests that the evolutionary origin of the Xin gene may have played a key role in vertebrate origins. Diverse vertebrates including mammals possess two paralogous genes, Xinα (or Xirp1) and Xinβ (or Xirp2), and this review focuses on the role of their encoded proteins in cardiac muscles. Complete loss of mouse Xinβ (mXinβ) results in the failure of forming ICD, severe growth retardation, and early postnatal lethality. Deletion of mouse Xinα (mXinα) leads to late-onset cardiomyopathy with conduction defects. Molecular studies have identified three classes of mXinα-interacting proteins: catenins, actin regulators/modulators, and ion-channel subunits. Thus, mXinα acts as a scaffolding protein modulating the N-cadherin-mediated adhesion and ion-channel surface expression. Xin expression is significantly upregulated in early stages of stressed hearts, whereas Xin expression is downregulated in failing hearts from various human cardiomyopathies. Thus, mutations in these Xin loci may lead to diverse cardiomyopathies and heart failure.
1. INTRODUCTION
The heart is the first functional organ to develop during embryogenesis, and it continues for a lifetime to pump the blood supply for all organs. Its proper formation and normal pumping action are essential for animal growth and survival. Aberrations in cardiac development and function would lead to congenital and acquired heart diseases.
1.1. Heart disease and intercalated disc (ICD)
Heart failure is a complex clinical syndrome that results from any structural or functional impairment of ventricular filling or ejection of blood. Heart failure due to cardiomyopathy, arrhythmias, and congenital heart diseases caused 275,000 deaths in 2009 in the United States (Go et al., 2013). Furthermore, heart failure is the only cardiovascular disease still with increasing incidence worldwide. In the United States, about 5.1 million persons have clinically manifest heart failure. The total health care costs for heart failure in 2013 in the United State are estimated to be 32 billion dollars and projected to increase almost 120% by 2030 (Go et al., 2013). Failing hearts generally experience a mechanical problem (systolic and/or diastolic dysfunction), but many of them also experience an electrical problem (arrhythmias), in which cellular excitability is inadequately regulated. As a consequence, the myocardium of failing heart is unable to pump sufficient volumes of blood to meet metabolic demands.
The intercalated disc (ICD), a unique structure of adult cardiac muscle, is located at the termini of the rod-shaped cardiomyocytes. An increasing line of evidence suggests that the ICD is responsible for mechanical and electrical coupling as well as transducing signals among cardiomyocytes. Disruption of the ICD structure and/or function is one of hallmarks observed in the progression of many acquired and congenital heart diseases to heart failure (Dupont et al., 2001; Kostin et al., 2002, 2004; Maron and Ferrans, 1973; Perriard et al., 2003). Conversely, mutations or deficiencies in ICD components have been shown to lead to many types of cardiomyopathy, arrhythmias, and heart failure in human patients and in various genetically engineered animal models (Delmar and McKenna, 2010; Li et al., 2011; Lombardi and Marian, 2011; Noorman et al., 2009; Severs et al., 2008; Sheikh et al., 2009; Swope et al., 2012; van Tintelen et al., 2007; Wang et al., 2012). Clearly, the ICD is an important structure unique to the heart. Its structure and function could reflect the normal and pathological status of the hearts. Studies on how normal ICDs are formed and maintained may advance our understanding of cardiac disease pathogenesis. Because it has proved difficult to use conventional ion-channel antagonists and/or myocardial performance-enhancing drugs to treat arrhythmias and heart failure (Echt et al., 1991; Krell et al., 1986), the studies of ICD structure and function may potentially identify targets for developing novel and effective treatments of these diseases.
1.2. Structure of ICD and ICD-associated proteins
Structurally, three classical junctional complexes (adherens junctions, desmosomes, and gap junctions) can be identified within the ICD by electron microscopy (Forbes and Sperelakis, 1985). Molecularly, the gap junctions are small communicating channels formed by connexin molecules to allow electrical and chemical coupling between cardiomyocytes, whereas the adherens junctions and the desmosomes are formed by cadherin molecules to ligate two myocytes together to transmit contractile force and maintain mechanical integrity. In a classic view, the adherens junctions are assembled through homophilic interactions of N-cadherins of two adjacent cells, whose intracellular domains interact with the catenins (α-catenin, β-catenin, γ-catenin/plakoglobin, and p120-catenin) and actin filaments. On the other hand, the desmosomes are assembled by desmosomal cadherins, whose intracellular domains interact with plakophilin, desmoplakin, plakoglobin, and intermediate filaments. However, in adult mammalian hearts, the ICDs additionally contain a mixed type of junction composed of components of both adherens junctions and desmosomes (known as area composita) (Borrmann et al., 2006; Franke et al., 2006; Pieperhoff and Franke, 2007). Protein database (HPA cardiac immunohistochemical data and ExPASy protein-binding data) search and literature survey reveal that nearly 200 proteins are known to be associated with ICDs and can be functionally classified into six categories: (i) ion channels, for example, SCN5A (Nav1.5), KCNA5 (Kv1.5), KCND2 (Kv4.2); (ii) mechanoreceptors, for example, TJP1 (ZO1), NRAP (nebulin-related anchoring protein), and PTK2 (FAK, focal adhesion kinase); (iii) ligand and ligand receptors, for example, CXADR (coxsackie virus and adenovirus receptors or CAR) and FADD (apoptosis-associated death receptor Fas); (iv) adhesion, anchoring, and binding proteins, for example, CDH2 (N-cadherin), CTNNB1 (β-catenin), CTNND1 (p120-catenin), JUP (plakoglobin), and Xin repeat-containing proteins (Xinα and Xinβ); (v) enzymes, for example, CAPN1 (calpain); and (vi) proteins that maintain structure/function, for example, CAV3 (caveolin 3), TTN (titin), and DSP (desmoplakin) (Estigoy et al., 2009). Furthermore, it is found that about 40% of these ICD-associated proteins change their expression level and/or localization in various heart diseases including heart failure (Estigoy et al., 2009).
1.3. Xin repeat-containing and ICD-associated family of proteins (Xin proteins)
In this review, we will focus on a family of Xin repeat-containing proteins (Xinα and Xinβ), which are associated with the adherens junctions of ICDs and belong to adhesion, anchoring, and binding protein category. The gene encoding this protein family is originally discovered as a differentially expressed gene (21C) during chicken cardiac development (Wang et al., 1996). Subsequent cloning and characterization have revealed the presence of multiple copies of a conserved repeating unit of 16 amino acid residues in all proteins from chick and mouse hearts (Grosskurth et al., 2008; Lin et al., 2005; Wang et al., 1999, 2010, 2012). Treatment of chicken developing embryos with Xin antisense oligonucleotides results in collapse of heart chamber walls and alteration of cardiac morphogenesis, suggesting that this protein family plays important role in chamber integrity and cardiac development (Wang et al., 1999). Therefore, we called this chicken or mouse protein as chicken Xin (cXin) or mouse Xin (mXin), and the 16-amino acid residue repeating units as the Xin repeats (Wang et al., 1999). The word “Xin” is derived from the Chinese character for “heart” in pronunciation.
Phylogenetic studies reveal that the Xin repeat sequence is only found in vertebrates with true chambered hearts composed of complete endocardial and myocardial layers (Grosskurth et al., 2008). A single Xin repeat-containing gene first occurs in lamprey about 550 million years ago after which vertebrate whole genome duplication results in the appearance of Xinα and Xinβ (Grosskurth et al., 2008). In 2001, the human orthologs (hXinα and hXinβ) of Xinα and Xinβ genes have been identified as coexpressed cardiomyopathy-associated gene 1 (CMYA1) and 3 (CMYA3), respectively (Walker, 2001), suggesting that both Xin genes and their related signaling pathway may be candidate targets for developing therapeutic drugs. The hXinα and hXinβ proteins are also called XIRP1 and 2 (Xin actin-binding repeat-containing protein 1 and 2) because the recombinant proteins of their Xin repeat regions have been shown to bind to actin filaments (Pacholsky et al., 2004). In the literature, the mouse Xinα and Xinβ (mXinα and mXinβ) are also known as XinABC/Xirp1 (Otten et al., 2010) and myomaxin/Xirp2 (Huang et al., 2006), respectively. In this review, we will briefly summarize organization and regulation of mXin genes and then focus on recent advances in understanding Xin’s functions from characterizing knockout mouse lines and from analyzing their cellular and biochemical properties such as identifying their interacting partners. Specifically, we will discuss how Xin proteins link the actin cytoskeleton to the ICD and influence surface expression of ion channels such as transient K+ outward (Ito,f) and delayed K+ rectifier (Ik,slow1) channels in cardiomyocytes. The underlying molecular mechanisms may explain the pathogenesis of cardiomyopathy, arrhythmias, and heart failure observed in mXinα or mXinβ knockout hearts.
2. ORGANIZATION AND REGULATION OF XIN GENES
2.1. Chromosome location of Xin and synteny
In the mouse, mXinα and mXinβ genes are located on chromosomes 9 and 2, respectively. The chromosomal regions nearby to these Xin genes exhibit shared synteny both with each other and with their orthologous chromosomal neighborhoods in the human genome. Thus, the mouse mXinα neighborhood is syntenic with the human chromosomal region 3p21–3p22.2, which contains XIRP1, while the mouse mXinβ neighborhood is syntenic with the human chromosomal region 2q24–2q31.1, which contains XIRP2 (Fig. 3.1). Among other genes, the regions upstream of both the XIRP1/ mXinα and XIRP2/mXinβ genes possess a cluster of three sodium channel (SCN) α-subunit genes. Thus, these syntenic blocks containing Xinα and Xinβ from the postulated whole genome duplications that occurred during early vertebrate evolution (Grosskurth et al., 2008). Interestingly, the intra-chromosomal sodium channel Scn genes (e.g., Scn5a vs. Scn10a) are much more closely related to each other than interchromosomal Scn genes (e.g., Scn5a vs. Scn1a), suggesting that these Scn genes were created by tandem duplication after the genomic duplication that established the two syntenic blocks. Correspondingly, the two Scn plus Xin gene clusters can be traced back to more distantly related vertebrates. For example, the mammalian syntenic block containing XIRP1/mXinα and its cluster of Scn genes is present and intact in the chicken, where it is present in a syntenic block in chicken chromosome 2. The syntenic gene block containing XIRP2/mXinβ can be located on chicken chromosome 7, but this region possesses only two sets of the three Scn genes (it is missing Scn7a) and has also lost Xinβ. The partial gene losses in this second syntenic neighborhood are not specific to the chicken, because the losses apparently extend to other bird genomes, such as zebra finch. Given this shared synteny and its evolutionary maintenance, and the possible roles of Scn genes in cardiac conductance, we also speculate whether the Scn genes may be coevolving together with their respective Xin genes. Alternatively, the persistence of a shared synteny may reflect as yet unidentified regulatory mechanisms that coordinate gene expression within each gene cluster.
Figure 3.1.

Xin gene locations and nearby syntenic gene blocks in mouse, human, and chicken chromosomes. Top: The relative locations of Xirp1/Xinα ortholog gene and nearby syntenic genes on mouse chromosome #9, human chromosome #3, and chicken chromosome #2 were derived from NCBI genomic sequences NC_000075.6, NC_000003.11, and NC_006089.3, respectively (www.ncbi.nlm.nih.gov/gene). Each gene indicating by a line on chromosome represents the center of that gene defined in the database. The orthologous genes were connected by dotted lines. The mouse and human Ttc21a gene has not been found in chicken chromosome #2 between Gorasp1 and Csrnp1. The // on chicken chromosome #2 represents a break between sequences #4,954,000 and 44,977,000. Three voltage-gated sodium channel α-subunit genes (Scn5a, Scn10a, and Scn11a) located to the left of Xirp1/Xinα gene. Bottom: The relative locations of Xirp2/Xinβ ortholog gene and nearby syntenic genes on mouse chromosome #2, human chromosome #2, and chicken chromosome #7 were derived from NCBI genomic sequences NC_000068. 7, NC_000002.11, and NC_006094.3, respectively. Each gene indicating by a line on chromosome represents the center of that gene defined in the database. Five voltage-gated sodium channel α-subunit genes (Scn3a, Scn2a, Scn1a, Scn9a, and Scn7a) also located to the left of Xirp2/Xinβ gene, except that in chicken chromosome #7, a deletion of Scn7a and Xinβ results in a much shorter Xirp2 (called Xirp2-like) gene containing no Xin repeating sequences.
