

Abstract
Dopamine (DA) neurons in the ventral tegmental area have been implicated in psychiatric disorders and drug abuse. Understanding the mechanisms through which their activity is regulated via the modulation of afferent input is imperative to understanding their roles in these conditions. Here we demonstrate that endocannabinoids liberated from DA neurons activate cannabinoid CB1 receptors located on glutamatergic axons and on GABAergic terminals targeting GABAB receptors located on these cells. Endocannabinoid release was initiated by inhibiting either presynaptic type-III metabotropic glutamate receptors or postsynaptic calcium-activated potassium channels, two conditions that also promote enhanced DA neuron excitability and bursting. Thus, activity-dependent release of endocannabinoids may act as a regulatory feedback mechanism to inhibit synaptic inputs in response to DA neuron bursting, thereby regulating firing patterns that may fine-tune DA release from afferent terminals.
Keywords: marijuana, metabotropic glutamate receptors, SK channels, bursting, mesolimbic, GABA, dopamine neuron, GABA-B receptors
Introduction
The mesolimbic dopamine (DA) system represents an important neural substrate underlying the rewarding properties of abused drugs. This pathway originates with a population of DA neuron somata located in the ventral tegmental area (VTA), the axons of which project to forebrain areas such as nucleus accumbens (NAc) and prefrontal cortex (PFC) (Wise, 2002). DA neurons fire action potentials in both pacemaker (tonic) and bursting (phasic) patterns (Cooper, 2002). The latter pattern results in a transient increase in DA release in NAc, particularly in relation to environmental stimuli possessing strong reward salience (Schultz, 1998). The transition to bursting from tonic firing is governed by the patterns of synaptic input from glutamatergic and GABAergic afferents to the DA neurons (Grace and Bunney, 1985; Smith and Grace, 1992; Chergui et al., 1994; Tepper et al., 1995). Cannabinoids (CBs), like the psychoactive ingredient in marijuana, Δ9-tetrahydrocannabinol (Δ9-THC), also increase DA release in the NAc by enhancing VTA DA neuron firing and bursting (French et al., 1997), which may be relevant to the self-administration of Δ9-THC in primates (Gardner, 2002; Tanda and Goldberg, 2003). Although cannabinoids stimulate mesolimbic DA neurotransmission, uncertainty remains regarding the mechanisms involved; however, because phasic increases in extracellular DA depend highly on afferent input (Schultz, 2002), and systemic (French et al., 1997) but not intra-VTA injections of Δ9-THC stimulate DA neuron activity (Gardner, 2002) and terminal DA release (Chen et al., 1993), the rewarding properties of cannabinoids may depend on alterations in afferent input to the VTA.
The activity of VTA DA neurons is also regulated by various presynaptic and postsynaptic mechanisms. For example, presynaptic type-III metabotropic glutamate receptors (mGluR-IIIs) monitor neurotransmitter “spillover” (Conn and Pin, 1997; Isaacson, 2000) and limit further release (Bonci et al., 1997; Zheng and Johnson, 2003). Another site of regulation is via GABABRs located on VTA DA neurons (Seutin et al., 1994). Axons originating in the NAc (Walaas and Fonnum, 1980; Sugita et al., 1992), ventral pallidum (VP) (Kalivas et al., 1993), and pedunculopontine (PdP) nuclei (Charara et al., 1996) innervate these GABABRs (Sugita et al., 1992) to modify DA neuron firing rates (Erhardt et al., 2002), NAc DA release (Westerink et al., 1996), and reward-relevant behaviors (Yoshida et al., 1994). In addition, enhancement of glutamatergic activity in the VTA (Kita and Kitai, 1987; Tokuno et al., 1988; Sesack and Pickel, 1992) and blockade of postsynaptic calcium-activated potassium (SK) channels by apamin (APA) (Johnson et al., 1992; Seutin et al., 1993; Cooper, 2002) initiate bursting in VTA DA neurons that is blocked by glutamate receptor antagonists (Overton and Clark, 1997) and altered by GABABR ligands (Seutin et al., 1994; Erhardt et al., 2002). Therefore, GABAergic and glutamatergic inputs to the VTA regulate DA neuron activity, and the presynaptic modulation of these synaptic inputs can alter this activity.
