

Abstract
Parkinson’s Disease (PD) related genes PINK1, a protein kinase [1], and Parkin, an E3 ubiquitin ligase [2], operate within the same pathway [3-5], which controls, via specific elimination of dysfunctional mitochondria, the quality of the organelle network [6]. Parkin translocates to impaired mitochondria and drives their elimination via autophagy, a process known as mitophagy [6]. PINK1 regulates Parkin translocation through a not yet completely understood mechanism [7, 8]. Mitochondrial outer membrane proteins Mitofusin (MFN), VDAC, Fis1 and TOM20 were found to be targets for Parkin mediated ubiquitination [9-11]. By adding ubiquitin molecules to its targets expressed on mitochondria, Parkin tags and selects dysfunctional mitochondria for clearance, contributing to maintain a functional and healthy mitochondrial network. Abnormal accumulation of misfolded proteins and unfunctional mitochondria is a characteristic hallmark of PD pathology. Therefore a therapeutic approach to enhance clearance of misfolded proteins and potentiate the ubiquitin-proteosome system (UPS) could be instrumental to ameliorate the progression of the disease. Recently, much effort has been put to identify specific de-ubiquitinating enzymes (DUBs) that oppose Parkin in the ubiquitination of its targets. Similar to other post-translational modifications, such as phosphorylation and acetylation, ubiquitination is also a reversible modification, mediated by a large family of DUBs [12]. DUBs inhibitors or activators can affect cellular response to stimuli that induce mitophagy via ubiquitination of mitochondrial outer membrane proteins MFN, VDAC, Fis1 and TOM20. In this respect, the identification of a Parkin-opposing DUB in the regulation of mitophagy, might be instrumental to develop specific isopeptidase inhibitors or activators that can modulate the fundamental biological process of mitochondria clearance and impact on cell survival.
Keywords: Drosophila, DUB, Mitofusin, Mitophagy, Parkin, parkinson’s disease, PINK1, Ubiquitination
THE NUMBERS OF PD
Parkinson's disease (PD) is the second most common neurodegenerative disease for which there is no cure. It is characterized by selective loss of dopaminergic neurons (DA) in the substantia nigra (SN) pars compacta and specific hallmark include accumulation of aggregates and unfolded proteins in the form of Lewy bodies. PD is a movement disorders with patients developing resting tremors, bradikinesia, muscle rigidity, postural instability and gait problems. It affects 1-2% of the population over the age of 65 and this percentage increases by approximately 4% in those older that 85 years [13]. Life expectancy has risen in developed countries from about 47 at the beginning of the last century to about 80 today and it is likely to increase even more, thanks to improving medical care and intervention. However, with the increased life expectancy worldwide, an increasing number of people will develop PD, which will socially and economically impact public healthcare and the future of modern society.
Nowadays, most of the treatment strategies for PD are based on the administration of dopamine, to compensate the lack of dopamine release from DA neurons [14]. However, these treatment strategies can alleviate the symptoms of the disease, but they can not stop or slow down the neuronal degeneration.
FAMILIAR FORMS OF PD AND THEIR GENETICS
Although most PD cases are sporadic and the exact cause for the disease onset is unknown, a small percentage of PD cases is genetically linked and shows an earlier manifestation [15]. Since the phenotypes of both sporadic and familiar cases are indistinguishable at the level of DA degeneration, the genetic cases, although rare, can provide the basis for a better understanding of the molecular pathways underling the disease and be instrumental to tackle the disease and potentially find a cure [16].
After the identification of SNCA gene, encoding for α-synuclein, that causes familiar forms of PD, many other genes have been discovered, which cause inherited PD and account for 10% of PD cases. Until now, several loci responsible genes for PD have been identified, and for six of them, the corresponding genes have been characterized. Four loci (Park1/4, Park3, Park5 and Park8) have been associated with autosomal dominant forms of PD, whereas Park2, Park6, Park7 and Park9 have been associated with autosomal recessive forms. Although no corresponding gene is known for loci Park3 and Park9, the other loci have been associated to α-synuclein gene (Park1/4), Parkin (Park2), UCHL1 (Park5), PINK1 (Park6), DJ1 (Park7) and LRRK2 (Park8), respectively [17].
Dominant Genes
Park1/4, one of the most common inherited forms of PD, is linked to parkinsonism caused by missense mutations and amplifications of α-synuclein and has been associated with autosomal dominant forms of PD [18]. This protein is expressed throughout the brain and is involved in learning, synaptic plasticity, vesicle dynamics and dopamine synthesis. The wild type protein is a potent inhibitor of phospholipase D2, which is involved in signal transduction, membrane vesicle trafficking and cytoskeletal dynamics. Considering how neurons rely on vesicular trafficking for their survival, functional α-synuclein is crucial for neuronal survival. Interestingly, due to its hydrophobic central region, this protein has naturally a high propensity to aggregate that is accentuated in mutants. Mutant forms of these proteins easily aggregate in neuronal cells in vitro and in vivo, initially forming an intermediate annular structure, and ultimately forming insoluble polymers or fibrils, which are the main constituents of the Lewy bodies, one of the most common histological hallmarks of PD.
Park8 has been identified as the leucine rich repeat kinase 2 gene (LRRK2). This is the most common form of inherited PD and the clinical features are similar to those of sporadic PD, except for the earlier onset age [19]. Until now, 20 missense or nonsense mutations have been reported. This gene encodes for an extremely large protein of 250 kDa, containing many different functional domains and it is highly expressed in the brain. It was reported to interact with Parkin [20], but it also genetically interacts with PINK1 and DJ-1 [21]. The G2019S mutation in the LRRK2 gene has been reported a number of times and appears to be one of the most common LRRK2 related mutations, accounting for 3-7% familial PD and 1–1.6% of so-called ‘sporadic PD’. Mutations in this gene inhibit an endogenous peroxidase promoting dysregulation of mitochondrial function and oxidative damage.
Recessive Genes
Recessive form of parkinsonism is known to be caused by mutations in parkin (Park2), PINK1 (Park6) and DJ-1 (Park7) genes. These are all relatively rare loss-of-function mutations that result in an early age of onset and slow disease progression.
DJ-1 mutations were firstly found in an Italian and a Dutch family and linked to autosomal-recessive forms of PD. After that, only one other mutation in a Uruguayan family has been identified [22]. DJ-I is almost ubiquitously expressed in organs, and it is present in synaptic terminals, mitochondria and membranous organelles [23]. The normal function of DJ-1 and its role in dopamine cell degeneration is unknown, but this protein is linked to oxidative stress response and mitochondrial function [24]. It has also reported that this gene has a role as tumor suppressor [25]. DJ-I protein was detected around Lewy bodies, but not as part of these. Several evidences suggested that DJ-I function as a dimer and analysis of the pathogenic Lys166Pro mutation showed that the dimer is less stable and an ectopic expression of this mutant is rapidly degraded [26].
Park6 gene was identified as PINK1 (PTEN induced kinase 1), a serine/threonine protein kinase that contains a Mitochondria Targeting Sequence (MTS) and localizes to mitochondria [27]. Mutations in this gene are much less common than mutations in the DJ1 or parkin gene. This protein is ubiquitously expressed and contains a serine/threonine kinase domain. Its function is to regulate mitochondrial dynamics and respiratory functions [28]. Interestingly, mitochondrial shape and dynamic is affected in PINK1 lacking cells, although literature does not always agree on the effect of PINK1 downregulation or overexpression upon mitochondria shape. Mutations in PINK1 have differential effects on protein stability, localization and kinase activity [29]. PINK1 associated cases of PD show a broad phenotypic spectrum, spanning from an early manifestation with atypical symptoms to late manifestation with the typical clinical PD symptoms.
