

Abstract
Autophagy is a cellular quality control mechanism crucial for neuronal homeostasis. Defects in autophagy are critically associated with mechanisms underlying Parkinson´s disease (PD), a common and debilitating neurodegenerative disorder. Autophagic dysfunction in PD can occur at several stages of the autophagy/lysosomal degradative machinery, contributing to the formation of intracellular protein aggregates and eventual neuronal cell death. Therefore, autophagy inducers may comprise a promising new therapeutic approach to combat neurodegeneration in PD. Several currently available FDA-approved drugs have been shown to enhance autophagy, which may allow for their repurposing for use in novel clinical conditions including PD. This review summarizes our current knowledge of deficits in the autophagy/lysosomal degradation pathways associated with PD, and highlight current approaches which target this pathway as possible means towards novel therapeutic strategies.
Keywords: Parkinson´s disease, autophagy, α-synuclein, LRRK2, lysosome, mTOR, rapamycin, TFEB.
INTRODUCTION
Parkinson´s disease (PD) is a debilitating neuro-degenerative disorder without a cure. It is characterized by the relatively selective loss of dopaminergic neurons in the substantia nigra pars compacta and the presence of Lewy bodies (LBs) in surviving neurons. Neuronal loss is found in predilection sites for LBs, suggesting that they may be markers for neurodegeneration. Whilst LBs consist of a heterogeneous mixture of proteins, they are particularly rich in fibrillar forms of α-synuclein [1]. The presence of such α-synuclein-containing intracytoplasmic aggregates in affected areas suggests that protein misfolding and aggregation are intimately involved in the mechanisms underlying PD. If causal to disease pathogenesis, then enhancing the cellular pathways able to degrade such protein aggregates may prove a feasible therapeutic strategy.
Cellular homeostasis consists of a proper balance between protein synthesis and recycling/degradation. The two major eukaryotic degradative systems include the ubiquitin-proteasome system (UPS) and autophagy. The UPS is responsible for the degradation of most short-lived and misfolded proteins. Proteins are ubiquitinated, which allows for their subsequent recognition, unfolding, deubiquitination and translocation into the core of the proteasome, where they are degraded [2]. Some data indicate that downregulation of the UPS may contribute to the pathogenesis of PD [3].
Autophagy is a cellular catabolic process in which cytosolic components including long-lived proteins, protein aggregates and defunct organelles are transported to the lysosome for degradation. Once in the lysosome, these components are degraded by a set of hydrolases whose activity is optimal at the acidic intralysosomal pH, and building blocks are subsequently recycled to the cytosol for their reuse. Dependent on the mechanisms by which the distinct components are delivered to the lysosome, autophagy has been subdivided into three subtypes (Fig. 1) [4]. Macro-autophagy (often just called autophagy) is the most common form of autophagy, and involves the initiation and elongation of a double-membraned structure called the phagophore or pre-autophagosomal structure (PAS). As elongation continues, this structure then sequesters cytoplasmic components, and elongation terminates with the formation of a double-membraned vesicle called the autophagosome. Autophagosomes subsequently fuse with either endosomes (to form amphisomes) or directly with lysosomes (to form autolysosomes), and cytoplasmic contents are degraded.
Fig. (1).
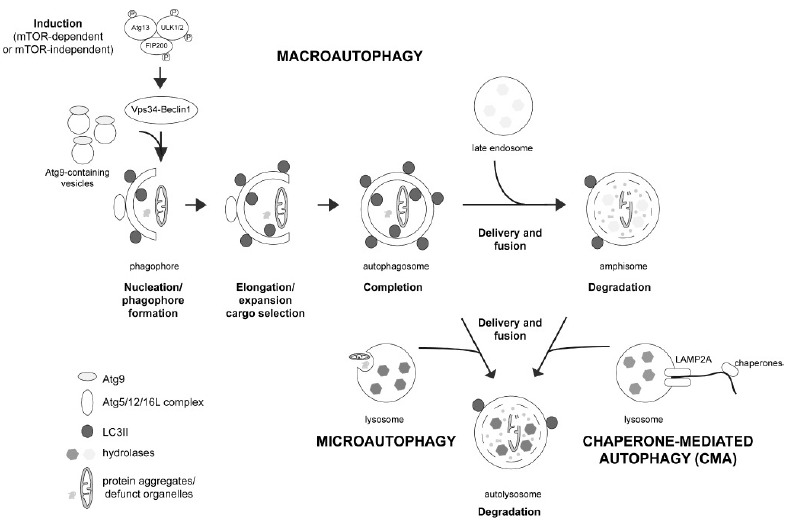
The three types of autophagy and their pathways. Macroautophagy (often referred to as just autophagy) is the most common pathway. Autophagy induction can be triggered by either mTORC1-dependent or -independent means, causing activation of the ULK protein complex, comprised of Atg13, ULK1/2 and FIP200, which assembles at the isolation membrane and causes activation of the Vps34-Beclin 1 complex at the PAS. Upon nucleation, phagophore formation and elongation is mediated by Atg9. This requires the Atg5-Atg12-Atg16L1 complex, which mediates the conjugation of phosphatidylethanolamine to LC3-I, generating LC3-II which becomes anchored on the surface of the nascent autophagosome. Upon completion, autophagosomes undergo microtubule-dependent transport to fuse with either late endosomes (to form amphisomes), or with lysosomes (to form autolysosomes), and contents are degraded in the acid environment with the help of acid hydrolases. During chaperone-mediated autophagy (CMA), substrates bearing a KFERQ-like motif are recognized by a chaperone/co-chaperone complex in the cytosol. This complex transfers the protein to the surface of the lysosomal membrane, where the binding to the cytosolic tail of the LAMP2A receptor takes place. Binding causes LAMP2A multimerization and substrate unfolding, followed by the translocation of the protein across the lysosomal membrane and its subsequent degradation by lysosomal hydrolases. Microautophagy is the least understood process, consisting of the direct invagination of the lysosomal membrane to form intraluminal vesicles loaded with cytosolic contents or organelles. Once inside the lysosome, these vesicles and their contents are then degraded. For further details see text.
Chaperone-mediated autophagy (CMA) is the most selective type of autophagy, and involves the translocation of cytosolic proteins containing a specific degradation signal (the KFERQ sequence motif) to the lysosomal lumen (Fig. 1). This motif, found in around 30 % of cytosolic proteins, is usually buried in the fully folded protein, but can be exposed upon partial unfolding. It is recognized by Hsc70 chaperone, which together with other co-chaperones targets the protein to the CMA adaptor (LAMP2A) localized on the lysosomal membrane. This causes unfolding and translocation of the protein into the lysosomal lumen, followed by degradation [5]. Finally, microautophagy is the least understood phenomenon, and is characterized by the direct invagination of the lysosomal membrane to sequester cytoplasmic components (Fig. 1) [6]. Autophagy can occur at basal levels, but is also known to be induced upon a whole variety of cellular stresses. In addition, the UPS and the different autophagy systems display significant crosstalk, where deficiency in one can be compensated by upregulation of the other one, thus further complicating the interpretations of causal relationships between deficits in specific proteostasis pathways and neurodegeneration in PD [7, 8].
DEFECTIVE AUTOPHAGY AND PD
The presence of LBs in sporadic PD patients suggests that defects in autophagic clearance of protein aggregates may contribute to disease pathomechanisms. In addition, accumulation of autophagosomes and/or lysosomal depletion have been observed in the substantia nigra pars compacta from postmortem brains of PD patients, as well as in animal models [9, 10], further hinting towards a deficit in proper autophagic-lysosomal clearance mechanisms.
The most compelling evidence for a link between aberrant autophagy and PD has come from genetic findings. Whilst PD is largely a sporadic disease with unknown etiology, around 10 % of cases are inherited. Mutations in several genes have been shown to cause familial forms of PD with either autosomal-dominant or recessive modes of inheritance. Strikingly, the dysfunction of all of those gene products is intimately linked to alterations in autophagic/lysosomal pathways, as further outlined below.
Autosomal Dominant PD Genes and their Links to Autophagy
α-synuclein is a naturally unfolded and aggregation-prone protein, and a major constituent of LBs in both sporadic and familial PD. The normal function of α-synuclein seems related to the regulation of vesicle fusion through modulation of SNARE proteins [11,12]. Interestingly, those proteins are also involved in autophagic vesicular fusion events [13]. Some studies indicate though that its normal physiological role may be divergent from its pathogenic role in disease [14].