2.2. Xin as a downstream target of Nkx2.5 and Mef2 transcription factors
Chicken sole Xin gene begins to express in the paired lateral plate mesoderm of the embryo at Hamburger–Hamilton (HH) stage 8 (Wang et al., 1999). Subsequent expression profiling in the developing hearts analyzed by Northern blot has revealed two peaks of cXin upregulation at HH stages 16–25 and posthatch days 12–14 (Wang et al., 2012), which coincide with the timing known for septa/chamber formation and postnatal heart growth/ICD formation, respectively. The first evidence to support a requirement of Xin gene in cardiac morphogenesis came from that cXin anti-sense oligonucleotide-treated embryos exhibit collapsing of chamber walls and disrupting of normal rightward looping (Wang et al., 1999). Furthermore, the expression of cXin can be induced by BMP2 (bone morphogenetic protein 2) cytokine on anterior medial mesoendoderm explants, which normally do not express cXin (Wang et al., 1999). The analysis of induction timing for various cardiac genes has further suggested that cXin participates in a BMP2–Nkx2.5–Mef2C–cXin–vMHC (ventricular myosin heavy chain) pathway to regulate cardiogenesis (Wang et al., 1999).
Xin as a downstream target of Nkx2.5 and Mef2 has been further observed in the mouse. Expression of Nkx2.5, Mef2C, or both (but not expression of Gata4) in nonmuscle cells can transactivate the reporter gene expression driven by the proximal promoter of mXinα (Lin et al., 2001; Wang et al., 1999). This has been further supported by a drastic reduction of mXinα messages detected in Nkx2.5-null embryos or in Mef2C-null hearts (Lin et al., 2005). Moreover, it is reported that mXinβ (myomaxin) is a transcriptional target of Mef2A. Transcription factor Mef2A can directly bind to the proximal promoter of mXinβ and drive the reporter gene expression under the control of mXinβ promoter (Huang et al., 2006).
2.3. Mouse Xinα (mXinα) gene organization, unusual intra-exonic splicing, and protein variants
Initial characterizations of mXinα cDNA and genomic clones have identified a stretch of 10 kb DNA region on mouse chromosome #9 containing 1848 bp proximal promoter, exon 1 (E1, 135 bp), intron 1 (I1, 3614 bp), E2 (E2a, 3395 bp), I2 (E2b, 391 bp), and E3 (E2c, 2279 bp) (Gustafson-Wagner et al., 2007; Wang et al., 1999). Subsequently, detailed RT-PCR analyses of mXinα cDNA variants further revealed that exon 2 is actually composed of E2a, E2b (I2), and E2c (E3) as a big exon. As can be seen in Fig. 3.2, an unusually alternative (intra-exonic) splicing of mXinα primary transcript to include or exclude E2b results in two mRNAs, which encode the Xin repeat-containing protein variants, mXinα-a (XinA) and mXinα (XinB), respectively (Gustafson-Wagner et al., 2007; Otten et al., 2010). In addition, splicing out of I1, E2a, and E2b gives rise to a smaller mRNA, XinC (Otten et al., 2010), which would use a new in-frame translational start site in E2c (ATG typed in red font in Fig. 3.2) to encode a smallest mXinα protein variant, XinC. Similar splicing events have also been shown in hXinα/XIRP1 gene to potentially generate three variants, human XinA, XinB, and XinC (van der Ven et al., 2006). XinC contains no Xin repeats, suggesting binding neither to actin filaments nor to β-catenin. However, XinC has sequence partially overlapping with the previously defined filamin c-binding domain on the large form of hXinα (XinA). Potentially, XinC may compete with XinA in modulating actin filament organization and dynamics. The predicted XinC protein has unusually higher isoelectric point (pI), and the expression of XinC in normal mouse or human heart can only be detected by RT-PCR but not by Western blot analysis. However, in hypertrophic human heart samples, a trace but significant amount of XinC protein can be detected, in addition to an upregulation of XinA (Otten et al., 2010). As the amino acid sequence of XinC is identical to the C-terminus of XinA, it is difficult to rule out the possibility that the detected XinC from hypertrophic human hearts may represent degraded product of XinA. Quantitative Western blot on developing postnatal mouse hearts has revealed that the expression levels of mXinα-a and mXinα are roughly equal at newborn to postnatal day 3.5 (P3.5). As development proceeds, mXinα expression level becomes significantly higher than mXinα-a (Wang et al., 2013a). The significance of these protein variants in the healthy and diseased hearts remains to be determined. It should be noted that mXinα seems to be more solubilized by buffer with physiological condition than mXinα-a.
Figure 3.2.
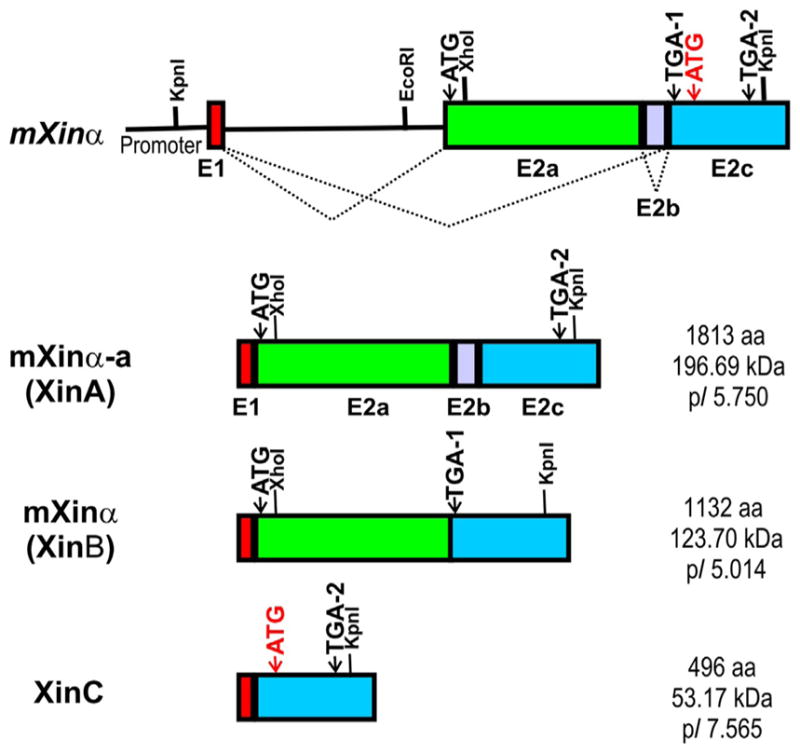
Schematic diagrams of mouse Xirp1/mXinα genomic organization and its encoded message and protein variants. The mouse Xinα gene contains a small exon E1 and a large exon E2. After removal of intron between E1 and E2, the E2 can further undergo an unusual intra-exonic splicing to give rise to two message or protein variants containing either whole E2 (mXinα-a or XinA) or E2 without E2b (mXinα or XinB) (Gustafson-Wagner et al., 2007; Otten et al., 2010). The predicted amino acid number, molecular mass, and pI value for each mouse Xinα protein variants are listed to the right of their respective message diagrams. Similar gene organization and splicing events including the intra-exonic splicing were also found in human XIRP1/hXinα gene (van der Ven et al., 2006). Another splicing event to remove E2a and E2b leading to a smallest message (XinC) has been detected by RT-PCR from normal mouse or human hearts. However, Western blot analysis with specific antibody barely detected the XinC protein from hypertrophic human hearts but not from normal mouse or human hearts (Otten et al., 2010). The predicted XinC protein has a relatively higher pI value and its sequence is identical to the C-terminus of mXinα-a/XinA. It is difficult to rule out that the detected XinC from hypertrophic human hearts may represent degraded product of XinA.
2.4. Mouse Xinβ (mXinβ) gene organization and protein variants
The mXinβ locates on a stretch of 85 kb DNA in mouse chromosome #2 containing at least 5 kb upstream promoter, E1 (153 bp), I1 (30,563 bp), E2 (149 bp), I2 (5379 bp), E3 (138 bp), I3 (22,650 bp), E4 (90 bp), I4 (379 bp), E5 (73 bp), I5 (1471 bp), E6 (134 bp), I6 (728 bp), E7 (9265 bp), I7 (2046 bp), E8 (134 bp), I8 (5418 bp), and E9 (1840 bp) (Huang et al., 2006; McCalmon et al., 2010; Wang et al., 2010). As can be seen in Fig. 3.3, the only alternatively spliced exon detected so far is E8. The exclusion and inclusion of this exon generate a larger mXinβ-a variant with 3300 residues and a smaller mXinβ variant with 3283 residues (Wang et al., 2010). Both variants have identical amino acid sequence from #1 to #3252 and divergence at their extreme C-terminal 31/48 residues. At the protein level, SDS-PAGE would not be able to differentiate them. However, estimated from the relative abundance of their messages, it was predicted that mXinβ is the major variant from mouse Xinβ gene (Wang et al., 2010). Again, the significance of these mXinβ variants in the heart remains to be determined.
Figure 3.3.

Schematic diagrams of mouse Xirp2/mXinβ genomic organization and its encoded message and protein variants. The mouse Xinβ gene contains eight small exons and one large exon (E7) (McCalmon et al., 2010; Wang et al., 2010). The inclusion of E8 together with the usage of different poly(A) sites gives rise to two distinct messages, which encode exactly the same protein with 3283 amino acid residues (mXinβ) (Wang et al., 2010). On the other hand, the exclusion of E8 leads to another message, which encodes protein with 3300 amino acid residues (mXinβ-a) (Wang et al., 2010). The predicted amino acid number, molecular mass, and pI value for each mouse Xinβ protein variants are listed to the right of their respective message diagrams. Estimated from relative abundance of messages, it was predicted that mXinβ is the major variants from mouse Xinβ gene (Wang et al., 2010). At the protein level, SDS-PAGE analysis could not effectively separate these two variants. However, specific antibody U1040 generated against the very C-terminal sequence (aa#3255–3278) of mXinβ reacts only with mXinβ, whereas common antibody U1013 generated against aa#1–532 of mXinα recognizes both mXinβ and mXinβ-a (Wang et al., 2010). Similar gene organization and splicing events were also found in human XIRP2/hXinβ gene (NCBI database).
Unlike mXinα, there is no evidence for the unusual intra-exonic splicing event occurring during the expression of mXinβ gene. Previously, evolutionary studies suggest that after vertebrate whole genome duplication, mXinα evolves faster than mXinβ in order to carry out more sophisticated and coordinated regulation of four-chambered heart functions (Grosskurth et al., 2008). Together, these findings imply that the intra-exonic splicing event may be a late evolved process.
2.5. Xin as a striated muscle-restricted gene
Northern blot analyses performed on adult chick and mouse multiple tissues reveal that cXin (9.0 kb), mXinα (5.8 kb), and mXinβ (12 kb) messages are preferentially expressed in striated muscle tissues with a weak expression in lung and no detectable expression in other tissues/organs (Huang et al., 2006; Lin et al., 2005; Wang et al., 1999). In situ hybridization further extends the striated muscle-restricted expression of cXin and mXinα in the developing chicken (Wang et al., 1996, 1999) and mouse embryos (Lin et al., 2001, 2005). The mXinβ message in developing mouse embryos has never been detected by in situ hybridization or Northern blot, suggesting that the expression level is extreme low, if expressed. Using RT-PCR, mXinβ transcripts are barely detected in proliferating C2C12 myoblasts (Huang et al., 2006). After 7 days of differentiation, C2C12 myotubes expressed a robust level of mXinβ messages (Huang et al., 2006). The striated muscle-restricted expression of Xin was further confirmed at the protein level by whole-mount immunofluorescence microscopy on developing mouse embryos (Sinn et al., 2002) and by Western blot analysis on various mouse tissues prepared from wild-type and mXinα-null mouse (Wang et al., 2010) with a broad species-specific anti-mXin antibody (U1013), which recognized all Xin proteins from frog, zebrafish, chicken, and mouse hearts.
2.6. Xin proteins preferentially localized at ICDs of cardiac muscle and myotendinous junctions of skeletal muscles
Immunofluorescence microscopy on frozen sections of adult mouse hearts and skeletal muscles with U1013 anti-mXin antibody revealed that both mXinα and mXinβ preferentially localize to the ICDs in cardiac muscle cells and to the myotendinous junctions in all of skeletal muscles tested (Feng et al., 2013; Gustafson-Wagner et al., 2007; Sinn et al., 2002; Wang et al., 1999). The ICD localization was also observed in the cryostat sections of adult human heart with mouse monoclonal antibody XR1, which was generated from mice immunized with the Xin repeat region (aa#83–285) of hXinα (XinA) (Otten et al., 2010; van der Ven et al., 2006). Contradictorily, by using another rabbit polyclonal antibody (BSU2) against aa#567–667 sequence within the Xin repeat region of myomaxin (mXinβ or Xirp2), Huang et al. showed a faint striated staining pattern with higher background. They claimed that myomaxin possibly localized to the Z-disc in mouse heart (Huang et al., 2006). It should be noted that all three antibodies (U1013, XR1, and BSU2) were raised against the Xin repeat region of Xin proteins and should cross-reacted with both Xinα and Xinβ. In cultured neonatal rat cardiomyocytes, both mXinα and mXinβ were detected by U1013 and XR1 or by peptide-specific antibodies U1697 and U1741, respectively. They were preferentially localized to the intercellular junctions (ICD-like structures) as well as nonstriated parts of myofibrils near the focal adhesions (Gustafson-Wagner et al., 2007; Lin et al., 2001; van der Ven et al., 2006).