The present study tests the hypothesis that CBs regulate synaptic inputs to VTA DA neurons and determines whether endogenous CBs (eCBs) are released after the manipulation of intrinsic mechanisms controlling DA neuron excitability. We find that activation of glutamatergic axons targeting VTA DA neurons initiates eCB release from these cells that then retrogradely stimulates CB1Rs on GABAergic and glutamatergic axon terminals to inhibit neurotransmitter release. This thereby provides a mechanism for VTA DA neurons to dynamically modulate their own synaptic inputs through activity-dependent eCB release.
Materials and Methods
All protocols were conducted under National Institutes of Health Guidelines using the NIH handbook Animals in Research and were approved by the Institutional Animal Care and Use Committee (National Institute on Drug Abuse, Intramural Research Program, Baltimore, MD).
Recordings. Midbrain horizontal slices (220 μm) were prepared from adult male rats (Charles River Laboratories, Raleigh, NC) (150-230 gm) as described previously (Williams et al., 1984). Horizontal slices were placed in a recording chamber (0.5 ml) and submerged in continuously flowing physiological saline (34°C) at a rate of 2 ml/min. The solution contained (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.4 CaCl2, 1.4 NaH2PO4, 25 NaHCO3, and 11 d-glucose equilibrated with 95% O2/5% CO2. Dopamine (principal) neurons of the VTA were identified by their large cell bodies, their characteristic pacemaker-like firing (1-5 Hz) observed in the cell-attached mode, and the presence of large (>200 pA) Ih currents (Johnson and North, 1992). Anatomical studies indicate that the VTA also contains a smaller fraction (10-15%) of non-DA neurons (Swanson, 1982), and subsequent work has demonstrated that only 13% of these non-DA neurons also show Ih (Cameron et al., 1997). Thus, although we recognize that the presence an Ih does not unequivocally identify DA neurons in midbrain slices, this represents a very small percentage (∼2%) of the total VTA neuron population. Recordings were made using an Axopatch 200B (Axon Instruments, Burlingame, CA), filtered at 2 kHz, and collected at 5 kHz to the hard-drive of a personal computer. Neuronal firing was monitored in some instances using the cell-attached mode. Cell-attached recordings used the same low-resistance pipettes as the whole-cell recordings (3 MΩ). The internal solution used for whole-cell voltage-clamp recordings contained (in mm): 115 K-methyl sulfate, 20 KCl, 1 MgCl2, 10 HEPES, 0.1 EGTA, 2 ATP, 0.3 GTP, and 10 creatine phosphate adjusted to pH 7.2 with 1 m KOH (270-280 mOsm).
All experiments were done in the presence of the DA D2 receptor antagonist eticlopride (300 nm). In addition, picrotoxin (100 μm), MK-801 (100 μm), and strychnine (1 μm) were used to block GABAA, NMDA, and glycine receptors, respectively. In some instances, the tissue was incubated with apamin (100 nm). In other instances, NBQX (5 μm) and CGP 35348 (200 μm) were included to block AMPA receptors and GABAB receptors, respectively. In experiments in which the Ca2+ dependence was studied, BAPTA (1 mm) was substituted for EGTA (0.1 mm) in the internal solution.
Evoked responses. Submaximal synaptic currents (30-50 pA, baseline) were evoked using bipolar tungsten stimulating electrodes with a tip separation of 300-400 μm. A train of six stimuli of 400 μsec duration at 0.3-1.5 mA was delivered at 50 Hz, every 60 sec. Stimulating electrodes were placed within 1 mm rostral of the recording site. Voltage-clamp and stimulation protocols were delivered using a pulse generator (Master 8, A.M.P.I., Jerusalem, Israel), and signals were acquired using a Laboratory PC 1200 or 6024E analog-to-digital converter (National Instruments, Austin, TX) and the Strathclyde electrophysiology software package (WCP, courtesy of Dr. John Dempster, Strathclyde University, Glasgow, UK; http://innovol.sibs.strath.ac.uk/physpharm). Ih currents were recorded by voltage clamping cells and stepping the membrane from -60 to -110 mV.