α-synuclein plays a role in familial PD, as both point mutations as well as triplications of the α-synuclein locus have been found to cause autosomal-dominant PD [15]. The latter indicates that elevated wildtype α-synuclein protein levels can cause cytotoxicity, and thus that enhanced clearance of this protein may confer neuroprotection. α-synuclein can be degraded by both the UPS as well as by autophagic-lysosomal pathways [16-18]. The relative efficacy by which it is degraded by one versus the other pathway seems to be dependent on its precise oligomeric state, and may be further modulated by various posttranslational modifications [19, 20]. In vivo studies using α-synuclein transgenic mice indicate that under normal conditions, degradation seems mainly mediated by the UPS, whilst under conditions of increased α-synuclein burden, autophagic-lysosomal degradation events may become more prominent [21]. Apart from being degraded by the main cellular proteolytic systems, it is also able to interfere with their normal functioning, and in this manner may contribute to neurodegeneration [20]. Whilst the precise mechanisms by which wildtype α-synuclein impairs autophagy remain unclear, it seems to depend on overall protein levels and/or aggregation state. On the one hand, overexpression of the protein compromises autophagy via inhibition of Rab1a and subsequent alterations of the very initial steps of autophagosome formation dependent on Atg9 (Fig. 2) [22]. On the other hand, aggregates of α-synuclein have been shown to be resistant to degradation and may impair autophagy via reduced autophagosome clearance (Fig. 2) [23]. Together, these data indicate that excess protein levels and/or distinct α-synuclein aggregates may interfere with autophagy at both early (induction) and late (clearance) steps, and this may further depend on additional factors such as metabolic activity [24].
Fig. (2).
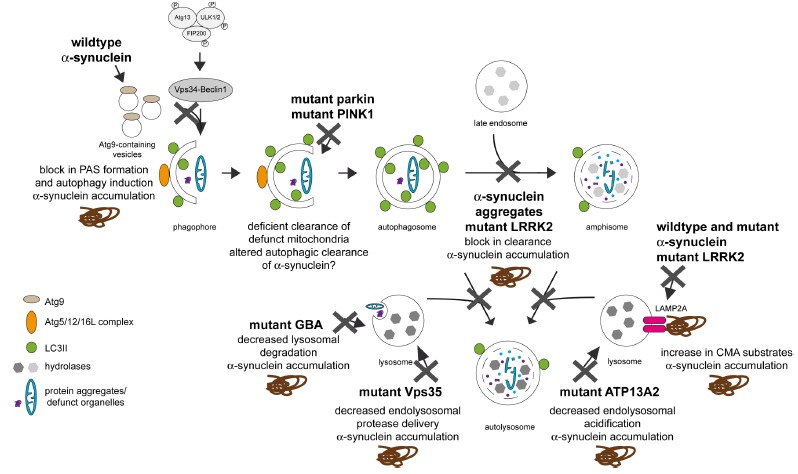
Effects of α-synuclein and other gene products linked to familial PD at various points along the autophagic pathway. Wildtype α-synuclein seems to interfere with autophagy induction by mislocalizing Atg9. In addition, α-synuclein aggregates can also impair autophagy at a later stage, and mutant α-synuclein can impair CMA. Mutant LRRK2 seems to interfere with autophagic clearance of autophagosomes, and also with CMA. Vps35 seems to interfere with the proper delivery of lysosomal hydrolases, thus causing reduced lysosomal functioning. Mutations in Parkin and PINK1 interfere with proper autophagic clearance of defunct mitochondria, causing a buildup of these organelles and failure to properly meet metabolic demands. Both mutant GBA and ATP13A2 decrease lysosomal degradative capacity. In all cases, this may lead to the toxic buildup of alphα-synuclein, causing further α-synuclein-mediated autophagic impairment and eventual cellular demise. See text for more details.
α-synuclein has also been reported to interfere with CMA [17,25]. Whilst wildtype α-synuclein is degraded by this pathway, mutant forms seem to tightly bind to the CMA receptor on the lysosomal membrane, without being able to be properly transported across into the lysosomal lumen. This not only interferes with the degradation of α-synuclein per se, but also with the degradation of other CMA substrates (Fig. 2) [17]. Posttranslational modifications or the aggregation state of α-synuclein also seem to interfere with their degradation by CMA, thereby causing an increase in α-synuclein levels followed by cytotoxicity [25]. Together, these data suggest a destructive feed-forward mechanism whereby α-synuclein triggers inappropriate autophagic degradation, leading to increased protein levels and/or aggregation, which further compromise autophagy and cause eventual cell death.
In contrast to mutations in α-synuclein, which are a relatively rare cause for familial PD, autosomal-dominant mutations in LRRK2 are the most common genetic cause of PD, and variations in the LRRK2 gene increase risk for PD, indicating that this protein is central to our understanding of mechanisms underlying PD pathogenesis. A large variety of reports support the notion that LRRK2 plays a role in autophagy [26]. Whilst knockdown or kinase inhibition seem to cause increased autophagic flux [27, 28], pathogenic mutant LRRK2 elicits phenotypes consistent with a block in autophagy, leading to accumulation of undegraded material, lysosome-like structures and lipid droplets in cell as well as animal model systems [29-38]. Whilst the precise mechanisms for the LRRK2-mediated alterations in autophagy remain unclear, they likely involve alterations in endolysosomal functioning (Fig. 2) [26]. In addition, LRRK2 can be degraded by CMA, and pathogenic mutants interfere with the CMA pathway, causing a buildup of α-synuclein [39], consistent with the observation that LRRK2-dependent neurodegeneration is α-synuclein-dependent (Fig. 2) [40]. Interestingly, brain-specific deletion of an essential autophagy gene causes accumulation of both endogenous α-synuclein and LRRK2 and delayed neurodegeneration, suggesting that the turnover of both proteins depends on proper autophagic clearance mechanisms [41]. Whilst the increased α-synuclein levels may in part be responsible for the heightened cellular toxicity, an increase in LRRK2 levels, possibly accompanied by an increase in overall cellular LRRK2 kinase activity interfering with proper autophagic flux, may contribute as well [41, 42].
Mutations in vacuolar protein sorting 35 (Vps35) cause a late-onset dominantly-inherited parkinsonian syndrome [43, 44]. Vps35 is part of the retromer complex which mediates retrograde transport from late endosomes to the trans-Golgi network. This may result in abnormal delivery of endolysosomal proteases and thus reduced degradation of proteins including α-synuclein (Fig. 2) [45]. Recent studies indicate a link between Vps35, eukaryotic translation initiation factor 4G (EIF4G1) and α-synuclein [46]. Whilst the precise mechanisms remain to be elucidated, EIF4G1 plays a crucial role in translation initiation, suggesting an involvement of proteotoxic stress, leading to the accumulation and misfolding of proteins, including α-synuclein, instigated by upregulation of EIF4G1 expression. This is reminiscent of findings that pathogenic LRRK2 may cause an overall increase in protein synthesis, which may lead to proteotoxic stress [47]. In addition, even though the precise mechanism(s) remain to be determined, recent studies suggest a link between pathogenic LRRK2, Vps35 and Rab7L1, a candidate gene for the PARK16 locus [48]. Altogether, autosomal dominant PD-related genes all seem to converge on altering late endosomal membrane trafficking, causing autophagic deficiencies associated with enhanced α-synuclein toxicity.
Autosomal Recessive PD Genes and their Links to Autophagy
Mutations in parkin and PINK1 (PTEN-induced kinase 1) cause autosomal-recessive familial PD [49]. Both seem to work together in mitochondrial quality control by mediating mitophagy, the autophagic degradation of mitochondria. PINK1 seems to accumulate on the outer membrane of damaged mitochondria and recruit parkin to those dysfunctional organelles in a manner requiring phosphorylation and ubiquitylation [50-56]. Once recruited to mitochondria, parkin then seems to ubiquitinate various outer mitochondrial membrane proteins to trigger selective mitophagy, even though additional receptor-mediated mitophagy mechanisms exist which are able to recruit LC3, promoting the sequestration of mitochondria into the autophagosomal isolation membrane, followed by autophagic mitochondrial clearance [57, 58].
Apart from the genetic links between mitochondrial clearance deficits and PD, studies of environmental toxins are consistent with the idea that mitochondrial stress is a central component of PD pathogenesis [59]. In addition, mitochondrial DNA deletions accumulate in substantia nigra neurons with age, and are more prominent in PD patients as compared to age-matched controls [60, 61]. The failure to clear damaged mitochondria by mitophagy may then lead to a buildup of those organelles, interefere with mitochondrial fission and/or biogenesis, associated with a failure to regulate changes in steady-state mitochondrial number to meet metabolic demands and causing cellular demise.