To further confirm the preferential localization of mXinα and mXinβ to the ICD, anti-mXin antibodies, U1013 for both mXinα and mXinβ, U1697 for mXinα, and U1040 for mXinβ, were used in immunofluorescence microscopy on heart cryosections prepared from wild-type (Wt), mXinα-null (AKO), mXinβ-null (BKO), and mXinα−/−;mXinβ −/− (DKO) mice. Similar to U1013 staining pattern (Fig. 3.4A), isoform-specific U1697 and U1040 antibodies detected preferential localizations of mXinα and mXinβ, respectively, to the ICDs of Wt hearts (Fig. 3.4B and C, respectively). In addition to ICD staining, the antibodies, U1013 and U1040, occasionally stained the transverse tubules (T-tubules) of AKO heart (arrowheads in Fig. 3.4D and F). In contrast, the antibody U1697 stained neither ICDs nor T-tubules of AKO hearts (Fig. 3.4E). Furthermore, both ICD and T-tubule staining patterns were not detected by U1040 on BKO heart sections (Fig. 3.4I). These results indicate that mXinβ locates majorly at ICDs and sometimes at T-tubules. As previously reported that BKO hearts failed to form mature ICDs (Wang et al., 2010, 2013a), mXinα variants detected by U1013 and U1697 on BKO sections remained as small puncta dispersed throughout cardiomyocytes (Fig. 3.4G and H). The specificity of U1013 antibody was further verified by double-label immunofluorescence with rabbit polyclonal U1013 and mouse monoclonal anti-N-cadherin antibody on DKO hearts. There was no staining in heart section stained with U1013 (Fig. 3.4J), whereas many small punta along the lateral membranes of DKO cardiomyocytes were observed on the same section counterstained with anti-N-cadherin antibody (Fig. 3.4K).
Figure 3.4.

Localization of mXinα and mXinβ in wild-type (Wt), mXinα-null (AKO), mXinβ-null (BKO), and mXinα−/−;mXinβ −/− double knockout (DKO) cardiomyocytes. Immunofluorescence microscopy was performed on cryosections of Wt, AKO, BKO, and DKO hearts with primary antibodies, including U1013 recognizing both mXinα and mXinβ (A, D, G, and J), U1697 specifically recognizing mXinα (B, E, and H), U1040 specifically recognizing mXinβ (C, F, and I), and 3B9 anti-N-cadherin (K). In Wt hearts (A–C), majority of mXinα and mXinβ proteins are localized to the ICDs. A small population of mXinα aggregates into small puncta around cardiomyocytes (A, adult heart and B, P24.5 heart), whereas very little or no mXinβ remains in small puncta (C, P24.5 heart). In AKO heart, both ICD and puncta staining patterns of mXinα are completely abolished (E, adult heart), whereas mXinβ remains localized to the ICDs (D and F, adult heart). In contrast, in the absence of mXinβ in BKO heart, mXinα puncta cannot redistribute to form ICDs (G–I, P16.5–19.5 hearts), as reported previously (Wang et al., 2010, 2013a). Double-label immunofluorescence microscopy on section of DKO heart with U1013 and anti-N-cadherin primary antibodies shows that in the absence of both mXinα and mXinβ (J, P19.5 heart), majority of N-cadherin puncta remain diffusely distributed along the lateral membranes of cardiomyocytes (K, P19.5 heart). Bar =20 μm.
In addition to ICD staining (indicated by arrowheads), U1697 but not U1040 labeled blood vessels (indicated by **) or muscle fibers near blood vessel (indicated by arrows) in the Wt heart sections (Fig. 3.5A and B), suggesting that mXinα but not mXinβ has additional localization associated with blood vessels. In contrast, blood vessels in mXinα-null (AKO) heart were not stained by this U1697 antibody (** in Fig. 3.5C) or by U1013 antibody (data not shown). The cavities of blood vessels in AKO heart were surrounded by β-catenin-positive cells and/or DAPI stained nuclei (** in Fig. 3.5D). In summary, both mXinα and mXinβ in cardiomyocytes have a similar staining pattern: preferentially at ICDs and weakly at T-tubules. However, mXinα but not mXinβ has an additional vessel-associated localization in myocardium. The vessel-associated localization of mXinα in the heart is very similar to the previous findings in various skeletal muscles (Feng et al., 2013) and may be accounted for a significant increase in perivascular fibrosis observed in mXinα-null mice (Otten et al., 2010).
Figure 3.5.

In addition to ICD localization, mXinα but not mXinβ is found in the blood vessel walls and in the cardiomyocytes closely contacted to the vessels. Immunofluorescence microscopy was performed on cryosections of P18.5 (A and C) and adult (B and D) hearts from Wt (A and B) and AKO (C and D) mice with primary antibodies including rabbit U1697 anti-mXinα (in red) and mouse anti-β-catenin antibody (in green). The heart sections were further stained with DAPI for nuclei (in blue). **, cavities of the blood vessels; arrows, contact sites of cardiomyocytes near to the blood vessels containing only mXinα stain; and arrowheads, ICDs containing mXinα and β-catenin stains. Bar=10 μm. These non-ICD localizations of mXinβ could not be detected by staining with U1040 anti-mXinβ specific antibody on Wt heart sections or with U1013 common anti-mXin antibody on AKO heart sections (data not shown).
2.7. Xin expression significantly upregulated in various stressed hearts at early stage and downregulated in failing hearts
In response to various abnormal stresses, hearts initially change their gene expression and exhibit compensatory hypertrophy to preserve pump function and subsequently progress to dilated cardiomyopathy and heart failure. Survey of microarray datasets in Gene Expression Omnibus (GEO, www.ncbi.nlm.nih.gov/sites/GDSbrowser) has revealed that Xinα (Xirp1) and Xinβ (Xirp2) are among those genes changing in response to stresses. In general, a significant upregulation of Xinα and/or Xinβ expression was detected in the early stage of various stresses, which include acute myocardial infarction (GDS2329 and GDS2330), ischemia–reperfusion (I–R) (GDS3662), pressure overload-induced cardiac hypertrophy (GDS2172, GDS2258, GDS2316, GDS3465, and GDS3736), and inflammatory dilated cardiomyopathy (GDS1032, GDS4311, and GDS2154). Conversely, a downregulation of Xinα and Xinβ expression was detected in failing hearts from human patients with diabetic or nondiabetic heart failure (GDS4314), and idiopathic dilated cardiomyopathy and ischemic cardiomyopathy (GDS651).
Time course studies of acute myocardial infarction induced by the ligation of left anterior descending coronary artery showed that both mXinα and mXinβ expressions were rapidly upregulated in the infarcted tissues with a maximum at 12 and 24 h postinfarction, respectively (Harpster et al., 2006). Subsequently, mXinα expression decreased rapidly and reached the basal expression level by 48 h postinfarction, whereas mXinβ expression decreased slowly and was still significantly higher than the basal level at 48 h postinfarction. Ischemic preconditioning (IP) or opiodergic sustained ligand-activated proconditioning (SLP) induces cardioprotection against prolonged ischemia/I–R injury in the hearts (Ashton et al., 2013; Eckle et al., 2006). It has been shown that mouse hearts in response to either IP (GDS3662) or SLP (Ashton et al., 2013) significantly upregulated mXinα expression. Unfortunately, there were no analyses for mXinβ expression in these preconditioning studies. Interestingly, one of genes, Csrnp1, closely linked to the Xirp1/mXinα gene had similar expression profiles in response to acute myocardial infarction or to I–R injury as Xirp1/mXinα did.
In mice, pressure-overloaded stress to the heart by transverse aortic constriction (TAC) causes cardiac hypertrophy initially (compensatory response) and eventually leads to heart failure (decompensation) (Hill et al., 2000; Rockman et al., 1991, 1993). Both messages and proteins of mXinα and mXinβ have been previously reported to be greatly upregulated in the hearts at 3 weeks post-TAC (Wang et al., 2012). The N-cadherin-associated ICDs become larger and thicken. The increased mXin proteins appear to colocalize with N-cadherin to the ICDs (Wang et al., 2012), suggesting that mXin may play a compensatory response important for modulating ICD functions in cardiac hypertrophy. During the compensatory response period (7 days to 30 weeks post-TAC), many microarray analyses also confirmed that Xirp1/mXinα expression was significantly upregulated in TAC hearts from various mouse background (Bisping et al., 2006; Colston et al., 2007; Mirotsou et al., 2006; Smeets et al., 2008). Using mice subjected to banding and debanding of the ascending aorta to mimic aortic stenosis and subsequent aortic valve replacement, microarray analysis also revealed a significant upregulation of Xirp1/mXinα after 4 weeks of banding and subsequent return to normal expression level at 3 days after debanding (Bjornstad et al., 2011).
The upregulation of Xirp2/mXinβ has been also observed in the hypertensive and damaged hearts induced by a potent cardiotoxic hormone, angiotensin II (Ang II), infusion (Duka et al., 2006). This Xirp2 gene (also known as Cmya3, mXinβ, and myomaxin) has been shown to be a direct target of the Mef2a transcription factor and is severely downregulated in Mef2a-null hearts (Huang et al., 2006; Naya et al., 2002). Again, the Xirp2 upregulation induced by Ang II appears to be one of the earliest molecular events. As both mXinα and mXinβ are downstream targets of Mef2a and Mef2c transcription factors (Huang et al., 2006; Lin et al., 2005; Wang et al., 1999), it is likely that mXinα upregulation may also be detected in the Ang II-infused hypertensive hearts. Furthermore, mXinβ hypomorphic mice with only 20% of wild-type mXinβ expression level result in cardiac hypertrophy (McCalmon et al., 2010). Hearts from these hypomorphic mice display less myocardial damages when exposed to Ang II (McCalmon et al., 2010). These results suggest that mXinβ downstream of Ang II signaling pathway can modulate cardiac function in health and disease.
Recent quantitative phosphoproteomic study of pressure-overload heart has revealed a significant increase in the phosphorylation of mXinα at S295 site and S205/S208 sites in the acute TAC (Chang et al., 2013). A significant increase in the phosphorylation of mXinα at these sites was detected as early as at 10 min after TAC. These increases appeared to be transient. By 60 min post-TAC, the phosphorylation levels returned to about 1.3–1.5 times of that in the control. Western blot analyses with phosphopeptide-specific antibodies further confirmed this rapid and transient phosphorylation of mXinα proteins in response to TAC (Chang et al., 2013), which represents another regulatory mechanism underlying TAC-induced cardiac hypertrophy. Sequence comparisons suggest that mXinβ may also have such phosphorylation sites equivalent to mXinα at S295 and S208. Group-based prediction system (GPS) 2.0 (Xue et al., 2008) predicted that Ca2+/calmodulin-activated kinase 2 (CAMK2) and MAPK-activated protein kinase (MAPKAPK) might recognize the phosphorylation site at S295, whereas PKC, CAMK4, and p21-activated kinase 1 (PAK1) could phosphorylate the S208 site. The confirmation of the protein kinases specific for mXin proteins and the significance of mXin phosphorylation in modulating TAC-induced cardiac hypertrophy remain to be determined.
3. ROLES OF XIN PROTEINS IN ICD MATURATION
It has been shown that mXinβ-null hearts fail to form mature ICDs, leading to mislocalization of N-cadherin, mXinα, and other ICD components (Wang et al., 2010, 2013a). On the other hand, mature ICDs form normally in the mXinα-null hearts, but eventually lose close membrane apposition between cardiomyocytes occurring between 1 and 3 months of age, and progressively worsening by older aging (Chan et al., 2011; Gustafson-Wagner et al., 2007; Otten et al., 2010; Wang et al., 2010). These results identify that mXinα and mXinβ are novel regulators of ICD formation, integrity, and then function. The hypothesis is that mXinβ initially establishes the formation of the ICDs and mXinα further stabilizes them.