Mutations in the parkin gene (Park2) are the most common among the three recessive forms of parkinsonism, and this gene was the first associated with recessive form of PD [30]. The gene codifies for an E3 ubiquitin ligase that is normally expressed in the cytoplasm, but translocates to mitochondria upon specific stimulation. More than 40 mutations have been identified, but only a weak correlation between clinical manifestation and type of mutation has been pointed out [31]. Of note, mutations in Parkin are not typically associated with the formation of Lewy bodies and α-synuclein aggregates.
PARKINSON’S DISEASE AND MITOCHONDRIA DYSFUNCTION
Most of the proteins encoded by Parkinson’s related genes, are linked to mitochondria and they have a role in protecting against some form of mitochondrial dysfunction and oxidative stress [32]. Some of them, like PINK1, are expressed on mitochondria and actively regulates mitochondria activity and fitness [1]. Others, like Parkin, are targeted to mitochondria upon specific stimulation, and select a subset of dysfunctional mitochondria for degradation [6]. DJ1 localizes to mitochondria during oxidative stress [33]. α-synuclein affects mitochondria function by interacting with mitochondria and enhancing mitochondria susceptibility to toxins, like rotenone, that interfere with electron transport chain creating build-up of electrons in the matrix and formation of reactive oxygen species (ROS) [34, 35].
Indeed, mitochondrial dysfunction is strongly implicated in the etiology of the disease and impaired mitochondria are found in animal and cellular models of PD. Body of evidences suggests that mitochondria dysfunction and subsequent oxidative stress causes the onset of PD.
Fission to Segregate; Fusion to Mitigate
Mitochondria are double membrane-bound organelles, which are responsible for multiple cellular events, including energy conversion [36, 37], Calcium (Ca2+) signaling [38] and amplification of programmed cells death cascade [39]. The most intriguing thing about mitochondria is that they are extremely dynamic and they frequently divide (mitochondria fission) and fuse (mitochondria fusion), changing size and shape and subcellular location [40]. Intuitively, mitochondria undergo division to populate new cells with new organelles; mitochondria fusion is on the other hands required to preserve the mitochondria network and allow intermixing of mitochondria matrix content (including mitochondria DNA) to preserve mitochondria function [41].
Mitochondria shape and localization are not random and directly correlated to mitochondria activity and fitness. In this respect, regulation of mitochondria fission and fusion events is required to respond to changes in metabolism. This is supported by the observations that mitochondria elongate in times of nutrient deprivation [42] or to boost oxidative phosphorylation.
Mitochondria fission and fusion is particularly important under stressful conditions: fusion between damaged mitochondria blends oxidative stress into the mitochondrial network and functionally compensate for potential damage [43]; fission is required to facilitate the removal of dysfunctional, damaged mitochondria. A pivotal study demonstrated how mitochondria fission often generates an asymmetric division where one daughter exhibits higher membrane potential and has better probability to undergoes fusion, while the other has lower membrane potential, does not fuse and it is more likely to be eliminated via mitophagy [41]. This work suggested that fission followed by selective fusion segregates dysfunctional mitochondria for degradation. In this respect, impairment of the fission machinery inhibits mitophagy.
Core components of the fission and fusion machinery are pro-fusion members dynamin related GTPases optic atrophy 1 (Opa1) and Mitofusin (Mfn), and pro-fission members dynamin like protein 1 (Drp1) and Fis1 [44]. Mitochondria fusion is achieved upon the coordinated activity of Mfn and Opa1 [45]. Mfn is a transmembrane GTPase embedded in the outer mitochondrial membrane, which is required on adjacent organelles to mediate the fusion of outer mitochondrial membrane. Opa1 is expressed on the inner mitochondrial membrane and regulates inner membrane fusion [46]. Mfn and Opa1 are eclectic proteins that have broader functions, despite their involvement in mitochondria fusion. For example, in mammals, while Mfn1 participates in the mitochondrial fusion reaction, in coordination with Opa1, Mfn2 forms complexes that are capable of tethering mitochondria to endoplasmic reticulum (ER), a structural feature essential for lipid synthesis, mitochondrial energy metabolism, Calcium (Ca2+) transfer between the organelles and Ca2+ dependent cell death [47]. Opa1 also has genetically distinguishable functions in mitochondria fusion and mitochondria cristae remodeling [48, 49], an ultrastuctural feature that allows the intramitochondrial redistribution of cytochrome c that is contained inside the mitochondrial cristae pockets. Opa1 functions as a molecular staple that keeps the mitochondria cristae junctions tight and its activity is required in the control of cristae junctions size upon induction of apoptosis [48].
To oppose fusion, Drp1, MFF (mitochondrial fission factor) and Fis1 have been found to be key components of the mammalian mitochondrial fission machinery. The large GTPase Drp1 is a dynamin-related protein that is expressed in the cytosol. A fraction of this protein is localized in spots on mitochondria, and a subset of these spots mark future fission sites in coordination with the endoplasmic reticulum. Upon induction of mitochondria fission, intermolecular oligomerization of Drp1 into ring-like structures occurs at membrane constriction sites [50]. Fis1 and MFF operate as Drp1 receptor on mitochondria outer membrane [51-53].
Pathogenesis of the Disease: The Mitochondrial Hypothesis
The earliest hypothesis of PD pathogenesis was based on the finding that chemical inhibition of mitochondrial Complex I could reproduce Parkinsonism [54]. Indeed it results in selective dopaminergic neuron loss and it iswidely used to create PD animal models. For instance, mitochondrial toxin rotenone that inhibits electron transfer from Complex I to ubiquinone, causes Parkinsonism [55]. Also, injection of MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), causes PD [56]: the product of metabolized MPTP, MPP+, inhibits Complex I of the electron transport chain and results in electron build up. The inhibition of Complex I has two major consequences: depletion of ATP and the generation of free radicals, with subsequent ROS formation that is toxic for the cell.
Supporting the hypothesis of a network of pathways converging to mitochondrial dysfunction, clear evidence of oxidative stress in postmortem PD brain and a reduction of Complex I activity has been described in organs of idiopathic PD patients [54]. Moreover, a primary role of mitochondrial respiratory chain impairment and consequent oxidative stress has also emerged from the study of rare familial forms of PD. PINK1 deficiency or disease-related PINK1 mutations, affects Complex I activity resulting in mitochondria depolarization and increased susceptibility to apoptotic stimuli [57]. General impaired respiration has also been observed in PINK1 deficient cells as a consequence of impaired delivery of respiratory chain substrates due to ROS dependent inhibition of glucose transporter [58]. Recently, a direct interaction between PINK1 and Complex I activity was described by Morais and co. workers, who showed that PINK1 dependent phosphorylation of Complex I on Serine-250 is a prerequisite for ubiquitinone reduction, thus unrevealing the biochemical link between PINK1 dysfunction and impaired respiration [59].
Of note, Parkin mutant fibroblasts from PD patients have also shown to have lower mitochondrial Complex I activity and ATP production, which was more markedly impaired when cells were forced to rely on oxidative phosphorylation rather than glycolysis to generate their ATP. These results are consistent with those seen in PINK1 deficient models and suggest that there might be a common pathway mediating recessive parkinsonism in humans as has been suggested from studies in Drosophila [60].
Interestingly, abnormalities in mitochondria shape, ultrastructure and subcellular localization, have been described in models of both PINK1 and Parkin deficient cells [9, 60, 61] and enlarged and swollen mitochondria have been found in postmortem tissues from biopsy of PD patients. However, it is still not clear whether mitochondria structure abnormalities are a consequence of respiratory chain impairment, perhaps to compensate for electron leak and decreased ATP synthesis, or directly contribute to the etiology of the disease and precede respiration impairment. Of note, deficits in Complex I driven respiration and specific Complex I activity impairment are observed before any mitochondrial morphology alteration manifests, suggesting that mitochondria morphology abnormalities are a consequence of respiration defects [57]. Considering that mitochondria respond to metabolism change by modulating their shape and dynamics, it is plausible that to compensate PINK1/Parkin respiratory defects and impaired ATP production, mitochondria elongate to boost oxidative phosphorylation. This might at least partially explain why enlarged and hyperfused mitochondria are found in cellular and animal models of PINK1/Parkin deficiency.