PD-Related Genes and Lysosomal Impairments
As autophagic cargo eventually needs to be degraded in the acidic environment of lysosomes, any deficiency in lysosomal clearance mechanisms will impact upon autophagic flux. For example, fusion of autophagosomes with endolysosomes requires a number of proteins including Rab7 and SNARE proteins [13, 62, 63]. Consistent with inducing a late autophagic deficit (Fig. 2), pathogenic mutant LRRK2 has recently been shown to decrease Rab7 activity [64]. Similarly, deficits in endolysosomal acidification, decreased lysosomal content or alterations in the activity of lysosomal hydrolases would negatively affect autophagic clearance.
Consistent with a link between lysosomal deficits and PD, at least two genes which encode for lysosomal proteins have been linked to parkinsonism [49]. Homozygous mutations in the lysosomal enzyme glucocerebrosidase (GBA) cause Gaucher´s disease, a lysosomal storage disorder. A subset of those patients have been shown to display parkinsonism, and heterozygous mutation carriers display an increased risk of developing PD [65]. As intralysosomal GBA catalyzes the conversion of glucosylceramide to glucose and ceramide, increased risk may be due to decreased lysosomal degradative capacity due to accumulation of glucosylceramide. Interestingly, this seems to promote α-synuclein oligomer formation (Fig. 2), further impairing trafficking of GBA from the ER and Golgi to lysosomes, thus comprising a positive feedback loop leading to neuronal cell death [66].
Mutations in ATP13A2 are responsible for a type of autosomal-recessive parkinsonism called Kufor-Rakeb syndrome [67]. ATP13A2 is a lysosomal P-type ATPase responsible for cation transport. ATP13A2 mutant cells display lysosomal impairments including lysosomal membrane instability, deficits in lysosomal acidification, defects in the clearance of autophagosomes and accumulation of α-synuclein (Fig. 2) [68-70]. Finally, a recent report indicates that mutations in DNAJC13 may cause PD [71]. As DNAJC13 is known to regulate the dynamics of endosomal clathrin coats, these findings again indicate that deficits in endo-lysosomal trafficking, recycling and degradation may underlie pathomechanisms in PD [72-74].
MECHANISTIC CONSIDERATIONS OF AUTOPHAGY
In most non-neuronal cells, autophagy takes place at a low basal level, and is activated upon various stresses such as nutrient starvation. Furthermore, autophagy is regulated under non-starved conditions by a variety of extracellular factors including growth factors, cytokines and chemokines. In contrast, as neurons depend almost exclusively on glucose to provide both energy and carbon chains for protein synthesis, autophagic responses in those cells are likely more related to the clearance of protein aggregates, damaged organelles or neuritic remodeling, rather than the generation of amino acids and energy. Indeed, neurons display high constitutive autophagic activity to maintain homeostasis, and genetic suppression of basal neuronal autophagy causes neurodegeneration in rodents [75, 76]. By analogy, the particular need of neurons for such high basal autophagy may explain why mutations in some PD genes which are not expressed in a neuron-specific manner do not cause wider systemic health problems. Furthermore, it is likely that different neuronal subtypes will display distinct susceptibilities to protein aggregation and other types of cellular stresses. Thus, they may display distinct autophagic needs and respond differently to autophagy modulators. It also needs to be considered that neuronal autophagy will be highly sensitive to deficits in vesicular trafficking events. This is because α-synuclein displays a largely presynaptic localization [77], and autophagosomes generated in response to abnormal α-synuclein burden or mutations in α-synuclein will require retrograde transport from the synapse back to the soma where they can fuse with the perinuclearly localized endolysosomes [78]. The high basal autophagic activity and the unique morphology of neurons needs to be taken into account when attempting to design strategies to modulate autophagic deficits associated with PD.
Autophagy can be subdivided into distinct steps including induction, vesicle nucleation, expansion/elongation, cargo recognition, and vesicle closure, followed by fusion of the autophagosome with endosomes or lysosomes, which results in autophagosome clearance (Fig. 1). All these steps are controlled by a series of genes called autophagy-related genes (ATGs) [4]. Autophagy induction can occur in a mammalian target of rapamycin (mTOR)-sensitive or insensitive manner, respectively. Starvation-induced autophagy is mainly regulated by mTOR, a phosphoinositide-3-kinase (PI3K)-related serine/threonine protein kinase. mTOR can form two distinct complexes (mTORC1 and mTORC2) involved in distinct signalling cascades [79]. The mTORC1 complex suppresses the autophagy pathway under nutrient-rich conditions, and is inactivated by several cellular signals including alterations in amino acid concentrations, ATP levels or growth factors. Its inactivation leads to changes in the phosphorylation state of a variety of proteins including ULK1. This causes activation of ULK1, resulting in the translocation of the Vps34 complex to the PAS (Fig. 1) [80]. The Vps34 complex is comprised of distinct proteins including Vps34, Vps15, Atg14L, AMBRA and beclin 1. Vps34 is a type III PI3K, allowing for generation of phosphatidylinositol-3-phosphate (PI3P) and formation of the PAS. This site may be formed at distinct intracellular locations including the ER, mitochondria and the plasma membrane. Generation of PI3P allows binding of additional proteins to the membrane including WIPI-1, WIPI-2 and DFCP1 in a process termed autophagy nucleation. Vesicle expansion/elongation and closure are mediated by two ubiquitin-like cascades, the Atg5-Atg12-Atg16L and LC3 cascades, with conjugation of phosphatidylethanolamine to LC3 being a crucial reaction (Fig. 1). On conjugation, the soluble LC3-I translocates to the autophagosome membrane where it is referred to as LC3-II. Autophagosomes then undergo microtubule-mediated transport to a perinuclear site where they fuse with endolysosomal structures. Efficient degradation of sequestered material in the lysosome requires proper functioning of endolysosomal hydrolases, whose activity is dependent on intralysosomal acidity. Upon lysosomal degradation, free amino acids, nucleotides, fatty acids and other components are released back into the cytosol and reused for biogenesis [4].
Autophagy is also controlled through mTOR-independent pathways. These pathways have been discovered by pharmacological screens to identify new molecular targets which enhance autophagy [81, 82]. They involve various signalling cascades implicating alterations in cAMP, calcium and inositol phosphate levels (Fig. 3). Inositol signalling seems to negatively regulate autophagy. G-protein-coupled, receptor-mediated activation of phospholipase C (PLC) causes the generation of IP3 and diacylglycerol. IP3 binds to IP3 receptors on the ER to release calcium into the cytosol. IP3 is degraded by a 5-phosphatase and inositol polyphosphate 1-phosphatase (IPPase) to form inositol phosphate, which is further hydrolyzed by inositol monophosphatase (IMPase) into free inositol. Increasing intracellular free inositol or IP3 levels inhibits autophagosome formation, whilst inositol-lowering agents induce autophagy. Importantly, most of the observed effects of altered inositol signalling on autophagic turnover may be due to altered intracellular calcium signalling, supported by findings that genetic or pharmacological inhibition of IP3 receptors stimulates autophagy. Indeed, intracellular calcium levels seem to play complex inhibitory effects on autophagy, which may depend on the precise locale of the calcium source. Calcium released from intracellular stores seems to block autophagic flux by increasing autophagosome numbers whilst decreasing autophagic clearance by blocking autophagosome-lysosome fusion, whereas influx of extracellular calcium seems to inhibit autophagy at the level of autophagosome synthesis. Calcium channel antagonists have been found to increase autophagosome synthesis and autophagic substrate clearance by decreasing cytosolic calcium levels, again supporting the notion that mTOR-independent autophagy is intimately linked to altered intracellular calcium concentrations (Fig. 3). Increased cytosolic calcium is also known to activate calpains, which are calcium-dependent cysteine proteases. Calpains seem to inhibit autophagy by cleaving the alpha-subunit of heterotrimeric G proteins (Gsalpha), which increases the activity of adenlyate cyclase and thus generates cAMP. Generation of cAMP is known to inhibit autophagy via activation of PLCepsilon, which generates IP3 and then inhibits autophagy, as described above (Fig. 3). Thus, a cAMP pathway seems to converge on an inositol signalling pathway to increase intracellular calcium levels, which negatively regulates autophagy, creating a negative inhibitory loop between the different pathways [82].
Fig. (3).