3.1. mXinβ playing essential roles in the second step of ICD formation
A two-step model for the development of ICD has been proposed in terms of the clustering of N-cadherin to the ICD (Wang et al., 2013a). During mouse embryogenesis, N-cadherin locates uniformly along the periphery of cardiomyocytes of developing heart tube, particularly enriched at the cell–cell contacts (Sinn et al., 2002). The uniform and peripheral localization of N-cadherin and its associated β-catenin in cardiomyocytes continues as heart tube becomes primitive four-chambered heart by septation and valvulogenesis. From embryonic day 15.5 (E15.5) to postnatal day 7.5 (P7.5), N-cadherin molecules through their cis and trans interactions cluster into small puncta localized along the lateral membranes of cardiomyocytes (the first step of ICD formation). Between P7.5 and P13.5, these lateral N-cadherin puncta redistribute into terminal ICD localization (the second step of ICD formation). Further maturation process of ICD including the formation of area composite in mouse takes more than a month. Studies with mXinβ-null hearts have revealed that mXinβ plays an essential role in the second step of ICD formation (Wang et al., 2010, 2013a). As such, mXinβ-null hearts failed to form terminal ICDs, N-cadherin puncta remained along the lateral membrane of cardiomyocytes, and mXinβ-null mice exhibited severe growth retardation and early postnatal lethality (Wang et al., 2010, 2013a). Other ICD components such as mXinα, desmoplakin, and connexin 43 (Cx43) also fail to be restricted to the ICD (termini of cardiomyocytes) in mXinβ-null hearts. Coincidently, a surged expression of mXinβ protein (from 2.17±0.09 pmol/heart at newborn to 28.51±5.73 pmol/heart at P13.5) was detected by quantitative Western blot analysis during this second-step period (Wang et al., 2013a). Confocal microscopy and subcellular fractionation provided further evidence for the preferential association of mXinβ but not mXinα with the forming/formed ICDs (Wang et al., 2013a). These results strongly imply that mXinβ initiates the terminal ICD formation. The timing of mXinβ upregulation in mouse heart is also very similar to that seen in the second peak of upregulation of cXin message (posthatch days 12–14) in chicken heart (Wang et al., 2012). In summary, unique temporal expression profile of mXinβ (Wang et al., 2013a), preferential association of mXinβ with forming ICD (Wang et al., 2013a), as well as failure to form ICDs in mXinβ-null heart (Wang et al., 2010) suggest that mXinβ plays essential roles in ICD formation/maturation during postnatal development. Although the molecular mechanisms underlying ICD formation and maturation remain largely unknown, a biphasic process of ICD maturation in terms of N-cadherin localization has been observed. Accumulated lines of evidence support that mXinβ initiates the formation of ICD by restricting N-cadherin molecules to the termini of cardiomyocytes. This restriction step by mXinβ may be fulfilled by its potential interaction with β-catenin (Choi et al., 2007), its regulation of Rac1 activity (Wang et al., 2010), and its ICD-localized messages (Wang et al., 2012).
3.2. mXinα as an actin-binding and catenin-binding protein linking actin cytoskeleton to adherens junctions of ICDs
As mentioned above, the alternative splicing events give rise to three Xinα variants and at least two Xinβ variants in human and mouse hearts. Amino acid sequence comparisons reveal that 15 and 28 Xin-repeating units are present within all Xinα (except XinC) and Xinβ variants, respectively. The consensus sequence of the Xin repeating unit is GDV(K/Q/R/S) XX(R/K/T)WLFET(Q/R/K/T)PLD (Grosskurth et al., 2008; Lin et al., 2005; Pacholsky et al., 2004). It has been shown that a minimum of three repeats is required for binding to actin filaments (Pacholsky et al., 2004). Thus, Xin repeat proteins should have multiple actin-binding domains/motifs. Similar to nebulin-like repeat proteins, Xin repeat proteins have been shown to bind to actin filaments in two distinct modes, which might provide a mechanism for these actin-binding proteins to stabilize actin filaments (Cherepanova et al., 2006). In addition, recombinant mXinα proteins are shown to be able to bundle actin filaments (Choi et al., 2007). With this actin-binding and -bundling ability, one would expect the in vivo localization of Xin proteins on the thin filaments and the cortical actin filaments in cardiomyocytes. However, as shown in Fig. 3.4 and several previous reports (Grosskurth et al., 2008; Gustafson-Wagner et al., 2007; Otten et al., 2010; Pacholsky et al., 2004; Sinn et al., 2002; Wang et al., 1999, 2010, 2013a), majority of mXinα and mXinβ proteins colocalize with β-catenin to the N-cadherin-mediated adherens junctions of the ICDs. This ICD localization of mXinα has been proved to be likely due to its direct interaction with β-catenin and its β-catenin-binding domain (aa#535–636) overlapping with the Xin repeats (Table 3.1; Choi et al., 2007). Moreover, this possibility is consistent with the fact that the in vitro actin-binding and -bundling activities of mXinα can be further enhanced by the addition of β-catenin (Choi et al., 2007). The results from analyses of stress fiber association of transfected mXinα and its various deletions in nonmuscle CHO cells further suggest that the C-terminal (aa#747–1129) of mXinα plays an inhibitory effect on actin association (Choi et al., 2007). These findings together imply that newly synthesized mXinα may be present in an auto-inhibited state, as far as actin binding is concerned, until the β-catenin-binding domain is occupied by the β-catenin. The binding of mXinα to β-catenin at the adherens junction would then change its conformation into an open state, which would enable subsequent actin binding and bundling. As summarized in Table 3.1, both the Xin repeats and the β-catenin-binding domain are highly conserved in all Xin repeat-containing proteins (Grosskurth et al., 2008). Recently, it has been shown that force-expressed mXinα can suppress p120-induced branching phenotype in CHO cells due to its ability to interact with both p120-catenin and cortactin (Wang et al., 2013b). Cotransfection and co-immunoprecipitation (Co-IP) experiments have revealed that mXinα possesses multiple p120-catenin-binding sites and multiple cortactin-binding sites, some of which appears to be distinct from β-catenin-binding domain. This suggests that mXinα can simultaneously bind to these proteins, particularly for β-catenin and p120-catenin, which are important regulators for the N-cadherin-mediated adhesion (Nelson, 2008; Pokutta and Weis, 2002; Reynolds and Carnahan, 2004). Using yeast two-hybrid screening and cotransfection and Co-IP/pull-down assays, it has been found that many mXinα-interacting proteins, such as cortactin, Mena/VASP, filamin b & c, tropomyosin, gelsolin, vinculin, and α-actinin, are indeed actin regulatory/binding proteins (Choi et al., 2007; Huang et al., 2006; van der Ven et al., 2006; Wang et al., 2013b) (Table 3.1). Therefore, mXinα is an integral component of adherens junction at the ICDs and provides a link between the adherens junction and the underlining actin cytoskeleton, which may stabilize adhesion. Supporting this idea, mXinα-null hearts develop abnormal ICD ultrastructure as early as 3 months of age and exhibit cardiac hypertrophy and cardiomyopathy with conduction defects (Gustafson-Wagner et al., 2007). This structural alteration is accompanied by a disorganization of myofibrils at the ICD and by a significant decrease in the expression of N-cadherin, β-catenin, p120-catenin, and desmoplakin (Gustafson-Wagner et al., 2007), suggesting that hypertrophy may be due to impaired organization of the ICD and instability of cell–cell adhesion.
Table 3.1.
List of known binding domains on Xin proteins and known Xin-interacting proteins
| Interacting protein | Domain/motif mapped or unmapped (sequence homology) | Common or unique | References |
|---|---|---|---|
| Actin filaments (binding and bundling) | The Xin repeat region:
|
Common to both Xinα and Xinβ | Cherepanova et al. (2006), Choi et al. (2007), Grosskurth et al. (2008), Huang et al. (2006), Pacholsky et al. (2004), van der Ven et al. (2006), and Wang et al. (2010) |
| β-Catenin | Highly conserved β-catenin-binding domain (mapped to aa#535–636 of mXinα) (aa#808–905 of mXinβ, unmapped) | May be common to both Xinα and Xinβ | Choi et al. (2007) and Grosskurth et al. (2008) |
| p120-Catenin | Multiple p120-catenin-binding sites on mXinα (the strongest binding site locates in the first half of the Xin repeat region aa#68–371, mXinα1R, as detected by cotransfection and Co-IP from CHO cells) | May be common to both Xinα and Xinβ | Wang et al. (2013b) |
| Cortactin | Multiple cortactin-binding sites on mXinα; one of them locates in aa#1–71 (mXinαNTR), and the strongest binding site locates in the second half of the Xin repeat region aa#364–748, mXinα2R, as detected by cotransfection and Co-IP from CHO cells | ? | Wang et al. (2013b) |
| EVH1 (Ena/VASP homology-1) domain-containing family proteins, including Mena, VASP, and Ena-VASP-like (EVL) protein |
|
May be unique to XIRP1/hXinα and mXinα | Grosskurth et al. (2008) and van der Ven et al. (2006) |
| Filamin c (Ig domain 20, muscle-specific) | Filamin c-interacting region mapped to aa#1685–1843 of XinA large variant from XIRP1/hXinα (aa#1667–1820 of mXinα-a) | Unique to the large variant (XinA) of hXinα and mXinα-a | Grosskurth et al. (2008) and van der Ven et al. (2006) |
| Filamin b (aa#2533–2603) | Interaction with mXinα, detected by yeast two-hybrid screening unmapped | ? | Choi et al. (2007) |
| Tropomyosin (aa#1–284) | Interaction with mXinα, detected in yeast two-hybrid screening unmapped | ? | Choi et al. (2007) |
| Gelsolin (aa#497–780) | Interaction with mXinα, detected in yeast two-hybrid screening unmapped | ? | Choi et al. (2007) |
| Vinculin (aa#1–1066) | Interaction with mXinα, detected in yeast two-hybrid assay unmapped | ? | Choi et al. (2007) |
| α-Actinin | Multiple α-actinin-interacting fragments (aa#334–680 and #1340–1644) of myomaxin (mXinβ), as detected by cotransfection and Co-IP from COS cells | ? | Huang et al. (2006) |
| KChIP2 (potassium channel-interacting protein 2) | Interaction with mXinα, detected by yeast two-hybrid assay unmapped | ? | Chan et al. (2011) |
3.3. N-cadherin-mediated adherens junction is the primary determinant of ICD’s structural integrity
During heart development, redistributions of both N-cadherin (adherens junctions) and desmoplakin (desmosomes) puncta to cell termini (the second step of ICD formation) appear to occur at very similar time frame but earlier than that of Cx43 (gap junctions) (Angst et al., 1997; Hirschy et al., 2006; Perriard et al., 2003; Pieperhoff and Franke, 2007; Sinn et al., 2002). Although developmental temporal studies did not differentiate the assembly of adherens junctions and desmosomes to the ICDs, recent studies with cardiac-specific conditional knockout of N-cadherin (N-cad CKO) revealed that loss of N-cadherin led to total disassembly of ICDs, that is, not only adherens junctions but also desmosomes and gap junctions, and consequently, mice exhibit ventricular tachyarrhythmias and sudden death (Kostetskii et al., 2005; Li et al., 2005, 2008). These results suggest that N-cadherin-mediated adherens junction is the primary organizer responsible for maintaining the ICD structural integrity and function. Molecular mechanisms by which adherens junction/N-cadherin organizes the redistribution of intercellular junctions into maturing ICD remain unclear. It has been recently shown that mXinβ-null hearts fail to redistribute the N-cadherin and mXinα puncta to the cell’s termini to form mature ICDs (Wang et al., 2010). Similar to N-cad CKO hearts, the redistributions of both desmoplakin (desmosome) and Cx43 (gap junction) to form terminal ICDs also fail to occur in mXinβ-null hearts (Wang et al., 2013a). These results again support that N-cadherin-mediated adherens junction is a primary determinant of ICD structure, since mXinβ colocalizes with N-cadherin/β-catenin and possesses a highly conserved β-catenin-binding domain in the Xin repeat region (Grosskurth et al., 2008; Wang et al., 2010). The mXinβ-null hearts provide another opportunity to test whether the failure to restrict intercellular junctions to the cell’s termini could be a result of disrupted association between intercellular junction components. The results from double-label immunofluorescence staining to determine the relationships between N-cadherin and desmoplakin/Cx43 have revealed that intercellular junction components retain their close relationship in mXinβ-null hearts despite being mislocalized (Wang et al., 2013a). Therefore, mXinβ does not appear to be critical for these types of associations among intercellular junction components. In fact, accumulated lines of evidence suggest that linker molecules such as plakophilin 2 and ZO-1 may play cross-linking roles between different intercellular junctions in the heart (Wang et al., 2012).