PINK1 AND PARKIN AND THE MITOCHONDRIA QUALITY CONTROL PATHWAY: A HATE-LOVE RELATIONSHIP
In 2006, by using the Drosophila Melanogaster fruit fly model system, three independent groups showed that PD related genes PINK1 and Parkin operate within the same pathway, with PINK1 functioning upstream of Parkin [3-5]. These works highlighted the fruit fly as an extremely powerful model system to gain insight into PD etiology. A number of fly models have been developed (such as PINK1, Parkin and OMI mutant flies), which show dopaminergic neuronal loss, mitochondrial dysfunction and locomotor deficits. In 2008, by using a fruit fly-based genetic interaction screening, Poole et al. showed a strong genetic interaction between PINK1/Parkin pathway and mitochondrial fission and fusion machinery. In particular, in flies, loss of function mutations of pro-fission protein Drp1 is lethal in a PINK1 or Parkin mutant background. Furthermore, PINK1 and Parkin mutant phenotypes, such as muscle degeneration, locomotor deficits and mitochondrial morphology alterations, are suppressed by increased Drp1 gene dosage or decreased pro- fusion Mitofusin gene dosage [62]. This finding highlighted for the first time a potential role for mitochondrial dynamics in the PINK1/Parkin pathway and suggested that PINK1/ Parkin pathway might promote mitochondria fission (or inhibit mitochondria fusion).
Recently, new insights have emerged into the function of the PINK1/Parkin pathway. Upon prolonged mitochondrial intoxication, using CCCP, Parkin is selectively recruited to impaired mitochondria and promotes their elimination via autophagy, a process known as mitophagy [6]. Further, PINK1 is required for Parkin translocation through a yet not fully understood mechanism [8]. Extended studies to elucidate the potential molecular mechanisms of this pathway showed that in several cell model systems, upon mitochondria intoxication by CCCP, Parkin is recruited to impaired mitochondria where it selectively ubiquitinates pro fusion protein Mfn [9, 10]. Lack of Parkin (or PINK1, which acts upstream Parkin in a linear pathway) results in impaired Mfn ubiquitination and increased Mfn steady state levels in several in vitro cellular systems and in vivo. This finding provided a biochemical explanation for the in vivo genetic interaction observations that show how decreased pro- fusion Mitofusin gene dosage in flies could ameliorate PINK1 or Parkin mutant phenotype.
Extensive studies in recent years allowed the dissection of this pathway and further details of the molecular mechanism of action of PINK/Parkin in mitochondria quality control have emerged. In healthy mitochondria, PINK1 is sequentially imported to mitochondria through the TOM and TIM complexes, and then it is released to span the inner mitochondrial membrane [63]. It has been shown that the proteases PARL and MPP [64] are responsible for its cleavage and subsequent degradation in a proteasome dependent manner [45]. However, upon CCCP induced mitochondria depolarization, PINK1 fails to be cleaved, it is exposed on the outer mitochondrial membrane, where it drives the recruitment of Parkin [8]. Recent works suggest that PINK1 both phosphorylates Parkin [65], Ubiquitin [66] and Mfn [67]. PINK1-dependent phosphorylation of Parkin regulates Parkin E3 ubiquitin ligase activity [65], although phosphomimetic Parkin mutation does not bypass PINK1 requirement for Parkin recruitment. However, PINK1 dependent phosphorylation of Ubiquitin is a Parkin activator and, in combination with PINK1-dependent phosphorylation of Parkin, is sufficient to fully activate Parkin E3 activity [66]. PINK1-dependent phosphorylation of Mfn is required for Parkin translocation. In particular, phospho-Mfn works as a molecular tag for the recruitment of Parkin that, once on mitochondria, ubiquitinates its targets [67]. In 2013 the complete repertoire of Parkin targets (Parkin-dependent ubiquitylome) have been published, which includes Mfn, VDAC, TOM20, Fis1, the authophagy adaptor p62 and Miro [11]. Parkin dependent ubiquitination of Miro (a GTPase that senses Ca2+ and binds mitochondria to the cytoskeleton via Milton) results in proteasome dependent degradation of Miro and consequent disruption of mitochondrion mobility [68]. Also, Mfn ubiquitination has followed by chaperone-mediated extraction of the protein from the outer mitochondrial membrane and its degradation [10]. As a result of Parkin-dependent ubiquitination of its targets on the outer mitochondrial membrane, mitochondria both loose their ability to fuse and to move along the microtubules of the cytoskeleton. They are therefore isolated from the mitochondrial network and targeted for degradation via Parkin dependent recruitment of cytosolic factors, including p62, that are required for the activation of mitophagy.
Recently a new pathway in the regulation of mitochondria quality control has been described, that accounts for the formation of mitochondria-derived vesicles (MDVs) that bud off mitochondria. Depending on the cargoes, emerging MDVs promote the degradation of their contents by either fusing to a subpopulation of peroxisomes [69] or lysosomes [70]. The latter is independent of autophagy, and it is induced by oxidative stress. Latest works describe a role for Parkin/PINK1 in the biogenesis of these vesicles, suggesting an additional role for these two proteins in the control of damaged mitochondrial proteins degradation [71]. These data characterize a novel vesicles-based highway that direct damaged mitochondrial proteins to lysosomes. This pathway is distinct from canonical mitophagy and it is primarily activated upon oxidative stress. Thus, PINK1 and Parkin promote mitochondrial quality control via at least two distinct pathways, either by tagging the entire organelle for autophagy-dependent degradation, or by shuttling specific cargoes to lysosome in an LC3/ATG-independent manner. The existence of an autophagy-independent pathway in the activation of mitochondria quality control is indeed supported by in vivo evidences [72].
UBIQUITINATION AND DE-UBIQUITINATION: A REVERSIBLE MODIFICATION THAT REGULATES SIGNALING PATHWAYS
Ubiquitination has recently emerged as a powerful tool to modulate proteins activity, via regulation of their subcellular localization and ability to interact with other proteins to form signaling complexes. E1 (ubiquitin-activating), E2 (ubiquitin-conjugating) and E3 (ubiquitin-ligase) enzymes can regulate the activity of proteins, through conjugating Ubiquitin (Ub) monomers. This event is called “ubiquitination” and consists in the formation of an isopeptide bond between the carboxyl group of the Ub C-terminus and the free amine group of a lysine side-chain of a target protein. In some cases, “linear ubiquitination” can also occur, that means the E1/E2/E3 cascade promotes the formation of a bond between Ub and the first methionine of a target, without the formation of a chain. However, Ub itself contain seven lysine residues, thus allowing ubiquitin chains formation [73]. Ub can form polyubiquitin chains of eight different linkages that mediate distinct biological functions [74]. Thus, the biological outcome of ubiquitin linkage can be modulated, depending on the ubiquitin chain that is formed. The best-characterized type of Ub conjugation is the Lys48 linked Ub chain that typically leads to degradation of the target by the proteasome. In contrast, chains linked via one of the other six lysines in Ub can function as regulatory signal in a variety of cellular pathways, including trafficking, signaling and autophagy. In this context, a common regulatory mechanism for many E3 ligases is the ability to self-catalyze their own ubiquitination, by a so-called “auto-ubiquitination” process.