Drugs. Unless indicated otherwise, drugs were applied to the slice by superfusion using a calibrated syringe pump (Razel Scientific Instruments, Stamford, CT). WIN-55,212-2 and AM251 were prepared as concentrated (10 mm) stock solutions in DMSO and then diluted to 2 mg/ml in Tween 80 (1%), DMSO (2%), and saline (70%). Final (bath) concentrations were <0.01% DMSO. The following compounds were purchased from Tocris (Ballwin, MO): (5S,10 R)-(+)-5-methyl-10, 11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate, (RS)-α-methyl-4-carboxyphenylglycine (MCPG), α-methyl-3-methyl-4-phosphonophenylglycine (UBP1112), (hydroxyimino)cyclopropa[b-]chromen-1a-carboxylate ethyl ester (CPCCOEt), (2S)-a-ethylglutamic acid (Eglu), 2-methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP), (2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid (LY341495), 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide disodium salt (NBQX), BAPTA, and picrotoxin. The following compounds were purchased from Sigma (St. Louis, MO): apamin, eticlopride, and WIN55,212-2 (WIN). K-methyl sulfate was purchased from Fisher Chemicals (Fairlawn, NJ).
Data analysis. Group data are presented as the mean ± SE in all cases. All other statistical tests, including t tests and one-way ANOVAs, were performed using a critical probability of p < 0.05 (Prism version 4.01, GraphPad Software, San Diego, CA). Post hoc analysis (Newman-Keuls test) was performed only when an ANOVA yielded a significant (p < 0.05) main effect. Statistical significance of changes in EPSC and IPSC amplitude was assessed using a one-way ANOVA.
Results
Presynaptic CB1Rs inhibit GABAB IPSCs
Our first experiments were designed to determine whether CB1Rs were located on GABAergic axon terminals innervating GABABRs located on VTA DA neurons. Neurons were visually identified in horizontal brain slices containing the ventral mesencephalon using a Zeiss Axioskop (Oberkochen, Germany) upright microscope equipped with differential interference contrast optics and infrared illumination. DA neurons were electrophysiologically identified while in cell-attached mode by their pacemaker-like regular spontaneous firing and large inwardly rectifying Ih currents recorded immediately after intracellular access (Fig. 1A2) (Johnson and North, 1992). GABABR-mediated IPSCs were evoked in voltage-clamped (-60 mV) neurons by electrically stimulating the tissue (50 Hz, 6 stimuli) (Johnson and North, 1992). The currents were blocked by the selective GABAB antagonist CGP35348 (200 μm) (Fig. 1A1,A3) (Olpe et al., 1990). A previous study demonstrated that a similar stimulation paradigm may also generate currents mediated by mGluR-Is, coupled to apamin-sensitive SK channels (Fiorillo and Williams, 1998). Therefore, apamin (100 nm) was applied at the end of these experiments to assess the contribution of SK channels to the presumed GABAB IPSC. Apamin did not significantly alter the IPSCs, indicating that mGluR-I currents did not routinely contribute to the CGP35438-sensitive GABAB-mediated outward currents (Fig. 1 A1,A3).
