

To the Editor
Recently developed data-independent acquisition (DIA) approaches for mass spectrometry data collection are gaining traction in the proteomics field. We present MSPLIT-DIA (Mixture-Spectrum Partitioning using Libraries of Identified Tandem mass spectra) as a spectral matching tool for untargeted and sensitive peptide identification in DIA data (http://proteomics.ucsd.edu).
Despite its sensitivity on modern mass spectrometers, the semi-stochastic nature of Data-Dependent Acquisition (DDA) leads to sampling a different subset of peptides each time a sample is analyzed, resulting in missing peptide identifications and decreased reproducibility across multiple runs. DIA strategies aim to alleviate this problem by systematically isolating and fragmenting ions based only on their m/z and not their intensities. DIA strategies often segment the usable m/z range into wide isolation windows (e.g. 25 Da windows in SWATH1), generating complex spectra with multiple peptides that cannot be readily identified with DDA tools. Instead, DIA data analysis tools are mostly based on targeted extraction of quantitative information using SRM-inspired strategies1, though recent approaches published in Nature Methods, one of which3 was under review concurrently with this submission, extract pseudo-MS/MS spectra which are then searched with DDA database search tools2, 3 Nevertheless, computational tools that explore alternative strategies for identifying peptides in multiplex spectra are still needed.
We introduce MSPLIT-DIA (Mixture-Spectrum Partitioning using Libraries of Identified Tandem mass spectra), a spectral matching tool for untargeted peptide identification in DIA data (Fig. 1a; Supplementary Methods). Due to the likely presence of many peaks from co-eluting peptides in each multiplexed spectrum, MSPLIT-DIA uses spectrum projections to match library spectra to each DIA spectrum, and spectrum-spectrum match (SSM) similarity is then evaluated using the normalized dot product. Since peptides are acquired and analyzed throughout their elution profile, MSPLIT-DIA also evaluates the similarity of the matched peaks between library spectra and multiplexed spectra across multiple consecutive DIA spectra. Finally, the statistical significance of SSMs matches is assessed at 1% peptide-level false discovery rate (FDR) using the target-decoy approach. For each multiplexed spectrum, all SSMs with FDR≤1% are returned as matches. MSPLIT-DIA effectively identified up to 10 peptides per spectrum, with complex samples such as human lysate generating a predominance of spectra containing more than one peptide (Fig. 1b; Supplementary Fig. 1).
Fig. 1. MSPLIT-DIA identification of peptides, proteins and protein-protein interactions.
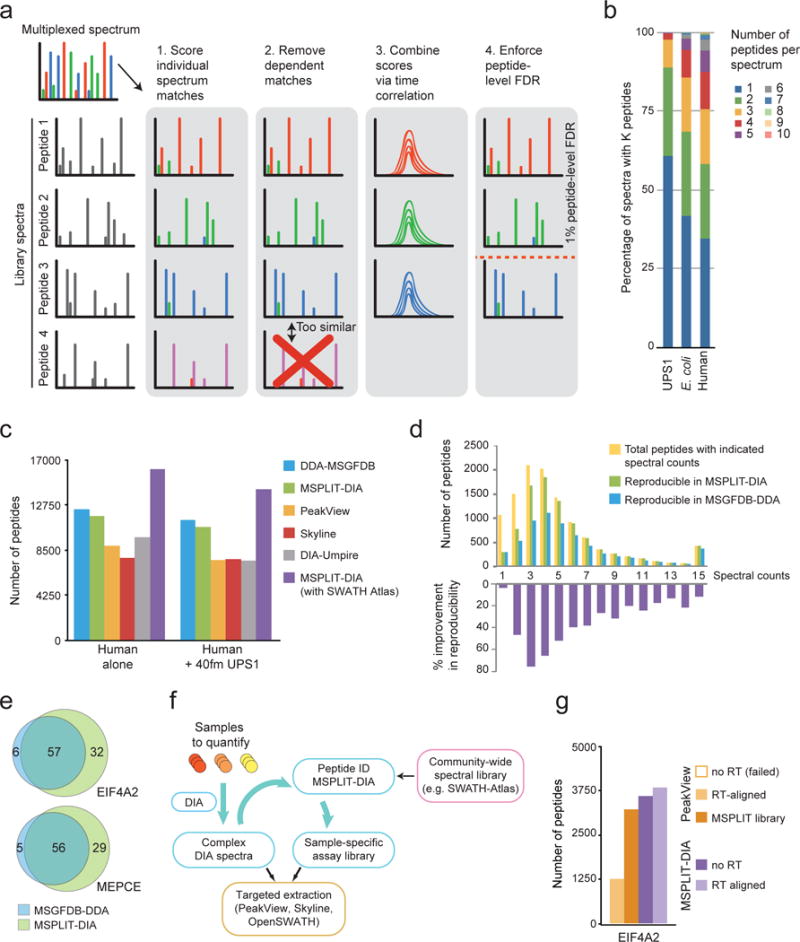
(a) Overview of the MSPLIT-DIA identification process (see also the Supplementary Note for details). (b) Peptide multiplicity as identified by MSPLIT-DIA in multiplexed spectra from DIA runs of varying complexity. (c) A human lysate was analyzed by itself (left) and with a spiked-in standard 48-protein mixture (UPS1, right); the number of unique peptides identified using six different data analysis approaches is shown. MSPLIT-DIA analysis was performed either with a library from paired Data Dependent Acquisition (DDA) runs of the same sample (green) or using