Similar to other post-translational modifications, such as phosphorylation and acetylation, ubiquitination is also a reversible modification, mediated by a large family of deubiquitinating enzymes (DUBs). Interestingly, a large set of DUBs has opposite role of the E1/E2/E3 activity. Most DUBs recognize and remove ubiquitin from conjugated proteins and/or shorten ubiquitin chains, although some DUBs can be cross-reactive for some Ubs. Clearly, this class of enzymes not only can regulate E1/E2/E3 ubiquitinated targets, but also auto-ubiquitinating proteins. Therefore, DUBs have been found to play an important role in the regulation of multiple processes, such as regulation of receptor trafficking, cell cycle progression, regulation of cell migration, regulation of intracellular signaling and transcriptional control [12]. Two different DUB enzyme mechanisms have been described: metalloproteases, classified as the JAMN/MPN+ domain superfamily, and the Cys-proteases. The Cys-proteases DUBs are further divided into four subclasses: the USP (ubiquitin-specific protease) superfamily, the OUT (ovarian tumour) superfamily, the UCH (ubiquitin C-terminal hydrolase) superfamily, and the Machado-Joseph disease domain superfamily (MJDs) [75]. Accumulating evidences indicate that DUBs mutations are involved with human diseases development.
Recent works identified DUBs interacting with and regulating proteins associated with familial forms of PD, α-synuclein such as [76] and Parkin [77-84]. In more detail, ataxin-3, USP8, USP15 and USP30 have been found to have a role in modulating Parkin auto-ubiquitination and Parkin-mediated mitophagy.
The MJDs superfamily member ataxin-3 is the first DUB reported to have a role in Parkin deubiquitination and stability. This enzyme, which mutations cause MJD, has been found to deubiquitinate Parkin, but no evidences indicated a role of wild-type ataxin-3 in Parkin stability regulation. However, the MJF-associated form of ataxin-3 promotes Parkin degradation in a proteasome-independent manner [78, 85]. Moreover, ataxin-3 resulted to be unable to hydrolyze preassembled Ub-conjugates on Parkin. Ataxin-3 acts through an unusual mechanism, stabilizing the interaction between Parkin and E2 that now cannot dissociate. When ataxin-3 is present, it can also interact with E2-Ub complex and redirect the Ub transfer from E2 onto itself, rather than onto Parkin [77]. Patients with MJD can exhibit symptoms similar to those with PD and show neurodegeneration in many of the same brain region. The interaction between ataxin-3 and Parkin could, at least partially, explain the similarities between these diseases.
Although ataxin-3 came out as a regulator of Parkin stability and turnover, its role on Parkin-mediated mitophagy was not investigated. Furthermore, as E3s could be regulated by multiple DUBs, the same research group performed an RNAi-screen in U2OS cells to identify other enzymes that could be involved in Parkin deubiquitination and Parkin-dependent mitophagy [79]. They report that Usp8, which was known to be associated with endosomal trafficking, is necessary for Parkin recruitment to mitochondria and mitophagy. Knock-down of Usp8 delayed but not abolished Parkin translocation to depolarized mitochondria, upon CCCP treatment. Counterintuitively, Parkin steady-state levels were increasing in Usp8 RNAi conditions, advocating the hypothesis that Usp8 specifically acts on Parkin by functional ubiquitination rather than degradative ubiquitination.Of interest, they found that Usp8 specifically removesK6-linked Ub conjugates, which in turns promotes Parkin recruitment to mitochondria [80]. Therefore, by impinging on the transcriptional levels of a Parkin-specific DUB, the authors showed how mitophagy could be inhibited.
Another research group used a different approach to reveal Parkin interacting protein. Tandem affinity purification coupled to mass spectrometry identified Usp11 and Usp15 as DUBs binding Parkin [84]. Further experiments, confirmed an interaction between Usp15 and overexpressed Parkin. Usp15 has an opposite role on mitophagy compared to Usp8, since Usp15 knockdown enhanced mitochondria elimination in SH-SY5Y cells. Moreover, RNAi-mediated knockdown of this DUB rescued mitophagy defects in fibroblasts both from PARK2 and PINK1 mutant PD patients. However, Usp15 does not act on Parkin protein itself, nor prevents Parkin translocation to impaired mitochondria. The targets of Usp15 are the Parkin-ubiquitinated proteins on mitochondria. Usp15 impairs mitophagy through deubiquitinating Parkin targets on depolarized mitochondria and in this respect, it counteracts Parkin ubiquitination activity over Parkin targets. These data were also confirmed in vivo in Drosophila, where silencing of Usp15 homolog rescued parkin mutant phenotype, thus showing a genetic interaction between the two genes [84].
Usp30 came out as a DUB inhibiting mitophagy during a human cDNA library screening in a mitochondrial degradation assay. Although more than 100 DUBs were tested, only two DUBs were able to robustly block mitophagy (USP30 and DUBA2) [83]. However, only Usp30 was reported to be localized in the outer mitochondrial membrane. Co-expression of Usp30 did not alter Parkin expression levels or its translocation to depolarized mitochondria, nevertheless it was able to reduce CCCP-induced recruitment of autophagy markers and ubiquitin signal on GFP-Parkin positive mitochondria. The genuine target of Usp30 deubiquitinating activity was found to be TOM20. Thus, Usp30 overexpression opposes Parkin ubiquitination of TOM20, blocking mitophagy. On the other hands, Usp30 downregulation enhances mitochondrial degradation in neurons via stabilization of ubiquitinated forms of TOM20, which work as mitophagy signal. Fruit fly in vivo models of Parkinson disease showed that knockdown of Usp30 was able to rescue defective mitophagy caused by Parkin mutation. Moreover, it improves mitochondrial integrity in Parkin- or PINK1-deficient flies and protects flies against paraquat toxicity in vivo. Parkin synthesizes Lys 6, Lys 11 and Lys 63 Ub chains on depolarized mitochondria. Usp30 opposes the induction of mitophagy through preferentially hydrolyzing Lys 6- and Lys 11-linked Ub chains on Parkin target TOM20 [82]. Lys 6- and Lys 11-linked Ub chains are fundamental mitophagy signals, since other DUBs targeted to mitochondria which are able to specifically hydrolyze these Ub chains are blocking mitochondrial degradation
All these studies, suggest a potential role for DUBs in modulating mitochondrial quality control and impact on cell survival. Specific DUBs inhibition or enhancement could, for instance, compensate for PINK1 or Parkin loss-of-function mutations in PD patients.
DUBs AS THERAPEUTIC TARGETS
Due to their involvement in the regulation of important signaling pathways, DUBs are emerging as attractive druggable candidates [86]. Clinical trials for specific inhibitors of the ubiquitin-proteasome system have already been approved in cancer therapy for the treatment of multiple myeloma [87-89]. Moreover, high-throughput screening of small chemical libraries identified non selective DUBs inhibitors as potent inducers of apoptosis in various cancer cells. For example, G5, a small molecule inhibitor of DUBs, was recently identified as a strong inhibitor of NLRP3 signaling pathway, thus affecting the NLRP3-dependent inflammatory response [90].
Recently, a number of reports identified additional syntetic small molecule as effective DUB inhibitors. Chemically diverse molecules have been reported to inhibit one or more of the UCH and USP family members. However, drug discovery against the ubiquitin system is still emerging and effective compounds are relatively limited. Often, these compounds target more than one DUB and the most promising chemicals showing selective inhibitions still have not been tested for all full length human DUBs. Here, we report the most promising inhibitors for DUBs of the USP superfamily, which hopefully, in the next future, could be used as therapy for different pathologies, including PD. For detailed information about the biochemical mechanism of action and other available inhibitors for DUBs, the reader is referred to excellent previews [91, 92].