3.4. Hierarchy of mXin proteins playing important roles in ICD structure and function
Mice without mXinα are viable and fertile, but eventually develop ICD ultrastructure defect and late-onset cardiomyopathy with conduction defects, despite a normal appearance of ICD at young (juvenile) ages (Chan et al., 2011; Gustafson-Wagner et al., 2007; Lai et al., 2007, 2008). An upregulation of mXinβ at both message and protein levels has been detected in mXinα-deficient hearts, suggesting a partial compensation for the loss of mXinα (Gustafson-Wagner et al., 2007). In contrast, complete loss of mXinβ in the heart results in the failure of forming ICD, severe growth retardation, diastolic dysfunction, ventricular septal defect, and early postnatal lethality (Wang et al., 2010). The mXinβ-null cardiomyocytes exhibit mislocalization of N-cadherin and mXinα, whereas the mXinα-null cardiomyocytes appear to have normal ICD localization of N-cadherin and mXinβ (Wang et al., 2010). As discussed above, the mXinβ plays an initiation role in the second step (redistribution of intercellular junction components) of the ICD formation, whereas the mXinα may play a stabilizing role for ICD structure integrity. As a consequence of the failure of forming ICD, mXinβ-null hearts show misaligned cardiomyocytes/myofibers, a significant reduction in the area of left ventricle compact myocardium and a trend of increase in the trabecular area (i.e., noncompaction phenotype) (Wang et al., 2010). In addition, misaligned mXinβ-null cardiomyocytes alter the engagement and clustering of N-cadherins (i.e., N-cadherin-mediated adhesion signaling), leading to upregulation of Stat3 activity and downregulation of surface receptor (IGF-IR and IL6Rα) and growth-related kinase (Akt and Erk1/2) activity (Wang et al., 2010). These findings not only consist with a critical role for N-cadherin-mediated adhesion in ICD integrity and cardiac function (Li et al., 2006) but also explain severe growth retardation and diastolic dysfunction phenotypes of mXinβ-null mice. The misaligned mXinβ-null cardiomyocytes during cardiac morphogenesis also likely develop into hearts with abnormal shape and/or ventricular septal defects. This chamber-defect phenotype in mXinβ-null hearts is also predicted from the phylogenetic studies showing that the evolving of Xin coincides with the occurrence of true chambered heart (Grosskurth et al., 2008).
Mislocalization of mXinα in mXinβ-null hearts and upregulation of mXinβ in mXinα-deficient hearts have been readily detected; however, the reversals have not been observed, suggesting a functional hierarchy between mXinα and mXinβ. If mXinα and mXinβ act in parallel in the same pathway, double knockout (DKO) mice should show more severe phenotypes than mXinβ single knockout (BKO) or mXinα single knockout (AKO). On the other hand, if they act in series, DKO may have similar phenotype as BKO. It has been found that the loss of both mXinα and mXinβ results in postnatal lethality and ICD defects (Fig. 3.4) indistinguishable from that caused by the loss of mXinβ alone (Wang et al., 2013a). Thus, mXin proteins act in series; that is, mXinβ initiates the second step of ICD formation and mXinα further stabilizes them. Molecular mechanisms how mXinα and mXinβ collaborate together toward the ICD maturation and function remain unclear. Many important questions remain to be answered such as whether mXinα and mXinβ can interact directly or indirectly and what the other mXinα- and mXinβ-interacting proteins are.
4. ROLES OF XIN PROTEINS IN ION-CHANNEL SURFACE EXPRESSION
As a functional unit, the ICD plays important roles in organizing and regulating surface receptor and ion-channel activities. As an example, mXinβ-null hearts fail to form the ICD and show downregulation of surface receptor IGF-1R and IL-6Rα activity (Wang et al., 2010). Accumulated lines of evidence suggest that ICD, through scaffolding/interacting/anchoring proteins, can spatially organize and maintain key ion-channel assemblies required for controlling the cardiac action potential. Defects in these processes can lead to arrhythmias and cardiac sudden death. Table 3.2 summarizes known ICD-associated channel subunits and their known interacting/scaffolding/anchoring proteins. Interestingly, synapse-associated protein 97 (SAP97), a member of membrane-associated guanylate kinase family of proteins, is preferentially localized to ICDs (Abriel, 2010; Abriel and Kass, 2005; Zimmer and Surber, 2008). Through its PDZ (shared domain first found in PSD95, Drosophila disc large tumor suppressor and ZO-1) domain, SAP97 interacts with the three last amino acid residues of Nav1.5 (Petitprez et al., 2011), Kv1.5 (Abi-Char et al., 2008; Murata et al., 2001), and Kv4.2/4.3 (El-Haou et al., 2009). Portions of the ion channels (INa, Ik,slow1, and Ito,f) assembled from these α-subunits are known to be ICD associated (Barry et al., 1995; Cheng et al., 2011; Kucera et al., 2002; Murata et al., 2001). Thus, it is likely that SAP97 plays partly anchoring role for these channel assemblies to the ICD. In this regard, it has been shown that SAP97 but not PSD97 or ZO-1 interacts with Nav1.5C-terminal SIV residues and is responsible for anchoring the pool of Nav1.5 channel at the ICD (Petitprez et al., 2011). Another scaffolding protein, ankyrin-G, has been shown to interact with Nav1.5, and this interaction is required for targeting Nav1.5 to the ICD and T-tubules of cardiomyocytes. Human Nav1.5 E1053K missense mutation disrupting this interaction leads to Brugada syndrome (Mohler et al., 2004). Conversely, ankyrin-G-deficient cardiomyocytes show reduced Nav1.5 surface expression and localization as well as reduced INa current density (Lowe et al., 2008). In addition to ankyrin-G, many sodium channel-interacting proteins, such as caveolin-3 (Vatta et al., 2006), α1-syntrophins (Cheng et al., 2009; Ueda et al., 2008), Nedd4-2 (Na+ channel degradation machinery protein) (Abriel and Kass, 2005), as well as GPD1-L (Na+ channel trafficking protein) (London et al., 2007; Van Norstrand et al., 2007; Weiss et al., 2002), are shown to regulate/influence Na+channel surface expression. Mutations in these proteins lead to sudden cardiac death syndrome, long QT syndrome, and Brugada syndrome.
Table 3.2.
List of known ICD-associated channel subunits and their interacting/scaffolding proteins
| Interacting/scaffolding protein | ICD-associated channel subunits | ||
|---|---|---|---|
| Nav1.5 (INa) | Kv1.5 (Ik,slow1) | Kv4.2/4.3 (Ito,f) | |
| SAP97 (synapse-associated protein 97) preferentially localizes to ICD and some T-tubules and plasma membrane (PM) A member of the MAGUK (membrane-associated guanylate kinase) family of proteins (Abriel, 2010; Abriel and Kass, 2005; Zimmer and Surber, 2008) | Nav1.5C-terminal SIV interacts with PDZ of SAP97 but not PSD95 or ZO1 SAP97 is responsible for anchoring the pool of Nav1.5 channels at ICD SAP97 colocalizes with Nav1.5 (Kucera et al., 2002; Petitprez et al., 2011; Verkerk et al., 2007) |
Kv1.5C-terminal TDV interacts with PDZ of SAP97 (Abi-Char et al., 2008; Cheng et al., 2011; Murata et al., 2001) | Kv4.2/4.3C-terminal SAL interacts with PDZ of SAP97 Kv4.x-SAP97-CaMKII complex is detected by pull-down and Co-IP SAP97 colocalizes with Kv4.2/4.3 (Barry et al., 1995; Chan et al., 2011; El-Haou et al., 2009) |
| Ankyrin-G localizes to ICD and T-tubules (lateral membranes) (Lowe et al., 2008; Mohler et al., 2004) | Ankyrin-G interacts with Nav1.5 (ankyrin-binding motif ) Missense mutation E1053K in the ankyrin-G binding domain of SCN5A (Nav1.5) causes Brugada syndrome |
||
| Cortactin | Cortactin Co-IP and colocalizes with Kv1.5 and Kcne2 (Cheng et al., 2011) | ||
| mXinα | The mXinα-null cardiomyocytes show drastic loss of the ICD-associated cortactin Pull-down, cotransfection, and Co-IP show mXinα interacts with cortactin (Wang et al., 2013b) | Yeast two-hybrid assay show mXinα interacts with KChIP2 and filamin (Chan et al., 2011) | |
Defective adhering junctions commonly observed in human patients and animal model hearts with mutations in adherens junctional components or desmosomal components generally lead to gap junction remodeling, that is, reduced Cx43 expression level and altered Cx43 localization (Wang et al., 2012). The gap junction remodeling has also been detected in mXinα-deficient hearts, which may partially explain the slower conduction phenotype seen in these mutant hearts (Gustafson-Wagner et al., 2007; Lai et al., 2008). In addition, whole-cell patch-clamp studies of cardiomyocytes prepared from juvenile (1-month-old) wild-type and mXinα-null hearts have revealed significant decreases in the transient K+ outward (Ito,f) and the delayed K+ rectifier (Ik,slow1) current density as well as prolonged action potential duration with high incidence of early afterdepolarization (EAD) (Chan et al., 2011). As no ICD structure defect can be detected in these juvenile mutant hearts, alterations in electrophysiological properties of the K+ outward channels would effectively lead to conduction defect in mXinα-deficient hearts. Studies of molecular mechanisms underlying these alterations further identify that mXinα and possibly mXinβ as scaffolding proteins together with their interacting proteins may be responsible for surface expression of the ICD-localized channels such as Ik,slow1, Ito,f, and possibly INa (Table 3.2).
4.1. mXinα via its interactions with K+ channel-interacting protein 2 (KChIP2) and filamin regulates surface expression of the transient K+ outward (Ito,f) channel
In many animal models of cardiac hypertrophy/cardiomyopathy and human heart failure, hypertrophied myocytes undergo K+ channel remodeling that causes a prolongation in action potential duration (Furukawa and Kurokawa, 2006; Knollmann et al., 2000; Mitarai et al., 2000; Sanguinetti, 2002; Tomaselli and Marban, 1999). A common target of K+ channel alteration is the depression of Ito in ventricular myocytes. Although Ito is known to consist of two components (Ito,f and Ito,s) (Brahmajothi et al., 1999), the Ito,f is a target of remodeling in these hypertrophied models (Furukawa and Kurokawa, 2006). In mice, the Ito,f channels reflect the assembly of Kv4.2/4.3 α-subunit, Kvβ1, and KChIP2 ancillary subunit (Fig. 3.6) (Guo et al., 2002; Nerbonne and Kass, 2005). The Kv4 pore-forming α-subunit is known to mediate the α-subunit interaction (Choe and Roosild, 2002) and the bindings of filamin (Petrecca et al., 2000), Kvβ1 (Nerbonne and Kass, 2005), and KChIP2 (An et al., 2000; Nerbonne and Kass, 2005). The association of KChIP2 with Kv4.2 greatly enhances the surface expression of Kv4.2 (Shibata et al., 2003), although the molecular mechanisms remain unclear. Furthermore, KChIP2-null hearts show a complete loss of Ito current, suggesting that KChIP2 is a major determinant for Ito,f current density (Kuo et al., 2001). As a consequence, the mutant mice become more susceptible to ventricular tachycardia (Kuo et al., 2001). It has been shown that mXinα can interact with KChIP2 and filamin (Choi et al., 2007), providing a novel mechanism to regulate surface expression of Ito,f channel. Subcellular fractionation studies have revealed that both KChIP2 and filamin associated with the membrane fraction of juvenile mXinα-null heart are significantly downregulated, as compared to that from both wild-type and heterozygous hearts (Chan et al., 2011). Although the Kv4.2 and Kv4.3 in this total membrane fraction show slightly or no reduction, a significant depression in the Ito current density from juvenile mXinα-null ventricular myocytes is readily detected (Chan et al., 2011). These results suggest that mXinα helps to recruit or/and stabilize Ito,f channel components through the interactions of mXinα with both KChIP2 and filamin.
Figure 3.6.

Proposed roles of mXinα in the structure and function at the ICD. Mouse hearts deficient in mXinα exhibit progressive ultrastructural defects in the ICDs and late-onset cardiac hypertrophy and cardiomyopathy with conduction defects (Chan et al., 2011; Gustafson-Wagner et al., 2007; Lai et al., 2008; Otten et al., 2010; Wang et al., 2013b). Through its interactions with catenins (p120-catenin and β-catenin) and many actin-binding proteins, mXinα could modulate N-cadherin-mediated adhesion for ICD structural integrity. Through its interactions with KChIP2, cortactin and Kv1.5, mXinα could influence surface expression of Ito,f (transient K+ outward current) and Ik,slow1 (delayed K+ rectifier) channels. Schematic diagram illustrating the molecular assembly of Ito,f and Ik,slow1 was adapted from previous review articles (Abbott et al., 2007; McCrossan and Abbott, 2004; Nerbonne and Kass, 2005).