the large generic SWATH-Atlas library (purple). (d) Number of peptides detected across four runs (top panel) and reproducibility analysis comparing MSPLIT-DIA and MSGFDB-DDA (bottom panel) in relation to peptide abundance (x-axis). (e) Comparison of MSGFDB-DDA and MSPLIT-DIA methods as applied in a semi-quantitative approach to detect protein-protein interactions from affinity-purified samples from two bait proteins (EIF4A2, MEPCE) and a negative control (GFP). (f) DIA analysis workflow using MSPLIT-DIA circumvents the need to use additional DDA runs, spike-in peptides or manual curation for generation of sample-specific assay libraries. (g) Results of PeakView and MSPLIT-DIA peptide quantification with or without retention-time alignment and with or without an MSPLIT-generated assay library.
Since DDA is the current standard for sensitive peptide identification, we compared MSPLIT-DIA with MSGFBD4 analysis of DDA data (Fig. 1c, Supplementary Figs. 2–4). While the performance was comparable when using spectral libraries generated from the same samples, MSPLIT-DIA with the SWATH-Atlas5 spectral library identified 26–31% more human peptides than the corresponding DDA analysis. MSPLIT-DIA also identified 66–89% more human peptides than DIA-Umpire2 and 81–88% and 86–107% more than PeakView6 and Skyline, respectively, in the same DIA runs. MSPLIT-DIA further enabled much more reproducible observations across 4 runs than DDA (Supplementary Fig. 5; 70% versus 50% at 1% FDR). The reproducibility gains were most pronounced for the 60% lower-abundance human peptides (Fig. 1d, Supplementary Fig. 5; 59% gains). This is important for comparative studies, where biological conclusions are drawn from the detection and non-detection of low-to-medium abundance peptides and proteins across samples.
We benchmarked MSPLIT-DIA for the analysis of protein-protein interactions (a major application of comparative proteomics), by re-analyzing DDA and DIA data for biological triplicates of the baits EIF4A2 and MEPCE compared to the negative control GFP6 (Supplementary Figs. 6,7). Since MSPLIT-DIA used in an untargeted manner yields spectral counts (Fig. 1d) that are not biased by precursor ion selection as in DDA dynamic exclusion protocols, we used these as a rough measure of abundance. The substantial ~3–4× gains in spectral counts synergized with the reproducibility improvements to yield better signal-to-noise for approaches relying on spectral counts, such as SAINT7. For MEPCE and EIF4A2, this resulted in the confident detection of ~33% more interacting proteins (Fig. 1e) that are consistent with the biological function of the bait proteins (Supplementary Tables 1,2; Supplementary Fig. 7). Thus, even when only coupled to rough abundance measurements, the sensitivity, reproducibility, and spectral count increases obtained with MSPLIT-DIA improve the sensitivity of detection of interactions. Since the generic SWATH-Atlas library5 was even better at detecting interactors than the sample-specific library (Supplementary Figs. 6,7; and more sensitive than DIA-Umpire2 for protein identifications on these samples), our results also suggest that time-consuming generation of tailored libraries or addition of external retention time (RT) calibration standards may become superfluous as more spectral libraries become publicly available.