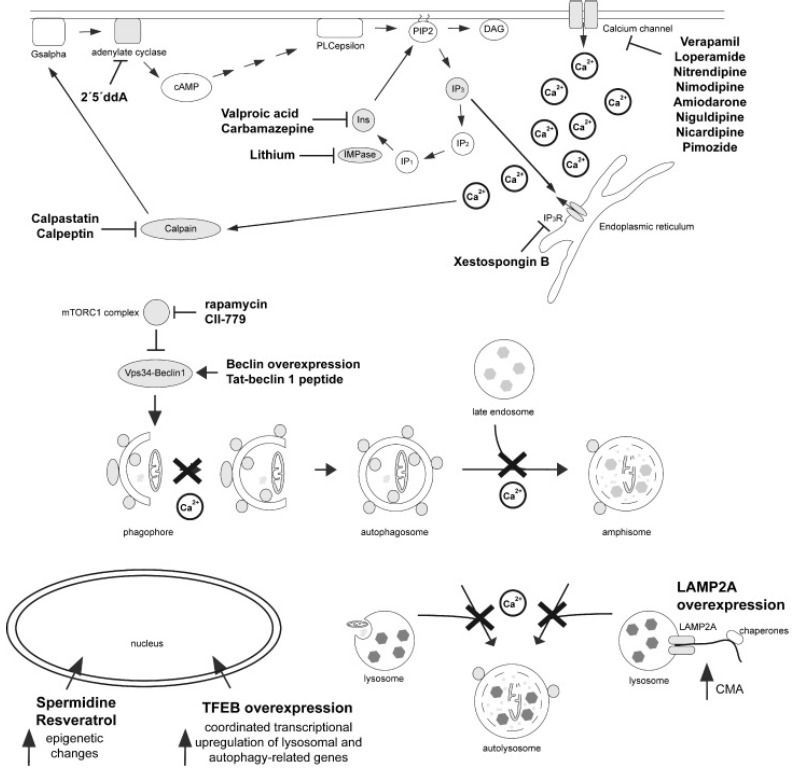
Regulation of autophagy by mTOR-dependent and -independent pathways. Rapamycin or its analog CCI-779 inhibit mTORC1, causing activation of the ULK complex and the Vps34-beclin 1 complex to induce autophagy. Autophagy can also be induced upon overexpression of beclin 1, or of a peptide derived from it, respectively. A variety of mTOR-independent means to induce autophagy have been described, which seem to work along a cyclical pathway involving changes in cAMP, inositol signalling and intracellular calcium levels. Increased cAMP levels seem to inhibit autophagy, and blocking adenylate cyclase induces autophagy. cAMP activates PLC, generating IP3, which seems to inhibit autophagy. IP3 causes calcium release from ER stores, and increased intracellular calcium inhibits autophagy. Therefore, decreasing cytosolic calcium levels by blocking calcium release from the ER, or by blocking calcium entry through calcium channels, enhances autophagy. Similarly, decreasing intracellular free inositol or IP3 levels induces autophagy. Calcium also activates calpain, which in turn activates adenylate cyclase through Gsalpha, thereby creating a negative feedback loop resulting in the generation of more IP3, more calcium release from internal stores, and more autophagy inhibition. The precise mechanism(s) by which calcium inhibits autophagy remain unclear, but involve both early steps by inhibiting autophagosome synthesis, as well as late steps by interfering with autophagosome-lysosome fusion. Apart from these pathways, autophagy can also be upregulated by expression of TFEB, causing a coordinated increase in the expression of autophagy-related and lysosomal genes, by spermidine and resveratrol, which cause varied epigenetic changes to enhance autophagic flux, and by LAMP2A, which causes an increase in CMA. For further details see text.
Apart from non-selective autophagy, various selective types have been described which are dependent on specific cargo recognition. This is achieved by adaptor proteins which recognize both the substrate (through ubiquitin-binding domains) and the autophagy machinery (through an LC3-interacting region). Those receptors include p62, as well as a set of other proteins implicated in different types of selective substrate degradation [4]. In summary, autophagy is comprised of a coordinated and complex set of reactions, with deficiencies in early steps able to cause autophagosome accumulation, and deficiencies in late steps able to cause accumulation of autolysosomes containing incompletely digested material and/or other endolysosomal alterations.
AUTOPHAGY: A PROCESS AMENABLE TO DRUG TARGETING STRATEGIES AGAINST PD
As discussed above, deficits in the autophagy pathway may be associated with both sporadic and familial forms of PD, resulting in the accumulation of protein aggregates and damaged organelles, eventually causing cellular demise. Thus, targeting autophagy by chemical or genetic means may prove beneficial to neuronal survival [82-84]. However, towards successfully applying such strategy, it is important to know which step(s) along the autophagy pathway are being compromised. Deficits in early steps may be compensated by autophagy inducers, but if late steps are affected, such as proper endolysosomal functioning, autophagy inducers may actually have deleterious effects, causing a further overload of the cellular degradation system.
A variety of studies have evaluated the effects of autophagy inducers in different PD model systems (Fig. 3, Table 1). This can be pharmacologically achieved in mTOR-dependent as well as -independent ways. Rapamycin and its analog CCI-779 are highly specific mTOR inhibitors, and have been shown to have beneficial effects in various in vitro and in vivo models of PD, leading to reduced accumulation of α-synuclein aggregates and attenuation of neuronal cell death [10, 85-87]. However, mTOR is also known to regulate other cellular processes, such that the reported beneficial effects may be triggered, at least in part, by autophagy-independent mechanisms. In addition, rapamycin is known to suppress some, but not all actions of mTOR. Indeed, torin1, a full catalytic mTOR inhibitor, seems not to be protective, but rather induces neuron death [85].
Table 1.
Autophagy enhancers, mode of action and evidence for beneficial effects in cellular and in vivo models of PD.
| Compound | Mode of Action | In vitro | In vivo | Refs. |
|---|---|---|---|---|
| rapamycin, CCI-779 | mTORC1 inhibitor | yes | yes | [10, 85-87] |
| latrepirdine | neuroactive reagent mTORC1 inhibitor (?) | yes | yes | [96] |
| lithium | IMPase inhibitor, reduction in inositol and IP3 levels | yes | unclear | [82] |
| valproic acid | MIPS inhibitor, reduction in inositol and IP3 levels | yes | n.d. | [82] |
| carbamazepine | anti-epileptic agent reduction in inositol and IP3 levels | yes | n.d. | [82] |
| verapamil loperamide nitrendipine nimodipine nicardipine pimozide | calcium channel antagonist reduction in cytosolic calcium | yes | n.d. | [82] |
| 2´5´ddA | adenylate cylase inhibitor reduction in cAMP levels | yes | n.d. | [82] |
| calpastatin | inhibition of calpain | yes | n.d. | [82] |
| spermidine | histone acetyltransferase inhibitor | yes | yes | [82] |
| resveratrol | sirtuin-1 activator | yes | yes | [82] |
| trehalose | unknown | yes | yes | [88-90] |
A whole range of FDA-approved compounds able to induce mTOR-independent autophagy have been described, even though they have not been extensively studied for their beneficial effects in PD models (Fig. 3, Table 1). These include inhibitors of inositol monophosphate phosphatase (IMPase) (lithium, L-690,330), inositol-lowering agents (carbamezipine, valproic acid), calcium channel blockers (verapamil, loperamide, nitrendipine, nimodipine, nicardipine, pimozide), calpain inhibitors (calpastatin, calpeptin), Gsalpha inhibitor (NF449), adenylate cyclase inhibitor (2´5´ddA), or IP3R antagonists (xestospongin B) [82]. Other molecules shown to induce autophagy include trehalose, a non-reducing disaccharide and an FDA-approved compound. Interestingly, trehalose seems to induce autophagic degradation of aggregate-prone proteins in cultured cells [88] and displays neuroprotective effects in various PD animal models (Fig. 3, Table 1) [89,90]. This may occur because of a trehalose-mediated transcriptional upregulation of a series of autophagy-related genes [91]. A major issue which will need to be addressed is the relative ability of these compounds to cross the blood-brain barrier. Whilst some compounds such as CCI-779, lithium, carbamazepine, dihydropyridines such as nimodipine and nitrendipine or phenylalkylamines such as verapamil easily cross the blood brain barrier, limited data are available for some of the other compounds including trehalose. When ingested, trehalase enzyme, which hydrolyzes trehalose to glucose, is present in significant amounts in the small intestine, such that little trehalose will enter the bloodstream. There is currently no convincing evidence that trehalose can cross the blood brain barrier, and cells are essentially impermeant to trehalose in the absence of its specific transporter [92], which has not been described in neurons, even though trehalose derivatives may comprise more suitable alternative compounds for future applications.
Additional molecules currently under investigation seem to work through indirect means, such as spermidine and resveratrol, which enhance autophagy by inducing epigenetic changes [93]. In addition, induction of mild endoplasmic reticulum (ER) stress by tunicamycin has been found to provide neuroprotection in a PD model due to upregulation of autophagy [94]. Thus, there may be significant crosstalk between the different proteostasis systems including autophagy, the UPS, the unfolded protein response in the ER, the heat-shock response and other quality control mechanisms [95]. Finally, some autophagy inducing molecules work through largely unknown mechanisms. For example, latrepirdine (dimebon), a neuroactive compound associated with neuroprotection and neurogenesis in mice, seems to cause a reduction of α-synuclein levels in vitro as well as in mice, possibly by inhibiting mTORC1 functioning (Fig. 3, Table 1) [96].