Figure 1.
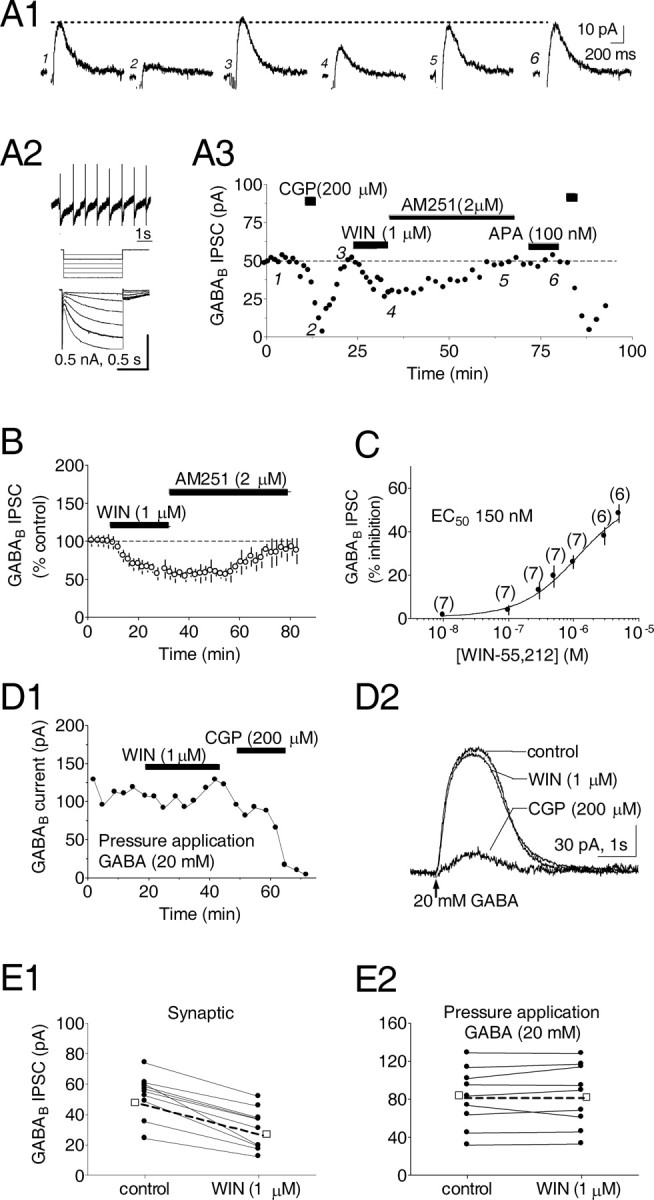
Activation of presynaptic cannabinoid receptors diminishes GABAB IPSCs recorded in VTA DA neurons. A1, Averaged traces of GABAB IPSCs during conditions indicated in A3. In this and subsequent figures, stimulus artifacts have been removed. A2, Electrophysiological properties of the same VTA DA neuron include (1) regular pacemaker firing in the cell-attached configuration, (2) Ih (sag) current activated by hyperpolarizing voltage steps (-60 to -100 mV). A3, Time course showing effect of the cannabinoid receptor agonist WIN55,212-2 (WIN) and the CB1R antagonist AM251. IPSCs were blocked by the GABAB antagonist CGP35348 (CGP) but not APA. B, Time course showing the WIN55,212-2-induced inhibition of IPSCs and reversal by AM251. C, Concentration-response relationship for the effect of WIN on GABAB IPSCs. The number of experiments for each point is indicated. D1, Time course showing the peak amplitude of currents produced by pressure application of GABA to a VTA DA neuron. D2, Averaged traces demonstrate that GABA currents are inhibited by CGP but not by WIN. E, Summary of WIN effects on GABAB currents resulting from synaptic stimulation (E1) or pressure application of GABA (E2) in all DA neurons tested. (-□-) denotes the mean response. All experiments were conducted in the presence of picrotoxin (100 μm); Vh = -60 mV.
The synthetic cannabinoid agonist WIN55,212-2 (1 μm), applied in the absence of apamin, inhibited GABABR IPSCs in a concentration-dependent manner (Fig. 1C), and its effect was reversed by the selective CB1R antagonist AM251 (2 μm) (Lan et al., 1999) (WIN = 48.4 ± 5.1% of control, n = 11; WIN plus AM251 = 89.8 ± 1.4% of control, n = 9) (Fig. 1B). The WIN55,212-2 inhibition of GABAB IPSCs was observed in ∼50% of the DA neurons, suggesting the presence of subpopulations of CB1R-expressing inhibitory inputs targeting these cells. Only those cells responding to WIN55,212-2 were included in the averaged results (Fig. 1B).
We next evaluated the presynaptic or postsynaptic locus of the CB1R inhibition of GABAB IPSCs. In the presence of picrotoxin (100 μm), pressure application of GABA (20 mm) to DA neuron dendrites produced outward currents that were blocked by CGP35348 (Fig. 1 D1,D2). Although WIN55,212-2 (1 μm) again inhibited electrically evoked GABAB IPSCs (59.3 ± 15.3% of control; n = 7) (Fig. 1 E1), it did not alter GABAB currents generated by dendritic GABA application (102.1 ± 13.6% of control; n = 9) (Fig. 1 E2), implicating a presynaptic locus for the CB1Rs in the VTA.