Usp7 is a deubiquitinase first identified as associated with a herpes-virus, and it facilitates lytic growth. It has also been shown as indirect regulator of p53, FOXO4 and PTEN proteins, able to destabilize and compromise the effectiveness of these tumor suppressor proteins [93]. Therefore, Usp7 can be considered as an oncogenic pro-survival protein. Since it was discovered, Usp7 antagonists have been deeply studied, as its inhibition could block tumor progression. At now, an high-throughput screening (HTS) identified HBX 19,818 and the related HBX 28,258 as inhibitors for Usp7 [94]. Using the same method, but in a parallel study, P5091 came out as selective antagonist of this DUB. These molecules were tested in multiple cell lines, thus proving their ability to stabilize p53, to inhibit tumor growth and to promote apoptosis in tumor cell lines. In vivo studies confirmed the effect of P22077, an optimized derivative of P5091, in several orthotopic neuroblastoma xenograft models [95].
Some DUBs have been found to have a role in DNA damage response, this is the case of Usp1. When it is associated to the co-factor UAF1 (USP1-associated factor 1), Usp1 deubiquitinates different targets involved in cell damage response and related to different pathologies. Usp1 deubiquitinates FANCD2 and FANCI in the Falconi anemia pathway, promoting DNA repair. This DUB participate in a similar process also in the translesion synthesis pathway, where deubiquitinates PCNA (proliferating cell nuclear antigen) in order to prevent low-fidelity polymerases recruitment and thereby preserve DNA integrity [96]. Through deubiquitinating inhibitors of DNA binding transcription factors, Usp1 has been found to maintain stem-cell characteristics in osteosarcoma cells [97]. An HTS using USP1/UAF1 complex identified different inhibitors for this deubiquitinating enzyme complex [98], and pimozide resulted to be the more efficient chemical against it. However, other DUBs resulted to be inhibited by this same compound, thus, the effects seen by using it in cells could not be imputed to the specific inhibition of USP1/UAF1 complex. However, other two independent HTS revealed other molecules that could be used as Usp1 inhibitors: ML323, and the two analogues SJB2-043 and SJB3-019A [99, 100]. This compounds resulted to be more selective for Usp1, interfering with the protein in the different pathways in which it controls DNA integrity maintenance. Different studies validated the potential therapeutic efficacy of Usp1 inhibition in different pathologies, such as leukemia and other cancers.
Usp14 and UCHL5 are two deubiquitinating enzymes found to be associated with the proteasome. These two proteins antagonize proteasomal degradation of substrate, by hydrolyzing poly-Ub chains from the substrate distal end, a process called trimming. Usp14 deletion is embryonic lethal in mice, since this enzyme is important in general homeostasis. Recently, IU1 has been reported as Usp14 inhibitor, confirming its role in Ub chain trimming. Inhibition of Usp14 has beneficial effects on cell viability, promoting proteasomal degradation of damaged proteins. On the other hand, overexpression of Usp14 is associated with cancer progression [101]. Moreover, in vivo and in vitro evidences showed the efficiency of inhibiting Usp14 and UCHL5 in tumor models, using b-AP15 [102].
Besides some exceptions, most of these inhibitor molecules are poorly characterized in terms of structure and mechanism of action. Further biochemical studies together with proteomic tools could better explain the features of these molecules, thus allowing to better understand their mechanism of action. Nevertheless, more DUBs inhibitors are under development, with the aim to discover new therapies for different human diseases.
AUREA MEDIOCRITAS: THE DESIRABLE MIDDLE BETWEEN TWO EXTREMES
In recent years, much effort has been put in order to identify specific DUBs that oppose Parkin in the ubiquitination of its targets. The identification of a Parkin-opposing DUB, counteracting Parkin activity in the regulation of mitophagy, might be instrumental to develop specific isopeptidase inhibitors or activators that can modulate the fundamental biological process of mitophagy and impact on cell survival.
Since one of the hallmark of PD consists of accumulation of misfolded proteins and unfunctional mitochondria as a result of impaired mitophagy, chemical or genetic intervention that suppress Parkin-opposing specific DUB, can potentially be used to eliminate these toxic compounds and improve viability (Fig. 1).
Fig. (1).
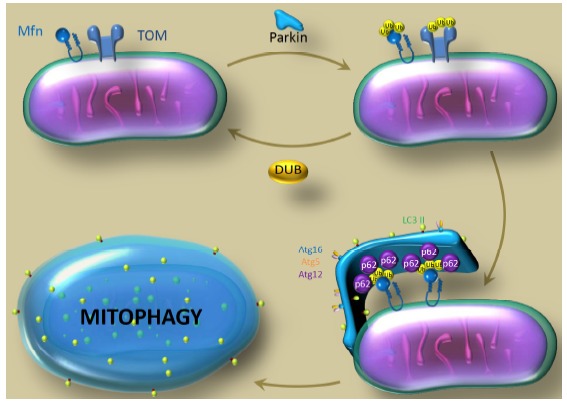
Counteracting Parkin. The cartoon illustrates the rational behind the potential therapeutic advantages of identifying DUBs that oppose Parkin in the ubiquitination of its target. In vivo evidences suggest that suppression of specific DUBs is sufficient to promote mitophagy in the absence of Parkin or PINK1, via stabilization of the ubiquitinated forms of Parkin substrates. Therefore, suppression of specific DUB activity might be instrumental for the activation of mitochondria clearance pathway downstream PINK1/Parkin and can offer a therapeutic approach to ameliorate PD phenotype.
Overall, the enhancement of proteasome activity may offer a strategy to reduce the levels of aberrant proteins in cells and in the whole organism under stress.
Of note, interference with the proteasome machinery has already been proved to be effective in cancer therapy and recently, high throughput screening resulted in the discovery of highly specific synthetic small molecules that target selective components of the proteasome machinery via enhancement or suppression of ubiquitin-conjugating enzymes and DUBs.
In vivo evidences suggest that suppression of specific DUBs is sufficient to promote mitophagy in the absence of Parkin or PINK1, via stabilization of the ubiquitinated forms of Parkin substrates. Suppression or enhancement of specific DUB activity might therefore be instrumental for the activation of mitochondria clearance pathway downstream PINK1/Parkin and can potentially be beneficial in ameliorating phenotypic deficits in PD.