Apart from pharmacological approaches, gene therapy and modified peptide approaches are being pursued as well, and display the added benefit that they can be employed in an organ-specific manner. For example, overexpression of beclin 1, part of the Vps34 complex described above, via gene transfer into the brains of α-synuclein transgenic mice significantly ameliorates the synaptic pathology and reduces the accumulation of α-synuclein (Fig. 3) [97]. Inducing autophagy by a peptide derived from beclin 1 similarly has been reported to be beneficial in clearing various protein aggregates [98]. In addition, apart from enhancing autophagy induction, enhancing lysosomal capacity may be another promising strategy to decrease toxic accumulation of proteins and protein aggregates. The currently most promising approach involves expression of transcription factor EB (TFEB), which translocates to the nucleus and coordinately upregulates the expression of genes involved in lysosome biogenesis as well as in autophagosome formation [99]. Overexpression of TFEB in an in vivo model of α-synuclein toxicity seems to cause neuroprotection by clearing α-synuclein oligomers in midbrain dopaminergic neurons (Fig. 3) [100]. As CMA comprises a pathway to eliminate α-synuclein, modulation of CMA may be a good therapeutic approach as well. Indeed, overexpression of LAMP2A, the CMA receptor on the lysosomal membrane, has been found to promote the clearance of α-synuclein in dopaminergic neurons and to reduce cell loss [101]. Unfortunately, there are no synthetic inducers or activators of beclin 1, TFEB or the CMA pathway known to date. Finally, if molecular pathways underlying neurodegeneration are shared between sporadic and familial PD, targetting GBA either by pharmacological chaperones or enzyme replacement therapies [102-104], or modulating the enzymatic activities of LRRK2 [105] may revert autophagic deficits common to the entire disease spectrum.
CONCLUSIONS
Pharmacological manipulations of autophagy may delay neurodegeneration associated with PD. However, as outlined above, the specific mechanisms for the autophagy defects may be distinct dependent on the underlying cause for the disease (e.g. mutations in one versus another specific gene causing familial PD, or causes underlying sporadic PD). Cellular responses associated with those autophagic alterations may further vary across the specific stages of the disease, such that autophagy enhancers may be detrimental in certain contexts and/or treatment windows. In addition, it will be important to define the dynamic range of autophagy enhancement within which it can be optimally exploited without adverse side effects due to over-degradation of cellular components.
Several small molecule autophagy modulators targeting both mTOR-dependent and -independent pathways have been investigated for their beneficial therapeutic effects. Some are already FDA-approved drugs, and several clinical trials are currently underway. However, it will remain a challenge to define chronic treatment regimens with adequate drug concentrations to avoid detrimental effects of overactivating autophagic pathways. In addition, it needs to be kept in mind that most available drugs also target other biological processes apart from autophagy, highlighting the need for novel pharmacological agents displaying higher specificity and improved pharmacokinetic and safety properties. Alternative approaches such as gene therapy, whilst more specific and able to be targeted to affected tissues, are associated with other safety issues. Importantly, there will be a need to develop sensitive biomarkers to evaluate the in vivo efficacy of autophagy modulators. Finally, since autophagy plays important roles for various cellular processes in non-neuronal tissues [106], secondary effects may be hard to control, and it may be necessary to engineer autophagy inducers to be targeted to specific cell types or tissues. Whilst much work is needed to assure successful implementation of autophagy modulators as a valid drug strategy against PD, current data indicate their potential as future therapeutic compounds.
ACKNOWLEDGEMENTS
Work in the laboratory is funded by FEDER, the Spanish Ministry of Economy and Competitiveness (SAF2014-58653-R), the Junta de Andalucia (CTS-6816), the BBVA Foundation and the Michael J. Fox Foundation. B.F. was funded by a Juan de la Cierva Fellowship (MINECO; JCI2010-07703).
List of ABBREVIATIONS
- CMA
chaperone-mediated autophagy
- ER
endoplasmic reticulum
- GBA
glucocerebrosidase
- LB
Lewy body
- PAS
pre-autophagosomal structure
- PD
Parkinson´s disease
- TFEB
transcription factor EB
- UPS
ubiquitin-proteasome system
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- 1.Wakabayashi K., Tanji K., Odagiri S., Miki Y., Mori F., Takahashi H. The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol. Neurobiol. 2013;47(2):495–508. doi: 10.1007/s12035-012-8280-y. [DOI] [PubMed] [Google Scholar]
- 2.Ciechanover A., Kwon Y.T. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp. Mol. Med. 2015;47:e147. doi: 10.1038/emm.2014.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dennissen F.J., Kholod N., van Leeuwen F.W. The ubiquitin proteasome system in neurodegenerative diseases: culprit, accomplice or victim? Prog. Neurobiol. 2012;96(2):190–207. doi: 10.1016/j.pneurobio.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Yang Z., Klionsky D.J. Eaten alive: a history of macroautophagy. Nat. Cell Biol. 2010;12(9):814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaushik S., Cuervo A.M. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 2012;22(8):407–417. doi: 10.1016/j.tcb.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahlberg J., Glaumann H. Uptake--microautophagy--and degradation of exogenous proteins by isolated rat liver lysosomes. Effects of pH, ATP, and inhibitors of proteolysis. Exp. Mol. Pathol. 1985;42(1):78–88. doi: 10.1016/0014-4800(85)90020-6. [DOI] [PubMed] [Google Scholar]
- 7.Nedelsky N.B., Todd P.K., Taylor J.P. Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochim. Biophys. Acta. 2008;1782(12):691–699. doi: 10.1016/j.bbadis.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaushik S., Massey A.C., Mizushima N., Cuervo A.M. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol. Biol. Cell. 2008;19(5):2179–2192. doi: 10.1091/mbc.E07-11-1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anglade P., Vyas S., Javoy-Agid F., Herrero M.T., Michel P.P., Marquez J., Mouatt-Prigent A., Ruberg M., Hirsch E.C., Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997;12(1):25–31. [PubMed] [Google Scholar]
- 10.Dehay B., Bové J., Rodríguez-Muela N., Perier C., Recasens A., Boya P., Vila M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010;30(37):12535–12544. doi: 10.1523/JNEUROSCI.1920-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burré J., Sharma M., Tsetsenis T., Buchman V., Etherton M.R., Südhof T.C. Alphα-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329(5999):1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burré J., Sharma M., Südhof T.C. Definition of a molecular pathway mediating α-synuclein neurotoxicity. J. Neurosci. 2015;35(13):5221–5232. doi: 10.1523/JNEUROSCI.4650-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nair U., Jotwani A., Geng J., Gammoh N., Richerson D., Yen W.L., Griffith J., Nag S., Wang K., Moss T., Baba M., McNew J.A., Jiang X., Reggiori F., Melia T.J., Klionsky D.J. SNARE proteins are required for macroautophagy. Cell. 2011;146(2):290–302. doi: 10.1016/j.cell.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burré J., Sharma M., Südhof T.C. Systematic mutagenesis of α-synuclein reveals distinct sequence requirements for physiological and pathological activities. J. Neurosci. 2012;32(43):15227–15242. doi: 10.1523/JNEUROSCI.3545-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singleton A.B., Farrer M., Johnson J., Singleton A., Hague S., Kachergus J., Hulihan M., Peuralinna T., Dutra A., Nussbaum R., Lincoln S., Crawley A., Hanson M., Maraganore D., Adler C., Cookson M.R., Muenter M., Baptista M., Miller D., Blancato J., Hardy J., Gwinn-Hardy K. alphα-synuclein locus triplication causes Parkinson’s disease. Science. 