SK channels and mGluRs regulate CB1R signaling
Although the above experiments did not reveal an alteration of GABAB IPSCs by apamin, stimulation similar that used in this study can produce rapidly desensitizing mGluR-I-mediated IPSCs (Fiorillo and Williams, 1998), which may overlap GABAB IPSCs. To minimize involvement of postsynaptic mGluR-Is, we blocked mGluR function and reevaluated the effects of WIN55,212-2 on the GABAB IPSCs; however, in the presence of either the nonselective mGluR antagonist MCPG (1 mm) or the small-conductance SK channel blocker apamin (100 nm), we found that the inhibition of GABAB IPSCs by WIN55,212-2 was lost (WIN55,212-2 = 52.2 ± 5.0% of control, n = 11, p < 0.001; WIN55,212-2 plus apamin = 102.2 ± 6.6% of control, n = 11, p > 0.05; WIN55,212-2 plus MCPG = 98.1 ± 6.9% of control, n = 9, p > 0.05) (Fig. 2A1,A2). Because SK channels contribute to calcium-dependent afterhyperpolarizations (AHPs) after single action potentials in midbrain DA neurons (Wolfart and Roeper, 2002), and because their blockade can dramatically reduce pacemaker precision in DA cells (Wolfart and Roeper, 2002), we hypothesized that the loss of CB1R effect in the presence of apamin might relate to an increase in DA neuron excitability. This would likely arise from changes occurring at distal DA neuron dendrites not adequately clamped during whole-cell recordings from DA neuron somata (Seutin et al., 1994). To test this notion, apamin (100 nm) was applied while spontaneous action potentials were recorded from VTA DA cells in the cell-attached configuration. Apamin greatly increased DA neuron firing rates in the absence (565.0 ± 85.8% increase; n = 6; p < 0.05) (Fig. 2B1) and presence of the 50 Hz, six-pulse electrical stimulus (651.0 ± 132.3% increase; n = 5; p < 0.05) (Fig. 2B1). Furthermore, in the presence of apamin, DA neurons exhibited prominent oscillations in the frequency of spontaneous action potential discharge after the electrical stimulation (Fig. 2B2d).
Figure 2.

Blockade of SK channels prevents CB1R activation by WIN55,212-2 and increases DA neuron activity. A, Cannabinoid receptor-mediated inhibition of GABAB IPSCs is prevented by blockade of SK channels with APA. A1, Mean time courses showing that the WIN-mediated inhibition is blocked by APA and MCPG, a nonselective mGluR antagonist. A2, Summary of the effects of WIN under different conditions. The number of experiments is indicated. ***p < 0.001 versus control. B, Effects of APA and electrical stimulation on DA neuron activity. B1, Summary of the percentage increase in DA neuron spontaneous action potential frequency during the indicated conditions. B2a, Individual time course showing the increase in cell firing rate during control (b), APA (100 nm) (c), and apamin during electrical stimulation (d). Activity was assessed in the cell-attached configuration. Asterisk represents electrical stimulation.
How might an increase in DA neuron excitability produced by apamin occlude the effect of WIN55,212-2? Because depolarization and neuronal excitation underlie the production of endogenous cannabinoids in several neuron phenotypes (Alger, 2002), we hypothesized that the apamin-induced increase in spike frequency enhanced eCB production, which then occluded the effect of WIN55,212-2 on GABAB IPSCs. We reasoned that if this were the case, then the eCB occupation of CB1Rs on GABAergic axon terminals would be revealed by an AM251-induced increase of GABAB IPSCs during apamin application. This hypothesized mechanism was supported by the data; that is, although AM251 alone had no effect on the GABAB IPSCs, during apamin (100 nm) application it significantly increased the amplitudes of these currents (AM251 = 18.8 ± 5.7% increase, n = 7, p > 0.05; AM251 plus apamin = 35.9 ± 8.3% increase, n = 7, p < 0.05) (Fig. 3A-C). This suggests that the blockade of postsynaptic SK channels and the concomitant increase in neuronal excitability likely stimulated the production of an eCB that then acted on CB1Rs to occlude the presynaptic effects of WIN55,212-2 on GABAergic terminals.
Figure 3.

Blockade of SK channels or mGluRs stimulates eCB production. A1, Three individual time courses during bath application of the CB1R-selective antagonist AM251. In the presence of APA or the nonselective mGluR antagonist LY341495 (LY), AM251 increased IPSC amplitudes. A2, Averaged GABAB IPSCs collected before and during AM251, as shown in A1. Calibration is identical in all traces. B, Mean time courses illustrating effects of SK channel or mGluR blockade on AM251-induced changes in GABAB IPSC amplitude. C, Summary showing combined blockade of both SK channels and mGluRs during AM251 additively increases IPSCs. Numbers above each bar indicate the number of experiments. *p < 0.01; **p < 0.001.