While we contrasted above the sensitivity of targeted extraction tools with MSPLIT-DIA, these are in fact complementary approaches (Fig. 1f, Supplementary Fig. 8). Although targeted extraction tools performed relatively well on libraries generated from the paired DDA samples (with matched complexity, instrument parameters and chromatographic resolution; Supplementary Fig. 2), this was not the case with the large generic SWATH-Atlas library5 (Fig. 1g; Supplementary Fig. 9). First, we showed that MSPLIT-DIA greatly facilitated targeted extraction by assisting in RT alignment without the need for spike-in standards as peptides identified by MSPLIT-DIA in the DIA run served as markers for alignment (“MSPLIT-DIA-assisted RT alignment” in Fig. 1g and Supplementary Fig. 9). Second, restricting the targeted quantification search space to only MSPLIT-DIA-identified peptides yielded much smaller assay libraries that either enabled (Skyline) or systematically improved (PeakView6, OpenSWATH1) targeted extraction results (“MSPLIT-DIA library” in Supplementary Fig. 9). Altogether, these processes substantially simplified the targeted extraction of quantitative data for up to 88% of the peptides identified by MSPLIT-DIA without affecting the reproducibility in the quantification of these newly identified peptides (Supplementary Fig. 10). Assay libraries for targeted extraction tools are automatically generated by MSPLIT-DIA to facilitate coupling of sensitive identification with accurate quantification from DIA data.
Supplementary Material
Acknowledgments
We thank B. MacLean, H. Röst and C.-C. Tsou and A. Nesvizhskii for their help with running Skyline, OpenSWATH and DIA-Umpire, respectively. This work was supported by the US National Institutes of Health Grant 2 P41 GM103484-06A1 from the National Institute of General Medical Sciences (to N.B., J.W.), the Government of Canada through Genome Canada and the Ontario Genomics Institute (to A.-C.G.) and the Canadian Institutes of Health Research (Foundation grant to A.-C.G). N.B. is an Alfred P. Sloan Research Fellow. A.-C.G. is the Canada Research Chair in Functional Proteomics and the Lea Reichmann Chair in Cancer Proteomics. J.-P.L was supported by a post-doctoral fellowship from CIHR and by a TD Bank Health Research Fellowship at the Lunenfeld-Tanenbaum Research Institute.
Abbreviations
- MS/MS
Tandem mass spectrometry
- DIA
Data independent acquisition
- DDA
Data dependent acquisition
- MSPLIT
Mixture-Spectrum Partitioning using Libraries of Identified Tandem mass spectra
- PSM
Peptide-Spectrum Match
- SSM
Spectrum-Spectrum Match
- FDR
False Discovery Rate
- SRM
Selected Reaction Monitoring
Footnotes
COMPETING FINANCIAL INTERESTS
S.T. is an employee of SCIEX. N.B. has an equity interest in Digital Proteomics, LLC, a company that may potentially benefit from the research results; Digital Proteomics LLC was not involved in any aspects of this research. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies.
AUTHOR CONTRIBUTION
J.W. and N.B. developed MSPLIT-DIA; J.W. implemented the software; B.L. and M.T. acquired mass spectrometry data; J.D.R.K., B.L., S.T. and J.-P.L. helped with data analysis; N.B. and A.-C.G. supervised the project; J.W., A.-C.G. and N.B. wrote the manuscript with input from all authors.
References
- 1.Rost HL, et al. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat Biotechnol. 2014;32:219–223. doi: 10.1038/nbt.2841. [DOI] [PubMed] [Google Scholar]
- 2.Tsou CC, et al. DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat Methods. 2015;12:258–264. doi: 10.1038/nmeth.3255. 257 p following 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Y, et al. Group-DIA: analyzing multiple data-independent acquisition mass spectrometry data files. Nat Methods. 2015 doi: 10.1038/nmeth.3593. [DOI] [PubMed] [Google Scholar]
- 4.Kim S, et al. The generating function of CID, ETD, and CID/ETD pairs of tandem mass spectra: applications to database search. Mol Cell Proteomics. 2010;9:2840–2852. doi: 10.1074/mcp.M110.003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenberger G, et al. A repository of assays to quantify 10,000 human proteins by SWATH-MS. Sci Data. 2014;1:140031. doi: 10.1038/sdata.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lambert JP, et al. Mapping differential interactomes by affinity purification coupled with data-independent mass spectrometry acquisition. Nat Methods. 2013;10:1239–1245. doi: 10.1038/nmeth.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Teo G, et al. SAINTexpress: improvements and additional features in Significance Analysis of INTeractome software. J Proteomics. 2014;100:37–43. doi: 10.1016/j.jprot.2013.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.