2003;302(5646):841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 16.Vogiatzi T., Xilouri M., Vekrellis K., Stefanis L. Wild type alphα-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008;283(35):23542–23556. doi: 10.1074/jbc.M801992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cuervo A.M., Stefanis L., Fredenburg R., Lansbury P.T., Sulzer D. Impaired degradation of mutant alphα-synuclein by chaperone-mediated autophagy. Science. 2004;305(5688):1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 18.Webb J.L., Ravikumar B., Atkins J., Skepper J.N., Rubinsztein D.C. Alphα-synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003;278(27):25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 19.Rott R., Szargel R., Haskin J., Bandopadhyay R., Lees A.J., Shani V., Engelender S. α-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc. Natl. Acad. Sci. USA. 2011;108(46):18666–18671. doi: 10.1073/pnas.1105725108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lashuel H.A., Overk C.R., Oueslati A., Masliah E. The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013;14(1):38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ebrahimi-Fakhari D., Cantuti-Castelvetri I., Fan Z., Rockenstein E., Masliah E., Hyman B.T., McLean P.J., Unni V.K. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of α-synuclein. J. Neurosci. 2011;31(41):14508–14520. doi: 10.1523/JNEUROSCI.1560-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winslow A.R., Chen C.W., Corrochano S., Acevedo-Arozena A., Gordon D.E., Peden A.A., Lichtenberg M., Menzies F.M., Ravikumar B., Imarisio S., Brown S., O’Kane C.J., Rubinsztein D.C. α-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J. Cell Biol. 2010;190(6):1023–1037. doi: 10.1083/jcb.201003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanik S.A., Schultheiss C.E., Volpicelli-Daley L.A., Brunden K.R., Lee V.M. Lewy body-like α-synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem. 2013;288(21):15194–15210. doi: 10.1074/jbc.M113.457408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu W.H., Dorado B., Figueroa H.Y., Wang L., Planel E., Cookson M.R., Clark L.N., Duff K.E. Metabolic activity determines efficacy of macroautophagic clearance of pathological oligomeric alphα-synuclein. Am. J. Pathol. 2009;175(2):736–747. doi: 10.2353/ajpath.2009.080928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinez-Vicente M., Talloczy Z., Kaushik S., Massey A.C., Mazzulli J., Mosharov E.V., Hodara R., Fredenburg R., Wu D.C., Follenzi A., Dauer W., Przedborski S., Ischiropoulos H., Lansbury P.T., Sulzer D., Cuervo A.M. Dopamine-modified alphα-synuclein blocks chaperone-mediated autophagy. J. Clin. Invest. 2008;118(2):777–788. doi: 10.1172/JCI32806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gómez-Suaga P., Fdez E., Fernández B., Martínez-Salvador M., Blanca Ramírez M., Madero-Pérez J., Rivero-Ríos P., Fuentes J.M., Hilfiker S. Novel insights into the neurobiology underlying LRRK2-linked Parkinson’s disease. Neuropharmacology. 2014;85:45–56. doi: 10.1016/j.neuropharm.2014.05.020. [DOI] [PubMed] [Google Scholar]
- 27.Alegre-Abarrategui J., Christian H., Lufino M.M., Mutihac R., Venda L.L., Ansorge O., Wade-Martins R. LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum. Mol. Genet. 2009;18(21):4022–4034. doi: 10.1093/hmg/ddp346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manzoni C., Lewis P.A. Dysfunction of the autophagy/lysosomal degradation pathway is a shared feature of the genetic synucleinopathies. FASEB J. 2013;27(9):3424–3429. doi: 10.1096/fj.12-223842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacLeod D., Dowman J., Hammond R., Leete T., Inoue K., Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52(4):587–593. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 30.Plowey E.D., Cherra S.J., III, Liu Y.J., Chu C.T. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J. Neurochem. 2008;105(3):1048–1056. doi: 10.1111/j.1471-4159.2008.05217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tong Y., Yamaguchi H., Giaime E., Boyle S., Kopan R., Kelleher R.J., III, Shen J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alphα-synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. USA. 2010;107(21):9879–9884. doi: 10.1073/pnas.1004676107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiong Y., Coombes C.E., Kilaru A., Li X., Gitler A.D., Bowers W.J., Dawson V.L., Dawson T.M., Moore D.J. GTPase activity plays a key role in the pathobiology of LRRK2. PLoS Genet. 2010;6(4):e1000902. doi: 10.1371/journal.pgen.1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramonet D., Daher J.P., Lin B.M., Stafa K., Kim J., Banerjee R., Westerlund M., Pletnikova O., Glauser L., Yang L., Liu Y., Swing D.A., Beal M.F., Troncoso J.C., McCaffery J.M., Jenkins N.A., Copeland N.G., Galter D., Thomas B., Lee M.K., Dawson T.M., Dawson V.L., Moore D.J. Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS Genet. 2011;6(4) doi: 10.1371/journal.pone.0018568. e1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gómez-Suaga P., Luzón-Toro B., Churamani D., Zhang L., Bloor-Young D., Patel S., Woodman P.G., Churchill G.C., Hilfiker S. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum. Mol. Genet. 2012;21(3):511–525. doi: 10.1093/hmg/ddr481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sánchez-Danés A., Richaud-Patin Y., Carballo-Carbajal I., Jiménez-Delgado S., Caig C., Mora S., Di Guglielmo C., Ezquerra M., Patel B., Giralt A., Canals J.M., Memo M., Alberch J., López-Barneo J., Vila M., Cuervo A.M., Tolosa E., Consiglio A., Raya A. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol. Med. 2012;4(5):380–395. doi: 10.1002/emmm.201200215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tong Y., Giaime E., Yamaguchi H., Ichimura T., Liu Y., Si H., Cai H., Bonventre J.V., Shen J. Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. 2012. [DOI] [PMC free article] [PubMed]
- 37.Bravo-San Pedro J.M., Niso-Santano M., Gómez-Sánchez R., Pizarro-Estrella E., Aiastui-Pujana A., Gorostidi A., Climent V., López de Maturana R., Sanchez-Pernaute R., López de Munain A., Fuentes J.M., González-Polo R.A. The LRRK2 G2019S mutant exacerbates basal autophagy through activation of the MEK/ERK pathway. Cell. Mol. Life Sci. 2013;70(1):121–136. doi: 10.1007/s00018-012-1061-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schapansky J., Nardozzi J.D., Felizia F., LaVoie M.J. Membrane recruitment of endogenous LRRK2 precedes its potent regulation of autophagy. Hum. Mol. Genet. 2014;23(16):4201–4214. doi: 10.1093/hmg/ddu138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Orenstein S.J., Kuo S.H., Tasset I., Arias E., Koga H., Fernandez-Carasa I., Cortes E., Honig L.S., Dauer W., Consiglio A., Raya A., Sulzer D., Cuervo A.M. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 2013;16(4):394–406. doi: 10.1038/nn.3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Skibinski G., Nakamura K., Cookson M.R., Finkbeiner S. Mutant LRRK2 toxicity in neurons depends on LRRK2 levels and synuclein but not kinase activity or inclusion bodies. J. Neurosci. 2014;34(2):418–433. doi: 10.1523/JNEUROSCI.2712-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Friedman L.G., Lachenmayer M.L., Wang J., He L., Poulose S.M., Komatsu M., Holstein G.R., Yue Z. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of α-synuclein and LRRK2 in the brain. J. Neurosci. 2012;32(22):7585–7593. doi: 10.1523/JNEUROSCI.5809-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Plotegher N., Civiero L. Neuronal autophagy, α-synuclein clearance, and LRRK2 regulation: a lost equilibrium in parkinsonian brain. J. Neurosci. 2012;32(43):14851–14853. doi: 10.1523/JNEUROSCI.3588-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vilariño-Güell C., Wider C., Ross O.A., Dachsel J.C., Kachergus J.M., Lincoln S.J., Soto-Ortolaza A.I., Cobb S.A., Wilhoite G.J., Bacon J.A., Behrouz B., Melrose H.L., Hentati E., Puschmann A., Evans D.M., Conibear E., Wasserman W.W., Aasly J.O., Burkhard P.R., Djaldetti R., Ghika J., Hentati F., Krygowska-Wajs A., Lynch T., Melamed E., Rajput A., Rajput A.H., Solida A., Wu R.M., Uitti R.J., Wszolek Z.K., Vingerhoets F., Farrer M.J. VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 2011;89(1):162–167. doi: 10.1016/j.ajhg.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zimprich A., Benet-Pagès A., Struhal W., Graf E., Eck S.H., Offman M.N., Haubenberger D., Spielberger S., Schulte E.C., Lichtner P., Rossle S.C., Klopp N., Wolf E., Seppi K., Pirker W., Presslauer S., Mollenhauer B., Katzenschlager R., Foki T., Hotzy C., Reinthaler E., Harutyunyan A., Kralovics R., Peters A., Zimprich F., Brücke T., Poewe W., Auff E., Trenkwalder C., Rost B., Ransmayr G., Winkelmann J., Meitinger T., Strom T.M. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 2011;89(1):168–175. doi: 10.1016/j.ajhg.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Follett J., Norwood S.J., Hamilton N.A., Mohan M., Kovtun O., Tay S., Zhe Y., Wood S.A., Mellick G.D., Silburn P.A., Collins B.M., Bugarcic A., Teasdale R.D. The Vps35 D620N mutation linked to Parkinson’s disease disrupts the cargo sorting function of retromer. Traffic. 2014;15(2):230–244. doi: 10.1111/tra.12136. [DOI] [PubMed] [Google Scholar]