ACKNOWLEDGEMENTS
Declared none.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- 1.Silvestri L., Caputo V., Bellacchio E., Atorino L., Dallapiccola B., Valente E.M., Casari G. Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum. Mol. Genet. 2005;14(22):3477–3492. doi: 10.1093/hmg/ddi377. [DOI] [PubMed] [Google Scholar]
- 2.Shimura H., Hattori N., Kubo Si., Mizuno Y., Asakawa S., Minoshima S., Shimizu N., Iwai K., Chiba T., Tanaka K., Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000;25(3):302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- 3.Clark I.E., Dodson M.W., Jiang C., Cao J.H., Huh J.R., Seol J.H., Yoo S.J., Hay B.A., Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441(7097):1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 4.Yang Y., Gehrke S., Imai Y., Huang Z., Ouyang Y., Wang J.W., Yang L., Beal M.F., Vogel H., Lu B. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc. Natl. Acad. Sci. USA. 2006;103(28):10793–10798. doi: 10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park J., Lee S.B., Lee S., Kim Y., Song S., Kim S., Bae E., Kim J., Shong M., Kim J.M., Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441(7097):1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 6.Narendra D., Tanaka A., Suen D.F., Youle R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ziviani E., Tao R.N., Whitworth A.J. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc. Natl. Acad. Sci. USA. 2010;107(11):5018–5023. doi: 10.1073/pnas.0913485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Narendra D.P., Jin S.M., Tanaka A., Suen D.F., Gautier C.A., Shen J., Cookson M.R., Youle R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8(1):e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ziviani E., Tao R.N., Whitworth A.J. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc. Natl. Acad. Sci. USA. 2010;107(11):5018–5023. doi: 10.1073/pnas.0913485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanaka A., Cleland M.M., Xu S., Narendra D.P., Suen D.F., Karbowski M., Youle R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarraf S.A., Raman M., Guarani-Pereira V., Sowa M.E., Huttlin E.L., Gygi S.P., Harper J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496(7445):372–376. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burrows J.F., Johnston J.A. Regulation of cellular responses by deubiquitinating enzymes: an update. Front. Biosci. (Landmark Ed.) 2012;17:1184–1200. doi: 10.2741/3980. [DOI] [PubMed] [Google Scholar]
- 13.Moore D.J., West A.B., Dawson V.L., Dawson T.M. Molecular pathophysiology of Parkinson's disease. Annu. Rev. Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- 14.Alobaidi H., Pall H. The role of dopamine replacement on the behavioural phenotype of Parkinson’s disease. Behav. Neurol. 2013;26(4):225–235. doi: 10.3233/BEN-2012-120265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gasser T. Molecular pathogenesis of Parkinson disease: insights from genetic studies. Expert Rev. Mol. Med. 2009;11:e22. doi: 10.1017/S1462399409001148. [DOI] [PubMed] [Google Scholar]
- 16.Ryan B.J., Hoek S., Fon E.A., Wade-Martins R. Mitochondrial dysfunction and mitophagy in Parkinson's: from familial to sporadic disease. Trends Biochem. Sci. 2015;40:200–210. doi: 10.1016/j.tibs.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 17.Corti O., Lesage S., Brice A. What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol. Rev. 2011;91(4):1161–1218. doi: 10.1152/physrev.00022.2010. [DOI] [PubMed] [Google Scholar]
- 18.Ozansoy M., Başak A.N. The central theme of Parkinson’s disease: α-synuclein. Mol. Neurobiol. 2013;47(2):460–465. doi: 10.1007/s12035-012-8369-3. [DOI] [PubMed] [Google Scholar]
- 19.Esteves A.R., Swerdlow R.H., Cardoso S.M. LRRK2, a puzzling protein: insights into Parkinson’s disease pathogenesis. Exp. Neurol. 2014;261:206–216. doi: 10.1016/j.expneurol.2014.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith W.W., Pei Z., Jiang H., Moore D.J., Liang Y., West A.B., Dawson V.L., Dawson T.M., Ross C.A. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc. Natl. Acad. Sci. USA. 2005;102(51):18676–18681. doi: 10.1073/pnas.0508052102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sämann J., Hegermann J., von Gromoff E., Eimer S., Baumeister R., Schmidt E. Caenorhabditits elegans LRK-1 and PINK-1 act antagonistically in stress response and neurite outgrowth. J. Biol. Chem. 2009;284(24):16482–16491. doi: 10.1074/jbc.M808255200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Duijn C.M., Dekker M.C., Bonifati V., Galjaard R.J., Houwing-Duistermaat J.J., Snijders P.J., Testers L., Breedveld G.J., Horstink M., Sandkuijl L.A., van Swieten J.C., Oostra B.A., Heutink P. Park7, a novel locus for autosomal recessive early-onset parkinsonism, on chromosome 1p36. Am. J. Hum. Genet. 2001;69(3):629–634. doi: 10.1086/322996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagakubo D., Taira T., Kitaura H., Ikeda M., Tamai K., Iguchi-Ariga S.M., Ariga H. DJ-1, a novel oncogene which transforms mouse NIH3T3 cells in cooperation with ras. Biochem. Biophys. Res. Commun. 1997;231:509–213. doi: 10.1006/bbrc.1997.6132. [DOI] [PubMed] [Google Scholar]
- 24.Kim S.J., Park Y.J., Hwang I.Y., Youdim M.B., Park K.S., Oh Y.J. Nuclear translocation of DJ-1 during oxidative stress-induced neuronal cell death. Free Radic. Biol. Med. 2012;53(4):936–950. doi: 10.1016/j.freeradbiomed.2012.05.035. [DOI] [PubMed] [Google Scholar]
- 25.Cao J., Lou S., Ying M., Yang B. DJ-1 as a human oncogene and potential therapeutic target. Biochem. Pharmacol. 2015;93(3):241–250. doi: 10.1016/j.bcp.2014.11.012. [DOI] [PubMed] [Google Scholar]
- 26.Repici M., Straatman K.R., Balduccio N., Enguita F.J., Outeiro T.F., Giorgini F. Parkinson’s disease-associated mutations in DJ-1 modulate its dimerization in living cells. J. Mol. Med. 2013;91(5):599–611. doi: 10.1007/s00109-012-0976-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Narendra D.P., Jin S.M., Tanaka A., Suen D.F., Gautier C.A., Shen J., Cookson M.R., Youle R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8(1):e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsuda S., Kitagishi Y., Kobayashi M. Function and characteristics of PINK1 in mitochondria. Oxid. Med. Cell. longevity. 2013;91(5):599–611. doi: 10.1155/2013/601587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song S., Jang S., Park J., Bang S., Choi S., Kwon K.Y., Zhuang X., Kim E., Chung J. Characterization of PINK1 (PTEN-induced putative kinase 1) mutations associated with Parkinson disease in mammalian cells and Drosophila. J. Biol. Chem. 2013;288(8):5660–5672. doi: 10.1074/jbc.M112.430801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y., Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392(6676):605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 31.Morrison K.E. Parkin mutations and early onset parkinsonism. Brain. 2003;126(Pt 6):1250–1251. doi: 10.1093/brain/awg189. [DOI] [PubMed] [Google Scholar]
- 32.Büeler H. Impaired mitochondrial dynamics and function in the pathogenesis of Parkinson’s disease. Exp. Neurol. 2009;218(2):235–246. doi: 10.1016/j.expneurol.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 33.Bonifati V., Rizzu P., Squitieri F., Krieger E., Vanacore N., van Swieten J.C., Brice A., van Duijn C.M., Oostra B., Meco G., Heutink P. DJ-1( PARK7), a novel gene for autosomal recessive, early onset parkinsonism. Neurol. Sci. 2003;24(3):159–160. doi: 10.1007/s10072-003-0108-0. [DOI] [PubMed] [Google Scholar]
- 34.Manning-Bog A.B., McCormack A.L., Li J., Uversky V.N., Fink A.L., Di Monte D.A. The herbicide paraquat causes up-regulation and aggregation of alphα-synuclein in mice: paraquat and alphα-synuclein. J. Biol. Chem. 2002;277(3):1641–1644. doi: 10.1074/jbc.C10056020. [DOI] [PubMed] [Google Scholar]