4.2. mXinα via its interaction with cortactin influences surface expression of the delayed K+ rectifier (Ik,slow1) channel
Recent studies with cardiac-specific N-cadherin conditional knockout (N-cad CKO) mice have provided strong evidence for an essential role of N-cadherin in ICD integrity, cardiac conduction, and cardiac rhythms (Cheng et al., 2011; Kostetskii et al., 2005; Li et al., 2005, 2008). N-cad-CKO mouse hearts show complete dissolution of the ICD structure, resulting in gap junction remodeling and slow conduction of ventricles. The mutant mice are more susceptible to arrhythmias and cardiac sudden death. Similar to mXinα-null ventricular myocytes, cells prepared from N-cad CKO hearts also display prolonged action potential duration, higher incidence of EAD, and depressed Ik,slow1 current density (Cheng et al., 2011). Co-IP and colocalization experiments have revealed that cortactin associates with α-subunit Kv1.5 and accessory subunit Kcne2 of Ik,slow1 (Cheng et al., 2011). Cortactin is an actin-binding protein capable of interacting with other actin-associated proteins and influencing the organization of membrane cortical actin network (Ammer and Weed, 2008). It has been shown that the N-terminus of cortactin interacts and cooperates with p120-catenin to regulate lamellipodial dynamics and adhesion (Boguslavsky et al., 2007). These abilities of cortactin together with its associations with Ik,slow1 channel components, Kv1.5 and Kcne2, may render this protein a critical modulator for Ik,slow1 channel surface expression. However, since N-cadherin in the adult heart is exclusively localized to the ICDs, question as to why the loss of N-cadherin leads to a global reduction of cortactin at both ICD and lateral membranes remains to be answered. Further Co-IP experiments with anti-cortactin or anti-N-cadherin have not detected a direct association between cortactin and N-cadherin (Cheng et al., 2011). It is known that mXinα interacts not only with cortactin (Wang et al., 2013b) but also with p120-catenin (Wang et al., 2013b) and β-catenin (Choi et al., 2007), suggesting that mXinα may be required for the association of Kv1.5, cortactin, and N-cadherin. Supporting this idea, the ICD-localized fraction of cortactin has been found to be largely diminished in mXinα-null cardiomyocytes, as compared to that in wild-type cardiomyocytes (Wang et al., 2013b). Preliminary results from pull-down and cotransfection and Co-IP experiments showed that mXinα contained multiple cortactin-binding sites (Table 3.1), some of which were distinguished from β-catenin-binding site (Choi et al., 2007) and p120-catenin-binding sites (Wang et al., 2013b). These results may imply that mXinα could simultaneously bind to these proteins and play regulatory roles in N-cadherin-mediated adhesion and in surface expression of Ik,slow1 channel. Figure 3.6 shows a schematic diagram of mXinα at the ICD, regulating the N-cadherin-mediated adhesion, the transient outward K+ channel (Ito,f) activity and the delayed rectifier K+ channel (Ik,slow1) activity.
5. CONCLUDING REMARKS
The ICDs are the essential structures unique to cardiac muscle. Disruption of ICD structure and function is an important hallmark of many congenital and acquired heart diseases, including cardiomyopathy, arrhythmias, and heart failure. Several lines of evidence discussed in this review suggest that the involvement of ICD-associated, Xin repeat-containing protein family, mXinα (Xirp1 or CMYA1) and mXinβ (Xirp2 or CMYA3), in linking and transducing signals important for cardiac remodeling in either healthy or diseased state. The upregulation of mXinβ, progressively developing ICD structural defect in mXinα-deficient hearts, and the failure of ICD formation in mXinβ-null but not mXinα-null hearts infer that there exists a functional hierarchy between these Xin proteins. Whether mXinα and mXinβ can directly or indirectly interact to carry out their functions remain to be determined. Despite its importance in initiating the formation of terminal ICD, the question as to whether mXinβ is also important for maintaining mature ICDs remains to be answered by characterizing cardiac-specific inducible mXinβ knockout. In addition to roles of mXinα in linking N-cadherin-mediated adhesion to the underlining actin network and in regulating surface ion-channel expression, how Xin proteins help ICDs carrying out their functions in the physiology and pathology of the heart remain largely unclear. Toward this long-term goal, electrophysiologically characterizing wild-type and Xin knockout cardiomyocytes, identifying and characterizing more Xin-interacting proteins, as well as defining their binding domains on Xin proteins should be continuously carried out.
Using statistical methods for gene expression analysis to identify potential drug targets for cardiomyopathy, Walker (2001) has identified 5 genes coexpressed with the 13 known cardiomyopathy-associated (CMYA) genes (Walker, 2001). Two of these genes are human orthologs, CMYA1 and CMYA3, of mXinα and mXinβ, respectively. The CMYA1 is mapped to chromosome 3p22.2, which is near the loci for dilated cardiomyopathy with conduction defect 2 (Olson and Keating, 1996). The mXinα-deficient mice exhibit similar cardiomyopathy with conduction defect phenotypes (Gustafson-Wagner et al., 2007). Both left atrial-pulmonary vein (LA-PV) (Lai et al., 2008) and ventricle (Lai et al., 2007) preparations from mXinα-null hearts show a slower conduction velocity and many areas of conduction block. Furthermore, the induction of atrial fibrillation is attenuated in mXinα-null LA-PV preparations even under conditions that enhance its induction in wild-type preparations (Lai et al., 2008). However, the automatic and triggered rhythms are not suppressed in mXinα-null preparations. Therefore, mXinα-deficient mice should provide a good model to study the mechanisms of arrhythmias and to identify alternate therapeutic approaches. Recent preliminary screening may have identified missense mutations on the CMYA1 gene in human patients with left ventricular noncompaction cardiomyopathy. The ventricular noncompaction phenotypes have been also detected in E14.5 and newborn mXinβ-null hearts, as compared to age-matched wild-type hearts (Wang et al., 2010). The existence of a functional hierarchy between mXinα and mXinβ, particularly the fact that mislocalization of mXinα is readily detected in mXinβ-null hearts, should support the possibility that through its defective but uncharacterized binding domain, missense mutations of mXinα may lead to mislocalization of mXinα and other ICD components to the ICDs and consequently the noncompaction phenotype. The CMYA3 gene is mapped to 2q24.3 and human patients with monosomy 2q24 also exhibit low birth weight, severe growth retardation, and congenital heart defects (www.orpha.net/data/patho/GB/uk-2q24.pdf). Also, a large Kyrgyz family with premature hypertension phenotype has been linked to chromosome 2q24.3–31.3 region (Kalmyrzaev et al., 2006). The mXinβ upregulation in Ang II-induced hypertension and cardiac damage (Duka et al., 2006) further supports this linkage between mXinβ and hypertension. Studies with mXinβ hypomorhic mice have concluded that mXinβ plays an important role in modulating Ang II signaling pathway for cardiac function in healthy and diseased hearts (McCalmon et al., 2010). The mXinβ has been shown to be essential for the postnatal formation of ICDs and for the localization of mXinα to the ICDs (Wang et al., 2010, 2013a), whereas the mXinα interacts not only with cortactin (Wang et al., 2013b), which co-immunoprecipitates with Kv1.5 (Cheng et al., 2011), but also with KChIP2 and filamin, which are associated with Kv4.2/4.3 (Chan et al., 2011). Based on these converging lines of evidence, a compelling hypothesis is that mXinβ also plays a role in cardiac electrophysiology. Further studies are warranted to explore the roles of mXinβ in surface expression of ion channels.
Acknowledgments
This work was supported by Grants HL42266 (a SCOR in congenital heart disease), HL075015, and HL107383 to J. J. C. L., and DE023575 to A. J. E. from National Institutes of Health, USA; a grant to J. J. C. L. from the American Heart Association, USA; and Grants NSC96-2320-B016-013 and NSC98-2320-B016-00-MY3 to C. I. L. from National Science Council, Taipei, Taiwan, ROC.
References
- Abbott GW, Xu X, Roepke TK. Impact of ancillary subunits on ventricular repolarization. J Electrocardiol. 2007;40:S42–S46. doi: 10.1016/j.jelectrocard.2007.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abi-Char J, El-Haou S, Balse E, Neyroud N, Vranckx R, Coulombe A, Hatem SN. The anchoring protein SAP97 retains Kv1.5 channels in the plasma membrane of cardiac myocytes. Am J Physiol Heart Circ Physiol. 2008;294:H1851–H1861. doi: 10.1152/ajpheart.01045.2007. [DOI] [PubMed] [Google Scholar]
- Abriel H. Cardiac sodium channel Na(v)1.5 and interacting proteins: physiology and pathophysiology. J Mol Cell Cardiol. 2010;48:2–11. doi: 10.1016/j.yjmcc.2009.08.025. [DOI] [PubMed] [Google Scholar]
- Abriel H, Kass RS. Regulation of the voltage-gated cardiac sodium channel Nav1.5 by interacting proteins. Trends Cardiovasc Med. 2005;15:35–40. doi: 10.1016/j.tcm.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Ammer AG, Weed SA. Cortactin branches out: roles in regulating protrusive actin dynamics. Cell Motil Cytoskeleton. 2008;65:687–707. doi: 10.1002/cm.20296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An WF, Bowlby MR, Betty M, Cao J, Ling HP, Mendoza G, Hinson JW, Mattsson KI, Strassle BW, Trimmer JS, Rhodes KJ. Modulation of A-type potassium channels by a family of calcium sensors. Nature. 2000;403:553–556. doi: 10.1038/35000592. [DOI] [PubMed] [Google Scholar]
- Angst BD, Khan LU, Severs NJ, Whitely K, Rothery S, Thompson RP, Magee AI, Gourdie RG. Dissociated spatial patterning of gap junctions and cell adhesion junctions during postnatal differentiation of ventricular myocardium. Circ Res. 1997;80:88–94. doi: 10.1161/01.res.80.1.88. [DOI] [PubMed] [Google Scholar]
- Ashton KJ, Tupicoff A, Williams-Pritchard G, Kiessling CJ, See Hoe LE, Headrick JP, Peart JN. Unique transcriptional profile of sustained ligand-activated preconditioning in pre- and post-ischemic myocardium. PLoS One. 2013;8:e72278. doi: 10.1371/journal.pone.0072278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry DM, Trimmer JS, Merlie JP, Nerbonne JM. Differential expression of voltage-gated K+channel subunits in adult rat heart. Relation to functional K+channels? Circ Res. 1995;77:361–369. doi: 10.1161/01.res.77.2.361. [DOI] [PubMed] [Google Scholar]
- Bisping E, Ikeda S, Kong SW, Tarnavski O, Bodyak N, McMullen JR, Rajagopal S, Son JK, Ma Q, Springer Z, Kang PM, Izumo S, Pu WT. Gata4 is required for maintenance of postnatal cardiac function and protection from pressure overload-induced heart failure. Proc Natl Acad Sci U S A. 2006;103:14471–14476. doi: 10.1073/pnas.0602543103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornstad JL, Sjaastad I, Nygard S, Hasic A, Ahmed MS, Attramadal H, Finsen AV, Christensen G, Tonnessen T. Collagen isoform shift during the early phase of reverse left ventricular remodelling after relief of pressure overload. Eur Heart J. 2011;32:236–245. doi: 10.1093/eurheartj/ehq166. [DOI] [PubMed] [Google Scholar]