- 46.Dhungel N., Eleuteri S., Li L.B., Kramer N.J., Chartron J.W., Spencer B., Kosberg K., Fields J.A., Stafa K., Adame A., Lashuel H., Frydman J., Shen K., Masliah E., Gitler A.D. Parkinson’s disease genes VPS35 and EIF4G1 interact genetically and converge on α-synuclein. Neuron. 2015;85(1):76–87. doi: 10.1016/j.neuron.2014.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martin I., Kim J.W., Lee B.D., Kang H.C., Xu J.C., Jia H., Stankowski J., Kim M.S., Zhong J., Kumar M., Andrabi S.A., Xiong Y., Dickson D.W., Wszolek Z.K., Pandey A., Dawson T.M., Dawson V.L. Ribosomal protein s15 phosphorylation mediates LRRK2 neurodegeneration in Parkinson’s disease. Cell. 2014;157(2):472–485. doi: 10.1016/j.cell.2014.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.MacLeod D.A., Rhinn H., Kuwahara T., Zolin A., Di Paolo G., McCabe B.D., Marder K.S., Honig L.S., Clark L.N., Small S.A., Abeliovich A. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson's disease risk. Neuron. 2013;77(3):425–439. doi: 10.1016/j.neuron.2012.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trinh J., Farrer M. Advances in the genetics of Parkinson disease. Nat. Rev. Neurol. 2013;9(8):445–454. doi: 10.1038/nrneurol.2013.132. [DOI] [PubMed] [Google Scholar]
- 50.Narendra D., Tanaka A., Suen D.F., Youle R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008;183(5):795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Narendra D.P., Jin S.M., Tanaka A., Suen D.F., Gautier C.A., Shen J., Cookson M.R., Youle R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8(1) doi: 10.1371/journal.pbio.1000298. e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kane L.A., Lazarou M., Fogel A.I., Li Y., Yamano K., Sarraf S.A., Banerjee S., Youle R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014;205(2):143–153. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pickrell A.M., Youle R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85(2):257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kazlauskaite A., Muqit M.M. PINK1 and Parkin – mitochondrial interplay between phosphorylation and ubiquitylation in Parkinson’s disease. FEBS J. 2015;282(2):215–223. doi: 10.1111/febs.13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koyano F., Okatsu K., Kosako H., Tamura Y., Go E., Kimura M., Kimura Y., Tsuchiya H., Yoshihara H., Hirokawa T., Endo T., Fon E.A., Trempe J.F., Saeki Y., Tanaka K., Matsuda N. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510(7503):162–166. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- 56.Okatsu K., Koyano F., Kimura M., Kosako H., Saeki Y., Tanaka K., Matsuda N. Phosphorylated ubiquitin chain is the genuine Parkin receptor. J. Cell Biol. 2015;209(1):111–128. doi: 10.1083/jcb.201410050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wei H., Liu L., Chen Q. Selective removal of mitochondria via mitophagy: distinct pathways for different mitochondrial stresses. Biochim. Biophys. Acta. 2015;S0167-4889(15):00114–00117. doi: 10.1016/j.bbamcr.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 58.Liu L., Sakakibara K., Chen Q., Okamoto K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014;24(7):787–795. doi: 10.1038/cr.2014.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schapira A.H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008;7(1):97–109. doi: 10.1016/S1474-4422(07)70327-7. [DOI] [PubMed] [Google Scholar]
- 60.Kraytsberg Y., Kudryavtseva E., McKee A.C., Geula C., Kowall N.W., Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006;38(5):518–520. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- 61.Bender A., Krishnan K.J., Morris C.M., Taylor G.A., Reeve A.K., Perry R.H., Jaros E., Hersheson J.S., Betts J., Klopstock T., Taylor R.W., Turnbull D.M. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006;38(5):515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 62.Jäger S., Bucci C., Tanida I., Ueno T., Kominami E., Saftig P., Eskelinen E.L. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004;117(Pt 20):4837–4848. doi: 10.1242/jcs.01370. [DOI] [PubMed] [Google Scholar]
- 63.Gutierrez M.G., Munafó D.B., Berón W., Colombo M.I. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 2004;117(Pt 13):2687–2697. doi: 10.1242/jcs.01114. [DOI] [PubMed] [Google Scholar]
- 64.Gómez-Suaga P., Rivero-Ríos P., Fdez E., Blanca Ramírez M., Ferrer I., Aiastui A., López De Munain A., Hilfiker S. LRRK2 delays degradative receptor trafficking by impeding late endosomal budding through decreasing Rab7 activity. Hum. Mol. Genet. 2014;23(25):6779–6796. doi: 10.1093/hmg/ddu395. [DOI] [PubMed] [Google Scholar]
- 65.Sidransky E., Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012;11(11):986–998. doi: 10.1016/S1474-4422(12)70190-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mazzulli J.R., Xu Y.H., Sun Y., Knight A.L., McLean P.J., Caldwell G.A., Sidransky E., Grabowski G.A., Krainc D. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146(1):37–52. doi: 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ramirez A., Heimbach A., Gründemann J., Stiller B., Hampshire D., Cid L.P., Goebel I., Mubaidin A.F., Wriekat A.L., Roeper J., Al-Din A., Hillmer A.M., Karsak M., Liss B., Woods C.G., Behrens M.I., Kubisch C. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006;38(10):1184–1191. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- 68.Dehay B., Ramirez A., Martinez-Vicente M., Perier C., Canron M.H., Doudnikoff E., Vital A., Vila M., Klein C., Bezard E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc. Natl. Acad. Sci. USA. 2012;109(24):9611–9616. doi: 10.1073/pnas.1112368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Usenovic M., Tresse E., Mazzulli J.R., Taylor J.P., Krainc D. Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity. J. Neurosci. 2012;32(12):4240–4246. doi: 10.1523/JNEUROSCI.5575-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schultheis P.J., Fleming S.M., Clippinger A.K., Lewis J., Tsunemi T., Giasson B., Dickson D.W., Mazzulli J.R., Bardgett M.E., Haik K.L., Ekhator O., Chava A.K., Howard J., Gannon M., Hoffman E., Chen Y., Prasad V., Linn S.C., Tamargo R.J., Westbroek W., Sidransky E., Krainc D., Shull G.E. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited α-synuclein accumulation and age-dependent sensorimotor deficits. Hum. Mol. Genet. 2013;22(10):2067–2082. doi: 10.1093/hmg/ddt057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vilariño-Güell C., Rajput A., Milnerwood A.J., Shah B., Szu-Tu C., Trinh J., Yu I., Encarnacion M., Munsie L.N., Tapia L., Gustavsson E.K., Chou P., Tatarnikov I., Evans D.M., Pishotta F.T., Volta M., Beccano-Kelly D., Thompson C., Lin M.K., Sherman H.E., Han H.J., Guenther B.L., Wasserman W.W., Bernard V., Ross C.J., Appel-Cresswell S., Stoessl A.J., Robinson C.A., Dickson D.W., Ross O.A., Wszolek Z.K., Aasly J.O., Wu R.M., Hentati F., Gibson R.A., McPherson P.S., Girard M., Rajput M., Rajput A.H., Farrer M.J. DNAJC13 mutations in Parkinson disease. Hum. Mol. Genet. 2014;23(7):1794–1801. doi: 10.1093/hmg/ddt570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Perrett R.M., Alexopoulou Z., Tofaris G.K. The endosomal pathway in Parkinson's disease. Mol. Cell. Neurosci. 2015;S1044-7431(15):00022–00026. doi: 10.1016/j.mcn.2015.02.009. [DOI] [PubMed] [Google Scholar]
- 73.Tofaris G.K. Lysosome-dependent pathways as a unifying theme in Parkinson’s disease. Mov. Disord. 2012;27(11):1364–1369. doi: 10.1002/mds.25136. [DOI] [PubMed] [Google Scholar]