- 35.Sherer T.B., et al. An in vitro model of Parkinson's disease: linking mitochondrial impairment to altered alphα-synuclein metabolism and oxidative damage. J. Neurosci. 2002;22:7006–7015. doi: 10.1523/JNEUROSCI.22-16-07006.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Attardi G., Schatz G. Biogenesis of mitochondria. Annu. Rev. Cell Biol. 1988;4:289–333. doi: 10.1146/annurev.cb.04.110188.001445. [DOI] [PubMed] [Google Scholar]
- 37.Saraste M. Oxidative phosphorylation at the fin de siecle. Science. 1999;283:1488–1493. doi: 10.1126/science.283.5407.1488. [DOI] [PubMed] [Google Scholar]
- 38.Butow R.A., Avadhani N.G. Mitochondrial signaling: the retrograde response. Mol. Cell. 2004;14(1):1–15. doi: 10.1016/S1097-2765(04)00179-0. [DOI] [PubMed] [Google Scholar]
- 39.Danial N.N., Korsmeyer S.J. Cell death: critical control points. Cell. 2004;116(2):205–219. doi: 10.1016/S0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 40.Chen H., Chan D.C. Critical dependence of neurons on mitochondrial dynamics. Curr. Opin. Cell Boil. 2006;18:453–459. doi: 10.1016/j.ceb.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 41.Twig G., Elorza A., Molina A.J., Mohamed H., Wikstrom J.D., Walzer G., Stiles L., Haigh S.E., Katz S., Las G., Alroy J., Wu M., Py B.F., Yuan J., Deeney J.T., Corkey B.E., Shirihai O.S. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gomes L.C., Di Benedetto G., Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011;13(5):589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soubannier V., Rippstein P., Kaufman B.A., Shoubridge E.A., McBride H.M. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS One. 2012;7(12):e52830. doi: 10.1371/journal.pone.0052830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chan D.C. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 2006;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- 45.Meeusen S.L., Nunnari J. How mitochondria fuse. Curr. Opin. Cell Biol. 2005;17(4):389–394. doi: 10.1016/j.ceb.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 46.McNiven M.A., Cao H., Pitts K.R., Yoon Y. The dynamin family of mechanoenzymes: pinching in new places. Trends Biochem. Sci. 2000;25(3):115–120. doi: 10.1016/S0968-0004(99)01538-8. [DOI] [PubMed] [Google Scholar]
- 47.de Brito O.M., Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456(7222):605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 48.Frezza C., Cipolat S., Martins de Brito O., Micaroni M., Beznoussenko G.V., Rudka T., Bartoli D., Polishuck R.S., Danial N.N., De Strooper B., Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 49.Cogliati S., Frezza C., Soriano M.E., Varanita T., Quintana-Cabrera R., Corrado M., Cipolat S., Costa V., Casarin A., Gomes L.C., Perales-Clemente E., Salviati L., Fernandez-Silva P., Enriquez J.A., Scorrano L. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160–171. doi: 10.1016/j.cell.2013.08.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smirnova E., Griparic L., Shurland D.L., van der Bliek A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell. 2001;12(8):2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoon Y., Krueger E.W., Oswald B.J., McNiven M.A. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell. Biol. 2003;23(15):5409–5420. doi: 10.1128/MCB.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gandre-Babbe S., van der Bliek A.M. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol. Biol. Cell. 2008;19(6):2402–2412. doi: 10.1091/mbc.E07-12-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Otera H., Wang C., Cleland M.M., Setoguchi K., Yokota S., Youle R.J., Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. cells. J. Cell Biol. 2010;191:1141–1158. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schapira A.H., Cooper J.M., Dexter D., Jenner P., Clark J.B., Marsden C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet. 1989;1(8649):1269. doi: 10.1016/S0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- 55.Betarbet R., Sherer T.B., MacKenzie G., Garcia-Osuna M., Panov A.V., Greenamyre J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000;3(12):1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 56.Langston J.W., Ballard P., Tetrud J.W., Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219(4587):979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- 57.Morais V.A., Verstreken P., Roethig A., Smet J., Snellinx A., Vanbrabant M., Haddad D., Frezza C., Mandemakers W., Vogt-Weisenhorn D., Van Coster R., Wurst W., Scorrano L., De Strooper B. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med. 2009;1(2):99–111. doi: 10.1002/emmm.200900006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gandhi S., Wood-Kaczmar A., Yao Z., Plun-Favreau H., Deas E., Klupsch K., Downward J., Latchman D.S., Tabrizi S.J., Wood N.W., Duchen M.R., Abramov A.Y. PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell. 2009;33:627–638. doi: 10.1016/j.molcel.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morais V.A., Haddad D., Craessaerts K., De Bock P.J., Swerts J., Vilain S., Aerts L., Overbergh L., Grünewald A., Seibler P., Klein C., Gevaert K., Verstreken P., De Strooper B. PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science. 2014;344(6180):203–207. doi: 10.1126/science.1249161. [DOI] [PubMed] [Google Scholar]
- 60.Mortiboys H., Thomas K.J., Koopman W.J., Klaffke S., Abou-Sleiman P., Olpin S., Wood N.W., Willems P.H., Smeitink J.A., Cookson M.R., Bandmann O. Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann. Neurol. 2008;64(5):555–565. doi: 10.1002/ana.21492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Klein P., Müller-Rischart A.K., Motori E., Schönbauer C., Schnorrer F., Winklhofer K.F., Klein R. Ret rescues mitochondrial morphology and muscle degeneration of Drosophila Pink1 mutants. EMBO J. 2014;(5):341–355. doi: 10.1002/embj.201284290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Poole A.C., Thomas R.E., Andrews L.A., McBride H.M., Whitworth A.J., Pallanck L.J. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. USA. 2008;105(5):1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jin S.M., Lazarou M., Wang C., Kane L.A., Narendra D.P., Youle R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010;191(5):933–942. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Greene A.W., Grenier K., Aguileta M.A., Muise S., Farazifard R., Haque M.E., McBride H.M., Park D.S., Fon E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012;13(4):378–385. doi: 10.1038/embor.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sha D., Chin L.S., Li L. Phosphorylation of parkin by Parkinson disease-linked kinase PINK1 activates parkin E3 ligase function and NF-kappaB signaling. Hum. Mol. Genet. 2010;19(2):352–363. doi: 10.1093/hmg/ddp501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Koyano F., Okatsu K., Kosako H., Tamura Y., Go E., Kimura M., Kimura Y., Tsuchiya H., Yoshihara H., Hirokawa T., Endo T., Fon E.A., Trempe J.F., Saeki Y., Tanaka K., Matsuda N. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510(7503):162–166. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- 67.Chen Y., Dorn G.W., II PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340(6131):471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang X., Winter D., Ashrafi G., Schlehe J., Wong Y.L., Selkoe D., Rice S., Steen J., LaVoie M.J., Schwarz T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Neuspiel M., Schauss A.C., Braschi E., Zunino R., Rippstein P., Rachubinski R.A., Andrade-Navarro M.A., McBride H.M. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr. Biol. 2008;18(2):102–108. doi: 10.1016/j.cub.2007.12.038. [DOI] [PubMed] [Google Scholar]
- 70.Soubannier V., McLelland G.L., Zunino R., Braschi E., Rippstein P., Fon E.A., McBride H.M. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 2012;22(2):135–141. doi: 10.1016/j.cub.2011.11.057. [DOI] [PubMed] [Google Scholar]