- Boguslavsky S, Grosheva I, Landau E, Shtutman M, Cohen M, Arnold K, Feinstein E, Geiger B, Bershadsky A. p120 catenin regulates lamellipodial dynamics and cell adhesion in cooperation with cortactin. Proc Natl Acad Sci U S A. 2007;104:10882–10887. doi: 10.1073/pnas.0702731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrmann CM, Grund C, Kuhn C, Hofmann I, Pieperhoff S, Franke WW. The area composita of adhering junctions connecting heart muscle cells of vertebrates. II Colocalizations of desmosomal and fascia adhaerens molecules in the intercalated disk. Eur J Cell Biol. 2006;85:469–485. doi: 10.1016/j.ejcb.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Brahmajothi MV, Campbell DL, Rasmusson RL, Morales MJ, Trimmer JS, Nerbonne JM, Strauss HC. Distinct transient outward potassium current (Ito) phenotypes and distribution of fast-inactivating potassium channel alpha subunits in ferret left ventricular myocytes. J Gen Physiol. 1999;113:581–600. doi: 10.1085/jgp.113.4.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan FC, Cheng CP, Wu KH, Chen YC, Hsu CH, Gustafson-Wagner EA, Lin JL, Wang Q, Lin JJ, Lin CI. Intercalated disc-associated protein, mXin-alpha, influences surface expression of ITO currents in ventricular myocytes. Front Biosci (Elite Ed) 2011;3:1425–1442. doi: 10.2741/e344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YW, Chang YT, Wang Q, Lin JJ, Chen YJ, Chen CC. Quantitative phosphoproteomic study of pressure-overloaded mouse heart reveals dynamin-related protein 1 as a modulator of cardiac hypertrophy. Mol Cell Proteomics. 2013;12:3094–3107. doi: 10.1074/mcp.M113.027649. http://dx.doi.org/10.1074/mcp.M113.027649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Van Norstrand DW, Medeiros-Domingo A, Valdivia C, Tan BH, Ye B, Kroboth S, Vatta M, Tester DJ, January CT, Makielski JC, Ackerman MJ. Alpha1-syntrophin mutations identified in sudden infant death syndrome cause an increase in late cardiac sodium current. Circ Arrhythm Electrophysiol. 2009;2:667–676. doi: 10.1161/CIRCEP.109.891440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Yung A, Covarrubias M, Radice GL. Cortactin is required for N-cadherin regulation of Kv1.5 channel function. J Biol Chem. 2011;286:20478–20489. doi: 10.1074/jbc.M111.218560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanova O, Orlova A, Galkin VE, van der Ven PF, Furst DO, Jin JP, Egelman EH. Xin-repeats and nebulin-like repeats bind to F-actin in a similar manner. J Mol Biol. 2006;356:714–723. doi: 10.1016/j.jmb.2005.11.082. [DOI] [PubMed] [Google Scholar]
- Choe S, Roosild T. Regulation of the K channels by cytoplasmic domains. Biopolymers. 2002;66:294–299. doi: 10.1002/bip.10309. [DOI] [PubMed] [Google Scholar]
- Choi S, Gustafson-Wagner EA, Wang Q, Harlan SM, Sinn HW, Lin JL, Lin JJ. The intercalated disc protein, mXinα, is capable of interacting with β-catenin and bundling actin filaments. J Biol Chem. 2007;282:36024–36036. doi: 10.1074/jbc.M707639200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colston JT, Boylston WH, Feldman MD, Jenkinson CP, de la Rosa SD, Barton A, Trevino RJ, Freeman GL, Chandrasekar B. Interleukin-18 knockout mice display maladaptive cardiac hypertrophy in response to pressure overload. Biochem Biophys Res Commun. 2007;354:552–558. doi: 10.1016/j.bbrc.2007.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmar M, McKenna WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res. 2010;107:700–714. doi: 10.1161/CIRCRESAHA.110.223412. [DOI] [PubMed] [Google Scholar]
- Duka A, Schwartz F, Duka I, Johns C, Melista E, Gavras I, Gavras H. A novel gene (Cmya3) induced in the heart by angiotensin II-dependent but not salt-dependent hypertension in mice. Am J Hypertens. 2006;19:275–281. doi: 10.1016/j.amjhyper.2005.08.017. [DOI] [PubMed] [Google Scholar]
- Dupont E, Matsushita T, Kaba RA, Vozzi C, Coppen SR, Khan N, Kaprielian R, Yacoub MH, Severs NJ. Altered connexin expression in human congestive heart failure. J Mol Cell Cardiol. 2001;33:359–371. doi: 10.1006/jmcc.2000.1308. [DOI] [PubMed] [Google Scholar]
- Echt DS, Liebson PR, Mitchell LB, Peters RW, Obias-Manno D, Barker AH, Arensberg D, Baker A, Friedman L, Greene HL, et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N Engl J Med. 1991;324:781–788. doi: 10.1056/NEJM199103213241201. [DOI] [PubMed] [Google Scholar]
- Eckle T, Grenz A, Kohler D, Redel A, Falk M, Rolauffs B, Osswald H, Kehl F, Eltzschig HK. Systematic evaluation of a novel model for cardiac ischemic preconditioning in mice. Am J Physiol Heart Circ Physiol. 2006;291:H2533–H2540. doi: 10.1152/ajpheart.00472.2006. [DOI] [PubMed] [Google Scholar]
- El-Haou S, Balse E, Neyroud N, Dilanian G, Gavillet B, Abriel H, Coulombe A, Jeromin A, Hatem SN. Kv4 potassium channels form a tripartite complex with the anchoring protein SAP97 and CaMKII in cardiac myocytes. Circ Res. 2009;104:758–769. doi: 10.1161/CIRCRESAHA.108.191007. [DOI] [PubMed] [Google Scholar]
- Estigoy CB, Ponten F, Odeberg J, Herbert B, Guilhuas M, Charleston M, Ho JWK, Cameron D, dos Remedies CG. Intercalated discs: multiple proteins perform multiple functions in non-failing and failing human hearts. Biophys Rev. 2009;1:43–49. doi: 10.1007/s12551-008-0007-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng HZ, Wang Q, Reiter RS, Lin JL, Lin JJ, Jin JP. Localization and function of Xinalpha in mouse skeletal muscle. Am J Physiol Cell Physiol. 2013;304:C1002–C1012. doi: 10.1152/ajpcell.00005.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes MS, Sperelakis N. Intercalated discs of mammalian heart: a review of structure and function. Tissue Cell. 1985;17:605–648. doi: 10.1016/0040-8166(85)90001-1. [DOI] [PubMed] [Google Scholar]
- Franke WW, Borrmann CM, Grund C, Pieperhoff S. The area composita of adhering junctions connecting heart muscle cells of vertebrates. I Molecular definition in intercalated disks of cardiomyocytes by immunoelectron microscopy of desmosomal proteins. Eur J Cell Biol. 2006;85:69–82. doi: 10.1016/j.ejcb.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Furukawa T, Kurokawa J. Potassium channel remodeling in cardiac hypertrophy. J Mol Cell Cardiol. 2006;41:753–761. doi: 10.1016/j.yjmcc.2006.07.021. [DOI] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB American Heart Association Statistics Committee Stroke Statistics Subcommittee. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosskurth SE, Bhattacharya D, Wang Q, Lin JJ. Emergence of Xin demarcates a key innovation in heart evolution. PLoS One. 2008;3:e2857. doi: 10.1371/journal.pone.0002857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Li H, Aimond F, Johns DC, Rhodes KJ, Trimmer JS, Nerbonne JM. Role of heteromultimers in the generation of myocardial transient outward K+ currents. Circ Res. 2002;90:586–593. doi: 10.1161/01.res.0000012664.05949.e0. [DOI] [PubMed] [Google Scholar]
- Gustafson-Wagner EA, Sinn HW, Chen YL, Wang DZ, Reiter RS, Lin JL, Yang B, Williamson RA, Chen J, Lin CI, Lin JJ. Loss of mXinα, an intercalated disk protein, results in cardiac hypertrophy and cardiomyopathy with conduction defects. Am J Physiol Heart Circ Physiol. 2007;293:H2680–H2692. doi: 10.1152/ajpheart.00806.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harpster MH, Bandyopadhyay S, Thomas DP, Ivanov PS, Keele JA, Pineguina N, Gao B, Amarendran V, Gomelsky M, McCormick RJ, Stayton MM. Earliest changes in the left ventricular transcriptome postmyocardial infarction. Mamm Genome. 2006;17:701–715. doi: 10.1007/s00335-005-0120-1. [DOI] [PubMed] [Google Scholar]
- Hill JA, Karimi M, Kutschke W, Davisson RL, Zimmerman K, Wang Z, Kerber RE, Weiss RM. Cardiac hypertrophy is not a required compensatory response to short-term pressure overload. Circulation. 2000;101:2863–2869. doi: 10.1161/01.cir.101.24.2863. [DOI] [PubMed] [Google Scholar]
- Hirschy A, Schatzmann F, Ehler E, Perriard JC. Establishment of cardiac cytoarchitecture in the developing mouse heart. Dev Biol. 2006;289:430–441. doi: 10.1016/j.ydbio.2005.10.046. [DOI] [PubMed] [Google Scholar]
- Huang HT, Brand OM, Mathew M, Ignatiou C, Ewen EP, McCalmon SA, Naya FJ. Myomaxin is a novel transcriptional target of MEF2A that encodes a Xin-related alpha-actinin-interacting protein. J Biol Chem. 2006;281:39370–39379. doi: 10.1074/jbc.M603244200. [DOI] [PubMed] [Google Scholar]
- Kalmyrzaev B, Aldashev A, Khalmatov M, Polupanov A, Jumagulova A, Mamanova L, Wilkins MR, Town M. Genome-wide scan for premature hypertension supports linkage to chromosome 2 in a large Kyrgyz family. Hypertension. 2006;48:908–913. doi: 10.1161/01.HYP.0000244107.13957.2b. [DOI] [PubMed] [Google Scholar]
- Knollmann BC, Knollmann-Ritschel BE, Weissman NJ, Jones LR, Morad M. Remodelling of ionic currents in hypertrophied and failing hearts of transgenic mice overexpressing calsequestrin. J Physiol. 2000;525(Pt 2):483–498. doi: 10.1111/j.1469-7793.2000.t01-1-00483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostetskii I, Li J, Xiong Y, Zhou R, Ferrari VA, Patel VV, Molkentin JD, Radice GL. Induced deletion of the N-cadherin gene in the heart leads to dissolution of the intercalated disc structure. Circ Res. 2005;96:346–354. doi: 10.1161/01.RES.0000156274.72390.2c. [DOI] [PubMed] [Google Scholar]
- Kostin S, Klein G, Szalay Z, Hein S, Bauer EP, Schaper J. Structural correlate of atrial fibrillation in human patients. Cardiovasc Res. 2002;54:361–379. doi: 10.1016/s0008-6363(02)00273-0. [DOI] [PubMed] [Google Scholar]
- Kostin S, Dammer S, Hein S, Klovekorn WP, Bauer EP, Schaper J. Con-nexin 43 expression and distribution in compensated and decompensated cardiac hypertrophy in patients with aortic stenosis. Cardiovasc Res. 2004;62:426–436. doi: 10.1016/j.cardiores.2003.12.010. [DOI] [PubMed] [Google Scholar]
- Krell MJ, Kline EM, Bates ER, Hodgson JM, Dilworth LR, Laufer N, Vogel RA, Pitt B. Intermittent, ambulatory dobutamine infusions in patients with severe congestive heart failure. Am Heart J. 1986;112:787–791. doi: 10.1016/0002-8703(86)90475-8. [DOI] [PubMed] [Google Scholar]
- Kucera JP, Rohr S, Rudy Y. Localization of sodium channels in intercalated disks modulates cardiac conduction. Circ Res. 2002;91:1176–1182. doi: 10.1161/01.res.0000046237.54156.0a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo HC, Cheng CF, Clark RB, Lin JJ, Lin JL, Hoshijima M, Nguyen-Tran VT, Gu Y, Ikeda Y, Chu PH, Ross J, Giles WR, Chien KR. A defect in the Kv channel-interacting protein 2 (KChIP2) gene leads to a complete loss of I(to) and confers susceptibility to ventricular tachycardia. Cell. 2001;107:801–813. doi: 10.1016/s0092-8674(01)00588-8. [DOI] [PubMed] [Google Scholar]
- Lai YJ, Chen YY, Cheng CP, Lin JJ, Chudorodova SL, Roshchevskaya IM, Roshchevsky MP, Chen YC, Lin CI. Changes in ionic currents and reduced conduction velocity in hypertrophied ventricular myocardium of Xinα-deficient mice. Anatol J Cardiol. 2007;7(Suppl 1):90–92. [PMC free article] [PubMed] [Google Scholar]
- Lai YJ, Huang EY, Yeh HI, Chen YL, Lin JJ, Lin CI. On the mechanisms of arrhythmias in the myocardium of mXinalpha-deficient murine left atrial-pulmonary veins. Life Sci. 2008;83:272–283. doi: 10.1016/j.lfs.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Patel VV, Kostetskii I, Xiong Y, Chu AF, Jacobson JT, Yu C, Morley GE, Molkentin JD, Radice GL. Cardiac-specific loss of N-cadherin leads to alteration in connexins with conduction slowing and arrhythmogenesis. Circ Res. 2005;97:474–481. doi: 10.1161/01.RES.0000181132.11393.18. [DOI] [PubMed] [Google Scholar]
- Li J, Patel VV, Radice GL. Dysregulation of cell adhesion proteins and cardiac arrhythmogenesis. Clin Med Res. 2006;4:42–52. doi: 10.3121/cmr.4.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Levin MD, Xiong Y, Petrenko N, Patel VV, Radice GL. N-cadherin haploinsufficiency affects cardiac gap junctions and arrhythmic susceptibility. J Mol Cell Cardiol. 2008;44:597–606. doi: 10.1016/j.yjmcc.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Swope D, Raess N, Cheng L, Muller EJ, Radice GL. Cardiac tissue-restricted deletion of plakoglobin results in progressive cardiomyopathy and activation of {beta}-catenin signaling. Mol Cell Biol. 2011;31:1134–1144. doi: 10.1128/MCB.01025-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JJC, Wang D-Z, Reiter RS, Wang Q, Lin JLC, Williams HS. Differentially expressed genes and cardiac morphogenesis. In: Tomanek RJ, Runyan R, editors. Formation of the Heart and Its Regulation. Birkhauser; Boston, MA: 2001. pp. 75–96. [Google Scholar]
- Lin JJC, Gustafson-Wagner EA, Sinn HW, Choi S, Jaacks SM, Wang DZ, Evans S, Lin JLC. Structure, expression, and function of a novel intercalated disc protein, Xin. J Med Sci. 2005;25:215–222. doi: 10.1901/jaba.2005.25-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi R, Marian AJ. Molecular genetics and pathogenesis of arrhythmogenic right ventricular cardiomyopathy: a disease of cardiac stem cells. Pediatr Cardiol. 2011;32:360–365. doi: 10.1007/s00246-011-9890-2. [DOI] [PubMed] [Google Scholar]