- 74.Manzoni C., Lewis P.A. Dysfunction of the autophagy/lysosomal degradation pathway is a shared feature of the genetic synucleinopathies. FASEB J. 2013;27(9):3424–3429. doi: 10.1096/fj.12-223842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., Yokoyama M., Mishima K., Saito I., Okano H., Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441(7095):885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 76.Komatsu M., Waguri S., Chiba T., Murata S., Iwata J., Tanida I., Ueno T., Koike M., Uchiyama Y., Kominami E., Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441(7095):880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 77.Kahle P.J., Neumann M., Ozmen L., Muller V., Jacobsen H., Schindzielorz A., Okochi M., Leimer U., van Der Putten H., Probst A., Kremmer E., Kretzschmar H.A., Haass C. Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha -synuclein in human and transgenic mouse brain. J. Neurosci. 2000;20(17):6365–6373. doi: 10.1523/JNEUROSCI.20-17-06365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Maday S., Holzbaur E.L. Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev. Cell. 2014;30(1):71–85. doi: 10.1016/j.devcel.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Laplante M., Sabatini D.M. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Koyama-Honda I., Itakura E., Fujiwara T.K., Mizushima N. Temporal analysis of recruitment of mammalian ATG proteins to the autophagosome formation site. Autophagy. 2013;9(10):1491–1499. doi: 10.4161/auto.25529. [DOI] [PubMed] [Google Scholar]
- 81.Williams A., Sarkar S., Cuddon P., Ttofi E.K., Saiki S., Siddiqi F.H., Jahreiss L., Fleming A., Pask D., Goldsmith P., O’Kane C.J., Floto R.A., Rubinsztein D.C. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008;4(5):295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sarkar S. Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem. Soc. Trans. 2013;41(5):1103–1130. doi: 10.1042/BST20130134. [DOI] [PubMed] [Google Scholar]
- 83.Fleming A., Noda T., Yoshimori T., Rubinsztein D.C. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat. Chem. Biol. 2011;7(1):9–17. doi: 10.1038/nchembio.500. [DOI] [PubMed] [Google Scholar]
- 84.Vidal R.L., Matus S., Bargsted L., Hetz C. Targeting autophagy in neurodegenerative diseases. Trends Pharmacol. Sci. 2014;35(11):583–591. doi: 10.1016/j.tips.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 85.Malagelada C., Jin Z.H., Jackson-Lewis V., Przedborski S., Greene L.A. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 2010;30(3):1166–1175. doi: 10.1523/JNEUROSCI.3944-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Crews L., Spencer B., Desplats P., Patrick C., Paulino A., Rockenstein E., Hansen L., Adame A., Galasko D., Masliah E. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alphα-synucleinopathy. PLoS One. 2010;5(2):e9313. doi: 10.1371/journal.pone.0009313. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 87.Santini E., Heiman M., Greengard P., Valjent E., Fisone G. Inhibition of mTOR signaling in Parkinson’s disease prevents L-DOPA-induced dyskinesia. Sci. Signal. 2009;2(80):ra36. doi: 10.1126/scisignal.2000308. [DOI] [PubMed] [Google Scholar]
- 88.Sarkar S., Davies J.E., Huang Z., Tunnacliffe A., Rubinsztein D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alphα-synuclein. J. Biol. Chem. 2007;282(8):5641–5652. doi: 10.1074/jbc.M609532200. [DOI] [PubMed] [Google Scholar]
- 89.Rodríguez-Navarro J.A., Rodríguez L., Casarejos M.J., Solano R.M., Gómez A., Perucho J., Cuervo A.M., García de Yébenes J., Mena M.A. Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol. Dis. 2010;39(3):423–438. doi: 10.1016/j.nbd.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 90.Sarkar S., Chigurupati S., Raymick J., Mann D., Bowyer J.F., Schmitt T., Beger R.D., Hanig J.P., Schmued L.C., Paule M.G. Neuroprotective effect of the chemical chaperone, trehalose in a chronic MPTP-induced Parkinson’s disease mouse model. Neurotoxicology. 2014;44:250–262. doi: 10.1016/j.neuro.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 91.Castillo K., Nassif M., Valenzuela V., Rojas F., Matus S., Mercado G., Court F.A., van Zundert B., Hetz C. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy. 2013;9(9):1308–1320. doi: 10.4161/auto.25188. [DOI] [PubMed] [Google Scholar]
- 92.Kikawada T., Saito A., Kanamori Y., Nakahara Y., Iwata K., Tanaka D., Watanabe M., Okuda T. Trehalose transporter 1, a facilitated and high-capacity trehalose transporter, allows exogenous trehalose uptake into cells. Proc. Natl. Acad. Sci. USA. 2007;104(28):11585–11590. doi: 10.1073/pnas.0702538104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Morselli E., Mariño G., Bennetzen M.V., Eisenberg T., Megalou E., Schroeder S., Cabrera S., Bénit P., Rustin P., Criollo A., Kepp O., Galluzzi L., Shen S., Malik S.A., Maiuri M.C., Horio Y., López-Otín C., Andersen J.S., Tavernarakis N., Madeo F., Kroemer G. Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J. Cell Biol. 2011;192(4):615–629. doi: 10.1083/jcb.201008167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fouillet A., Levet C., Virgone A., Robin M., Dourlen P., Rieusset J., Belaidi E., Ovize M., Touret M., Nataf S., Mollereau B. ER stress inhibits neuronal death by promoting autophagy. Autophagy. 2012;8(6):915–926. doi: 10.4161/auto.19716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Balch W.E., Morimoto R.I., Dillin A., Kelly J.W. Adapting proteostasis for disease intervention. Science. 2008;319(5865):916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 96.Steele J.W., Ju S., Lachenmayer M.L., Liken J., Stock A., Kim S.H., Delgado L.M., Alfaro I.E., Bernales S., Verdile G., Bharadwaj P., Gupta V., Barr R., Friss A., Dolios G., Wang R., Ringe D., Protter A.A., Martins R.N., Ehrlich M.E., Yue Z., Petsko G.A., Gandy S. Latrepirdine stimulates autophagy and reduces accumulation of α-synuclein in cells and in mouse brain. Mol. Psychiatry. 2013;18(8):882–888. doi: 10.1038/mp.2012.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Spencer B., Potkar R., Trejo M., Rockenstein E., Patrick C., Gindi R., Adame A., Wyss-Coray T., Masliah E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alphα-synuclein models of Parkinson’s and Lewy body diseases. J. Neurosci. 2009;29(43):13578–13588. doi: 10.1523/JNEUROSCI.4390-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shoji-Kawata S., Sumpter R., Leveno M., Campbell G.R., Zou Z., Kinch L., Wilkins A.D., Sun Q., Pallauf K., MacDuff D., Huerta C., Virgin H.W., Helms J.B., Eerland R., Tooze S.A., Xavier R., Lenschow D.J., Yamamoto A., King D., Lichtarge O., Grishin N.V., Spector S.A., Kaloyanova D.V., Levine B. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494(7436):201–206. doi: 10.1038/nature11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Settembre C., Di Malta C., Polito V.A., Garcia Arencibia M., Vetrini F., Erdin S., Erdin S.U., Huynh T., Medina D., Colella P., Sardiello M., Rubinsztein D.C., Ballabio A. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Decressac M., Mattsson B., Weikop P., Lundblad M., Jakobsson J., Björklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc. Natl. Acad. Sci. USA. 2013;110(19):E1817–E1826. doi: 10.1073/pnas.1305623110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xilouri M., Brekk O.R., Landeck N., Pitychoutis P.M., Papasilekas T., Papadopoulou-Daifoti Z., Kirik D., Stefanis L. Boosting chaperone-mediated autophagy in vivo mitigates α-synuclein-induced neurodegeneration. Brain. 2013;136(Pt 7):2130–2146. doi: 10.1093/brain/awt131. [DOI] [PubMed] [Google Scholar]
- 102.de la Mata M., Cotán D., Oropesa-Ávila M., Garrido-Maraver J., Cordero M.D., Villanueva Paz M., Delgado Pavón A., Alcocer-Gómez E., de Lavera I., Ybot-González P., Paula Zaderenko A., Ortiz Mellet C., García Fernández J.M., Sánchez-Alcázar J.A. Pharmacological Chaperones and Coenzyme Q10 Treatment Improves Mutant β-Glucocerebrosidase Activity and Mitochondrial Function in Neuronopathic Forms of Gaucher Disease. Sci. Rep. 2015;5:10903. doi: 10.1038/srep10903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Benito J.M., García Fernández J.M., Ortiz Mellet C. Pharmacological chaperone therapy for Gaucher disease: a patent review. Expert Opin. Ther. Pat. 2011;21(6):885–903. doi: 10.1517/13543776.2011.569162. [DOI] [PubMed] [Google Scholar]
- 104.Schapira A.H. Glucocerebrosidase and Parkinson disease: Recent advances. Mol. Cell. Neurosci. 2015;66(Pt A):37–42. doi: 10.1016/j.mcn.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cookson M.R. LRRK2 Pathways Leading to Neurodegeneration. Curr. Neurol. Neurosci. Rep. 2015 Jul;15(7):564. doi: 10.1007/s11910-015-0564-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mizushima N., Levine B., Cuervo A.M., Klionsky D.J. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]