- 71.McLelland G.L., Soubannier V., Chen C.X., McBride H.M., Fon E.A. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014;33(4):282–295. doi: 10.1002/embj.201385902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vincow E.S., Merrihew G., Thomas R.E., Shulman N.J., Beyer R.P., MacCoss M.J., Pallanck L.J. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc. Natl. Acad. Sci. USA. 2013;110(16):6400–6405. doi: 10.1073/pnas.1221132110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kleiger G., Mayor T. Perilous journey: a tour of the ubiquitin-proteasome system. Trends Cell Biol. 2014;24(6):352–359. doi: 10.1016/j.tcb.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Komander D., Rape M. The ubiquitin code. Annu. Rev. Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 75.Komander D. Mechanism, specificity and structure of the deubiquitinases. Subcell. Biochem. 2010;54:69–87. doi: 10.1007/978-1-4419-6676-6_6. [DOI] [PubMed] [Google Scholar]
- 76.Rott R., Szargel R., Haskin J., Bandopadhyay R., Lees A.J., Shani V., Engelender S. α-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc. Natl. Acad. Sci. USA. 2011;108(46):18666–18671. doi: 10.1073/pnas.1105725108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Durcan T.M., Kontogiannea M., Bedard N., Wing S.S., Fon E.A. Ataxin-3 deubiquitination is coupled to Parkin ubiquitination via E2 ubiquitin-conjugating enzyme. J. Biol. Chem. 2012;287(1):531–541. doi: 10.1074/jbc.M111.288449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Durcan T.M., Kontogiannea M., Thorarinsdottir T., Fallon L., Williams A.J., Djarmati A., Fantaneanu T., Paulson H.L., Fon E.A. The Machado-Joseph disease-associated mutant form of ataxin-3 regulates parkin ubiquitination and stability. Hum. Mol. Genet. 2011;20(1):141–154. doi: 10.1093/hmg/ddq452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Durcan T.M., Tang M.Y., Pérusse J.R., Dashti E.A., Aguileta M.A., McLelland G.L., Gros P., Shaler T.A., Faubert D., Coulombe B., Fon E.A. USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J. 2014;33(21):2473–2491. doi: 10.15252/embj.201489729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Durcan T.M., Fon E.A. USP8 and PARK2/parkin-mediated mitophagy. Autophagy. 2015;11(2):428–429. doi: 10.1080/15548627.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dikic I., Bremm A. DUBs counteract parkin for efficient mitophagy. EMBO J. 2014;33(21):2442–2443. doi: 10.15252/embj.201490101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cunningham C.N., Baughman J.M., Phu L., Tea J.S., Yu C., Coons M., Kirkpatrick D.S., Bingol B., Corn J.E. USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat. Cell Biol. 2015;17(2):160–169. doi: 10.1038/ncb3097. [DOI] [PubMed] [Google Scholar]
- 83.Bingol B., Tea J.S., Phu L., Reichelt M., Bakalarski C.E., Song Q., Foreman O., Kirkpatrick D.S., Sheng M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014;510(7505):370–375. doi: 10.1038/nature13418. [DOI] [PubMed] [Google Scholar]
- 84.Cornelissen T., Haddad D., Wauters F., Van Humbeeck C., Mandemakers W., Koentjoro B., Sue C., Gevaert K., De Strooper B., Verstreken P., Vandenberghe W. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum. Mol. Genet. . 2014;23:5227–5242. doi: 10.1093/hmg/ddu244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Durcan T.M., Fon E.A. Mutant ataxin-3 promotes the autophagic degradation of parkin. Autophagy. 2011;7(2):233–234. doi: 10.4161/auto.7.2.14224. [DOI] [PubMed] [Google Scholar]
- 86.Salmena L., Pandolfi P.P. Changing venues for tumour suppression: balancing destruction and localization by monoubiquitylation. Nat. Rev. Cancer. 2007;7(6):409–413. doi: 10.1038/nrc2145. [DOI] [PubMed] [Google Scholar]
- 87.Brancolini C. Inhibitors of the Ubiquitin-Proteasome System and the cell death machinery: How many pathways are activated? Curr. Mol. Pharmacol. 2008;1(1):24–37. doi: 10.2174/1874467210801010024. [DOI] [PubMed] [Google Scholar]
- 88.Hussain S., Zhang Y., Galardy P.J. DUBs and cancer: the role of deubiquitinating enzymes as oncogenes, non-oncogenes and tumor suppressors. Cell Cycle. 2009;8(11):1688–1697. doi: 10.4161/cc.8.11.8739. [DOI] [PubMed] [Google Scholar]
- 89.Colland F. The therapeutic potential of deubiquitinating enzyme inhibitors. Biochem. Soc. Trans. 2010;38(Pt 1):137–143. doi: 10.1042/BST0380137. [DOI] [PubMed] [Google Scholar]
- 90.Py B.F., Kim M.S., Vakifahmetoglu-Norberg H., Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell. 2013;49(2):331–338. doi: 10.1016/j.molcel.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 91.Lill J.R., Wertz I.E. Toward understanding ubiquitin-modifying enzymes: from pharmacological targeting to proteomics. Trends Pharmacol. Sci. 2014;35(4):187–207. doi: 10.1016/j.tips.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 92.Ndubaku C., Tsui V. Inhibiting the deubiquitinating enzymes (DUBs). J. Med. Chem. 2015;58(4):1581–1595. doi: 10.1021/jm501061a. [DOI] [PubMed] [Google Scholar]
- 93.Li M., Chen D., Shiloh A., Luo J., Nikolaev A.Y., Qin J., Gu W. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature. 2002;416(6881):648–653. doi: 10.1038/nature737. [DOI] [PubMed] [Google Scholar]
- 94.Colland F., Formstecher E., Jacq X., Reverdy C., Planquette C., Conrath S., Trouplin V., Bianchi J., Aushev V.N., Camonis J., Calabrese A., Borg-Capra C., Sippl W., Collura V., Boissy G., Rain J.C., Guedat P., Delansorne R., Daviet L. Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol. Cancer Ther. 2009;8(8):2286–2295. doi: 10.1158/1535-7163.MCT-09-0097. [DOI] [PubMed] [Google Scholar]
- 95.Fan Y.H., Cheng J., Vasudevan S.A., Dou J., Zhang H., Patel R.H., Ma I.T., Rojas Y., Zhao Y., Yu Y., Zhang H., Shohet J.M., Nuchtern J.G., Kim E.S., Yang J. USP7 inhibitor P22077 inhibits neuroblastoma growth via inducing p53-mediated apoptosis. Cell death dis. 2013;4:e867. doi: 10.1038/cddis.2013.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.García-Santisteban I., Peters G.J., Giovannetti E., Rodríguez J.A. USP1 deubiquitinase: cellular functions, regulatory mechanisms and emerging potential as target in cancer therapy. Mol. Cancer. 2013;12:91. doi: 10.1186/1476-4598-12-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Williams S.A., Maecker H.L., French D.M., Liu J., Gregg A., Silverstein L.B., Cao T.C., Carano R.A., Dixit V.M. USP1 deubiquitinates ID proteins to preserve a mesenchymal stem cell program in osteosarcoma. Cell. 2011;146(6):918–930. doi: 10.1016/j.cell.2011.07.040. [DOI] [PubMed] [Google Scholar]
- 98.Chen J., Dexheimer T.S., Ai Y., Liang Q., Villamil M.A., Inglese J., Maloney D.J., Jadhav A., Simeonov A., Zhuang Z. Selective and cell-active inhibitors of the USP1/ UAF1 deubiquitinase complex reverse cisplatin resistance in non-small cell lung cancer cells. Chem. Biol. 2011;18(11):1390–1400. doi: 10.1016/j.chembiol.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liang Q., Dexheimer T.S., Zhang P., Rosenthal A.S., Villamil M.A., You C., Zhang Q., Chen J., Ott C.A., Sun H., Luci D.K., Yuan B., Simeonov A., Jadhav A., Xiao H., Wang Y., Maloney D.J., Zhuang Z. A selective USP1-UAF1 inhibitor links deubiquitination to DNA damage responses. Nat. Chem. Biol. 2014;10(4):298–304. doi: 10.1038/nchembio.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mistry H., Hsieh G., Buhrlage S.J., Huang M., Park E., Cuny G.D., Galinsky I., Stone R.M., Gray N.S., D’Andrea A.D., Parmar K. Small-molecule inhibitors of USP1 target ID1 degradation in leukemic cells. Mol. Cancer Ther. 2013;12(12):2651–2662. doi: 10.1158/1535-7163.MCT-13-0103-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.D’Arcy P., Linder S. Proteasome deubiquitinases as novel targets for cancer therapy. Int. J. Biochem. Cell Biol. 2012;44(11):1729–1738. doi: 10.1016/j.biocel.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 102.D’Arcy P., Brnjic S., Olofsson M.H., Fryknäs M., Lindsten K., De Cesare M., Perego P., Sadeghi B., Hassan M., Larsson R., Linder S. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat. Med. 2011;17(12):1636–1640. doi: 10.1038/nm.2536. [DOI] [PubMed] [Google Scholar]