- London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, Viswanathan PC, Pfahnl AE, Shang LL, Madhusudanan M, Baty CJ, Lagana S, Aleong R, Gutmann R, Ackerman MJ, McNamara DM, Weiss R, Dudley SC., Jr Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation. 2007;116:2260–2268. doi: 10.1161/CIRCULATIONAHA.107.703330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe JS, Palygin O, Bhasin N, Hund TJ, Boyden PA, Shibata E, Anderson ME, Mohler PJ. Voltage-gated Nav channel targeting in the heart requires an ankyrin-G dependent cellular pathway. J Cell Biol. 2008;180:173–186. doi: 10.1083/jcb.200710107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maron BJ, Ferrans VJ. Significance of multiple intercalated discs in hypertrophied human myocardium. Am J Pathol. 1973;73:81–96. [PMC free article] [PubMed] [Google Scholar]
- McCalmon SA, Desjardins DM, Ahmad S, Davidoff KS, Snyder CM, Sato K, Ohashi K, Kielbasa OM, Mathew M, Ewen EP, Walsh K, Gavras H, Naya FJ. Modulation of angiotensin II-mediated cardiac remodeling by the MEF2A target gene Xirp2. Circ Res. 2010;106:952–960. doi: 10.1161/CIRCRESAHA.109.209007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrossan ZA, Abbott GW. The MinK-related peptides. Neuropharmacology. 2004;47:787–821. doi: 10.1016/j.neuropharm.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Mirotsou M, Dzau VJ, Pratt RE, Weinberg EO. Physiological genomics of cardiac disease: quantitative relationships between gene expression and left ventricular hypertrophy. Physiol Genomics. 2006;27:86–94. doi: 10.1152/physiolgenomics.00028.2006. [DOI] [PubMed] [Google Scholar]
- Mitarai S, Reed TD, Yatani A. Changes in ionic currents and β-adrenergic receptor signaling in hypertrophied myocytes overexpressing G αq. Am J Physiol Heart Circ Physiol. 2000;279:H139–H148. doi: 10.1152/ajpheart.2000.279.1.H139. [DOI] [PubMed] [Google Scholar]
- Mohler PJ, Rivolta I, Napolitano C, LeMaillet G, Lambert S, Priori SG, Bennett V. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc Natl Acad Sci U S A. 2004;101:17533–17538. doi: 10.1073/pnas.0403711101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata M, Buckett PD, Zhou J, Brunner M, Folco E, Koren G. SAP97 interacts with Kv1.5 in heterologous expression systems. Am J Physiol Heart Circ Physiol. 2001;281:H2575–H2584. doi: 10.1152/ajpheart.2001.281.6.H2575. [DOI] [PubMed] [Google Scholar]
- Naya FJ, Black BL, Wu H, Bassel-Duby R, Richardson JA, Hill JA, Olson EN. Mitochondrial deficiency and cardiac sudden death in mice lacking the MEF2A transcription factor. Nat Med. 2002;8:1303–1309. doi: 10.1038/nm789. [DOI] [PubMed] [Google Scholar]
- Nelson WJ. Regulation of cell-cell adhesion by the cadherin-catenin complex. Biochem Soc Trans. 2008;36:149–155. doi: 10.1042/BST0360149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–1253. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- Noorman M, van der Heyden MA, van Veen TA, Cox MG, Hauer RN, de Bakker JM, van Rijen HV. Cardiac cell-cell junctions in health and disease: electrical versus mechanical coupling. J Mol Cell Cardiol. 2009;47:23–31. doi: 10.1016/j.yjmcc.2009.03.016. [DOI] [PubMed] [Google Scholar]
- Olson TM, Keating MT. Mapping a cardiomyopathy locus to chromosome 3p22-p25. J Clin Invest. 1996;97:528–532. doi: 10.1172/JCI118445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otten J, van der Ven PF, Vakeel P, Eulitz S, Kirfel G, Brandau O, Boesl M, Schrickel JW, Linhart M, Hayess K, Naya FJ, Milting H, Meyer R, Furst DO. Complete loss of murine Xin results in a mild cardiac phenotype with altered distribution of intercalated discs. Cardiovasc Res. 2010;85:739–750. doi: 10.1093/cvr/cvp345. [DOI] [PubMed] [Google Scholar]
- Pacholsky D, Vakeel P, Himmel M, Lowe T, Stradal T, Rottner K, Furst DO, van der Ven PFM. Xin repeats define a novel actin-binding motif. J Cell Sci. 2004;117:5257–5268. doi: 10.1242/jcs.01406. [DOI] [PubMed] [Google Scholar]
- Perriard JC, Hirschy A, Ehler E. Dilated cardiomyopathy: a disease of the intercalated disc? Trends Cardiovasc Med. 2003;13:30–38. doi: 10.1016/s1050-1738(02)00209-8. [DOI] [PubMed] [Google Scholar]
- Petitprez S, Zmoos AF, Ogrodnik J, Balse E, Raad N, El-Haou S, Albesa M, Bittihn P, Luther S, Lehnart SE, Hatem SN, Coulombe A, Abriel H. SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1.5 in cardiomyocytes. Circ Res. 2011;108:294–304. doi: 10.1161/CIRCRESAHA.110.228312. [DOI] [PubMed] [Google Scholar]
- Petrecca K, Miller DM, Shrier A. Localization and enhanced current density of the Kv4.2 potassium channel by interaction with the actin-binding protein filamin. J Neurosci. 2000;20:8736–8744. doi: 10.1523/JNEUROSCI.20-23-08736.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieperhoff S, Franke WW. The area composita of adhering junctions connecting heart muscle cells of vertebrates. IV: coalescence and amalgamation of desmosomal and adhaerens junction components—late processes in mammalian heart development. Eur J Cell Biol. 2007;86:377–391. doi: 10.1016/j.ejcb.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Pokutta S, Weis WI. The cytoplasmic face of cell contact sites. Curr Opin Struct Biol. 2002;12:255–262. doi: 10.1016/s0959-440x(02)00318-4. [DOI] [PubMed] [Google Scholar]
- Reynolds AB, Carnahan RH. Regulation of cadherin stability and turnover by p120ctn: implications in disease and cancer. Semin Cell Dev Biol. 2004;15:657–663. doi: 10.1016/j.semcdb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Rockman HA, Ross RS, Harris AN, Knowlton KU, Steinhelper ME, Field LJ, Ross J, Jr, Chien KR. Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc Natl Acad Sci U S A. 1991;88:8277–8281. doi: 10.1073/pnas.88.18.8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman HA, Knowlton KU, Ross JJ, Chien KR. In vivo murine cardiac hypertrophy. Circulation. 1993;87(Suppl VII):VII14–VII21. [Google Scholar]
- Sanguinetti MC. Reduced transient outward K+ current and cardiac hypertrophy: causal relationship or epiphenomenon? Circ Res. 2002;90:497–499. doi: 10.1161/01.res.0000014284.39141.8c. [DOI] [PubMed] [Google Scholar]
- Severs NJ, Bruce AF, Dupont E, Rothery S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc Res. 2008;80:9–19. doi: 10.1093/cvr/cvn133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh F, Ross RS, Chen J. Cell-cell connection to cardiac disease. Trends Cardiovasc Med. 2009;19:182–190. doi: 10.1016/j.tcm.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata R, Misonou H, Campomanes CR, Anderson AE, Schrader LA, Doliveira LC, Carroll KI, Sweatt JD, Rhodes KJ, Trimmer JS. A fundamental role for KChIPs in determining the molecular properties and trafficking of Kv4.2 potassium channels. J Biol Chem. 2003;278:36445–36454. doi: 10.1074/jbc.M306142200. [DOI] [PubMed] [Google Scholar]
- Sinn HW, Balsamo J, Lilien J, Lin JJ. Localization of the novel Xin protein to the adherens junction complex in cardiac and skeletal muscle during development. Dev Dyn. 2002;225:1–13. doi: 10.1002/dvdy.10131. [DOI] [PubMed] [Google Scholar]
- Smeets PJ, de Vogel-van den Bosch HM, Willemsen PH, Stassen AP, Ayoubi T, van der Vusse GJ, van Bilsen M. Transcriptomic analysis of PPARalpha-dependent alterations during cardiac hypertrophy. Physiol Genomics. 2008;36:15–23. doi: 10.1152/physiolgenomics.90296.2008. [DOI] [PubMed] [Google Scholar]
- Swope D, Cheng L, Gao E, Li J, Radice GL. Loss of cadherin-binding proteins beta-catenin and plakoglobin in the heart leads to gap junction remodeling and arrhythmogenesis. Mol Cell Biol. 2012;32:1056–1067. doi: 10.1128/MCB.06188-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270–283. doi: 10.1016/s0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- Ueda K, Valdivia C, Medeiros-Domingo A, Tester DJ, Vatta M, Farrugia G, Ackerman MJ, Makielski JC. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci U S A. 2008;105:9355–9360. doi: 10.1073/pnas.0801294105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Ven PF, Ehler E, Vakeel P, Eulitz S, Schenk JA, Milting H, Micheel B, Furst DO. Unusual splicing events result in distinct Xin isoforms that associate differentially with filamin c and Mena/VASP. Exp Cell Res. 2006;312:2154–2167. doi: 10.1016/j.yexcr.2006.03.015. [DOI] [PubMed] [Google Scholar]
- Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, Ackerman MJ. Molecular and functional characterization of novel glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) mutations in sudden infant death syndrome. Circulation. 2007;116:2253–2259. doi: 10.1161/CIRCULATIONAHA.107.704627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tintelen JP, Hofstra RM, Wiesfeld AC, van den Berg MP, Hauer RN, Jongbloed JD. Molecular genetics of arrhythmogenic right ventricular cardiomyopathy: emerging horizon? Curr Opin Cardiol. 2007;22:185–192. doi: 10.1097/HCO.0b013e3280d942c4. [DOI] [PubMed] [Google Scholar]
- Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, Tester DJ, Balijepalli RC, Foell JD, Li Z, Kamp TJ, Towbin JA. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114:2104–2112. doi: 10.1161/CIRCULATIONAHA.106.635268. [DOI] [PubMed] [Google Scholar]
- Verkerk AO, van Ginneken AC, van Veen TA, Tan HL. Effects of heart failure on brain-type Na+ channels in rabbit ventricular myocytes. Europace. 2007;9:571–577. doi: 10.1093/europace/eum121. [DOI] [PubMed] [Google Scholar]
- Walker MG. Pharmaceutical target identification by gene expression analysis. Mini Rev Med Chem. 2001;1:197–205. doi: 10.2174/1389557013407034. [DOI] [PubMed] [Google Scholar]
- Wang DZ, Hu X, Lin JL, Kitten GT, Solursh M, Lin JJ. Differential displaying of mRNAs from the atrioventricular region of developing chicken hearts at stages 15 and 21. Front Biosci. 1996;1:a1–a15. doi: 10.2741/a100. [DOI] [PubMed] [Google Scholar]
- Wang DZ, Reiter RS, Lin JL, Wang Q, Williams HS, Krob SL, Schultheiss TM, Evans S, Lin JJ. Requirement of a novel gene, Xin, in cardiac morphogenesis. Development. 1999;126:1281–1294. doi: 10.1242/dev.126.6.1281. [DOI] [PubMed] [Google Scholar]
- Wang Q, Lin JL, Reinking BE, Feng HZ, Chan FC, Lin CI, Jin JP, Gustafson-Wagner EA, Scholz TD, Yang B, Lin JJ. Essential roles of an intercalated disc protein, mXinbeta, in postnatal heart growth and survival. Circ Res. 2010;106:1468–1478. doi: 10.1161/CIRCRESAHA.109.212787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Lin JL, Wu KH, Wang DZ, Reiter RS, Sinn HW, Lin CI, Lin CJ. Xin proteins and intercalated disc maturation, signaling and diseases. Front Biosci. 2012;17:2566–2593. doi: 10.2741/4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Lin JL, Chan SY, Lin JJ. The Xin repeat-containing protein, mXinbeta, initiates the maturation of the intercalated discs during postnatal heart development. Dev Biol. 2013a;374:264–280. doi: 10.1016/j.ydbio.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Lu TL, Adams E, Lin JL, Lin JJ. Intercalated disc protein, mXinα, suppresses p120-catenin-induced branching phenotype via its interactions with p120-catenin and cortactin. Arch Biochem Biophys. 2013b;535:91–100. doi: 10.1016/j.abb.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss R, Barmada MM, Nguyen T, Seibel JS, Cavlovich D, Kornblit CA, Angelilli A, Villanueva F, McNamara DM, London B. Clinical and molecular heterogeneity in the Brugada syndrome: a novel gene locus on chromosome 3. Circulation. 2002;105:707–713. doi: 10.1161/hc0602.103618. [DOI] [PubMed] [Google Scholar]
- Xue Y, Ren J, Gao X, Jin C, Wen L, Yao X. GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol Cell Proteomics. 2008;7:1598–1608. doi: 10.1074/mcp.M700574-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer T, Surber R. SCN5A channelopathies—an update on mutations and mechanisms. Prog Biophys Mol Biol. 2008;98:120–136. doi: 10.1016/j.pbiomolbio.2008.10.005. [DOI] [PubMed] [Google Scholar]