Our next experiments were designed to determine whether eCB production occurred during other manipulations that increase DA neuron excitability. In the VTA (Bonci et al., 1997) as elsewhere in the brain, mGluRs located on glutamatergic axon terminals act as autoreceptors to limit synaptic excitation (Schoepp, 2001). We hypothesized that blockade of these presynaptic mGluRs might increase the glutamatergic excitation of postsynaptic neurons in the VTA by removing the inhibitory autoreceptor influence on glutamate release, thereby increasing the activation of ionotropic glutamate receptors. Furthermore, if this were the case, then blockade of presynaptic mGluRs might also increase eCB production. To test this we evaluated two structurally dissimilar, nonselective mGluR antagonists, LY341495 (200 μm) and MCPG (1 mm). Similar to the results obtained with apamin, AM251 increased GABAB IPSCs when applied in the presence of the mGluR antagonists (LY341495 = 63.0 ± 8.2% increase, n = 4, p < 0.01; MCPG = 49.1 ± 9.7% increase, n = 5, p < 0.05) (Fig. 3A1,A2,B,C), suggesting that the antagonism of mGluRs could increase eCB production and inhibit GABA release from afferents to the VTA.
If apamin and mGluR antagonists acted via different cellular mechanisms to liberate eCBs from DA neurons, then their combined application should additively increase the response to AM251. Consistent with this idea, combined blockade of SK channels and mGluRs produced an additive increase in the effect of AM251 on the GABAB IPSCs (apamin plus LY341495 = 113.0 ± 19.1% increase, n = 4, p < 0.01; apamin plus MCPG = 104.6 ± 19.7% increase, n = 5, p < 0.01) (Fig. 3A1,A2,B,C). Also, as expected from the effects of these manipulations on endocannabinoid release, baseline GABAB IPSC amplitudes in apamin and mGluR antagonist treatment groups were significantly smaller than control IPSCs (LY341495 plus apamin: 30.5 ± 2.5 pA, n = 4, p < 0.05; apamin: 34.8 ± 2.5 pA, n = 8, p < 0.05; LY341495: 37.5 ± 5.8 pA, n = 4, p < 0.05; MCPG: 39.6 ± 1.6 pA, n = 5, p < 0.05; MCPG plus apamin: 31.0 ± 10.0 pA, n = 5, p < 0.05; control: 61.7 ± 6.0 pA, n = 7; one-way ANOVA and post hoc Newman-Keuls comparison); however, the baseline control responses within each of these groups was not significantly different (p > 0.05). Together, the above data indicate that SK channels and mGluRs can independently regulate eCB production and, indirectly, the release of GABA onto VTA DA neurons.
eCB release is Ca2+ dependent
Because the VTA contains a heterogeneous population of neurons, it was important to determine the cellular source of the eCB in this preparation. Furthermore, because eCB production is dependent on a rise in intracellular Ca2+ in several neuronal phenotypes (Alger, 2002), an examination of this possible mechanism in the VTA DA neurons was necessary. Therefore, VTA DA neurons were loaded with the Ca2+ chelator BAPTA (1 mm), rather than EGTA (0.1 mm), during whole-cell recordings. When apamin (100 nm) and LY341495 (200 μm) were applied to the VTA slice to stimulate eCB production, we found that AM251 (1 μm) failed to alter the amplitudes of the GABAB IPSCs (EGTA = 132.0 ± 23.1% increase, n = 5; BAPTA = 3.6 ± 6.4% increase, n = 13; p < 0.001) (Fig. 4A1,A2). In addition, baseline IPSC amplitude and whole-cell input resistance were unaffected by the inclusion of BAPTA in the pipette. The absence of an AM251-induced increase in the GABAB IPSCs in the presence of BAPTA confirm that, similar to other neuronal systems, eCB production required increased intracellular Ca2+, and additionally, that the eCB was derived from the recorded VTA DA neurons.
Figure 4.

Endocannabinoid-mediated inhibition of GABA release involves intracellular Ca2+ and is initiated by blockade of presynaptic mGluR-IIIs. A1, Mean time courses during bath application of AM251, CGP, and LY. The AM251-induced increase in GABAB IPSC amplitude after mGluR antagonist treatment was prevented by substituting BAPTA for EGTA in the recording solution. A2, Summary of the AM251-induced change in GABAB IPSC amplitude with EGTA or BAPTA. The number of data points is indicated. p < 0.001; t test. All experiments were conducted in the presence of apamin (100 nm) and LY341495 (200 μm). B1, Individual time courses showing the AM251-induced increased in GABAB IPSCs during application of UBP1112 (UBP), a selective mGluR-III antagonist. For comparison, an additional time course for AM251 plus APA is also shown. B2, Averaged IPSC traces collected before and during AM251, as shown in B1. Calibration is identical in both traces. C, Mean time courses illustrating the AM251-induced increase in GABAB IPSCs during UBP1112 treatment. D, Summary of the effects of mGluR antagonists of various classes on the AM251-induced increase in GABAB IPSCs recorded in VTA DA neurons. The nonselective mGluR antagonists include LY341495 (LY2, 200 μm), the mGluR-I selective antagonist CPCCOET (CPC, 100 μm) and MPEP (30 μm), the mGluR-II selective antagonist LY341495 (LY5, 500 nm), and EGlu (200 μm). In all instances, apamin treatment occurred before mGluR antagonist application. **p < 0.01 versus control; one-way ANOVA and post hoc Newman-Keuls comparison.
Presynaptic mGluR-IIIs regulate eCBs
In the VTA and adjacent substantia nigra pars compacta, mGluRs are localized to both presynaptic and postsynaptic membranes (Testa et al., 1994, 1998; Kosinski et al., 1998). To better understand their role in eCB production and to confirm the locus of their action, we used antagonists selective for distinct mGluR subclasses while measuring GABAB IPSC amplitudes during AM251 application. In total, six different mGluR antagonists selective for mGluR-I, -II, and -III subclasses were evaluated (Fig. 4D); however, other than MCPG and LY341495 at nonselective concentrations, only the selective mGluR-III antagonist UBP1112 (Conway et al., 2001; Miller et al., 2003) was found to promote eCB formation, as determined by increased GABAB IPSCs during AM251 application (Fig. 4B-D). Furthermore, like the nonselective mGluR antagonists, apamin induced eCB production during UBP1112 application (UBP1112 alone = 70.4 ± 6.7% increase, n = 4, p < 0.01; UBP1112 plus apamin = 132.7 ± 22.4% increase, n = 4, p < 0.01) (Fig. 4B-D). These data are consistent with immunocytochemical studies localizing mGluR-IIIs to presynaptic terminals (Testa et al., 1998) and with electrophysiological evidence indicating that their activation preferentially decreases glutamatergic EPSCs in VTA neurons (Bonci et al., 1997).
Antagonism of mGluR-IIIs enhances EPSCs
One mechanism through which antagonism of mGluR-IIIs could enhance eCB production would be through the blockade of their presynaptic inhibition of glutamate release onto VTA DA neurons (Bonci et al., 1997). This would be expected to result in augmented glutamate release during the six-pulse, 50 Hz stimulus trains used to evoke GABAB IPSCs, and an increased depolarization of DA neurons through the activation of postsynaptic AMPA-kainate receptors. To test this hypothesis we examined the effect of the AMPA-kainate antagonist NBQX (5 μm) on the ability of AM251 to increase GABAB IPSCs. As in our previous experiments, AM251 (1 μm) increased the amplitudes of the GABAB IPSCs during coapplication of apamin and MCPG (104.6 ± 19.7% increase; n = 5) (Fig. 5A1,A2); however, this effect was completely blocked by NBQX (2.45 ± 4.9% increase; n = 9) (Fig. 5A), suggesting that functional AMPA-kainate receptors were necessary to observe mGluR antagonist-induced eCB release in the VTA.
Figure 5.
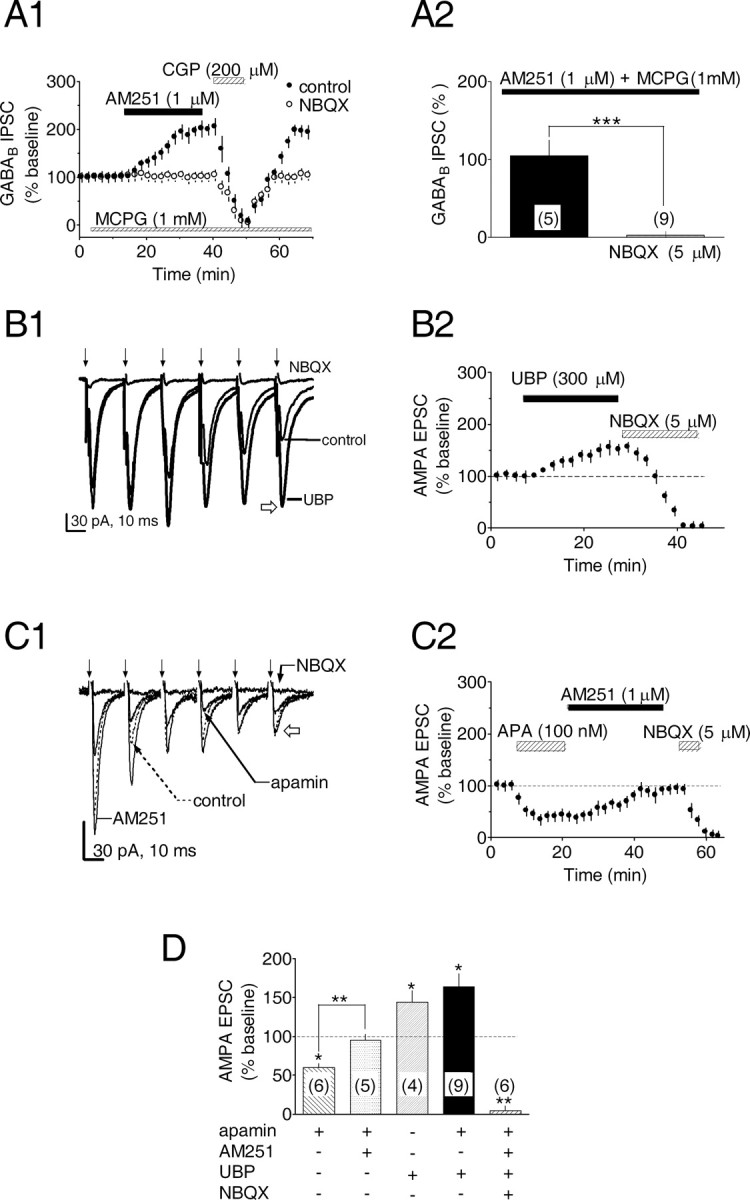
Blockade of eCB production by AMPA-kainate receptor antagonism and CB1R-mediated inhibition of EPSCs via SK channel and mGluR-dependent eCB release. A1, Mean time courses showing that the AMPA-kainate receptor antagonist NBQX blocks the AM251-induced increase in the GABAB IPSC amplitude in the presence of the mGluR antagonist MCPG. A2, Summary of the effect of NBQX on the AM251-induced increase in IPSC amplitude during MCPG application. The number of experiments is indicated. B, C, During trains of electrical stimuli, application of UBP (B), a selective mGluR-III antagonist, or APA (C), a blocker of SK channels, facilitates or attenuates EPSCs in VTA DA neurons, respectively. Sample traces from a single experiment show averaged EPSCs evoked by the same electrical stimulation (↓) used to elicit GABAB IPSCs (50 Hz, 6 stimuli) during UBP (B1) or apamin (C1) application. In both B and C, EPSCs were blocked by NBQX, as determined as amplitude of the sixth EPSC of the series (B1, C1, open arrows). In C2, the apamin-induced inhibition was reversed by AM251, indicating that apamin acted via eCB release. D, Summary demonstrating the mean change in the EPSCs during the conditions described above. The number of treatments for each combination is indicated. *p < 0.05 and **p < 0.01 versus control; one-way ANOVA and post hoc Newman-Keuls comparison. All experiments were conducted in the presence of the GABAB antagonist CGP35348 (200 μm).
To examine the role of the presynaptic mGluR-IIIs more directly in the regulation of glutamate release, we measured the effect of UBP1112 on AMPA-kainate-mediated EPSCs elicited by the same stimulus trains used to evoke GABAB IPSCs. The EPSCs were isolated from GABAB IPSCs by blocking these currents with CGP35348 (200 μm), and the EPSCs were completely blocked by NBQX (5 μm) (n = 6; p < 0.01) (Fig. 5B,C). Under baseline conditions the train-evoked EPSCs demonstrated a pronounced frequency-dependent inhibition, with the last (sixth) EPSP of the series measuring only ∼60% of the first EPSC (Fig. 5B1,C1). As predicted, the mGluR-III antagonist UBP1112 (300 μm) increased mean EPSC amplitudes and blocked the frequency-dependent inhibition of these responses measured at the end of the train (UBP1112 = 144.3 ± 14.3% of control; n = 4; p < 0.05) (Fig. 5B1,B2,D). These data suggest that the increase in extracellular glutamate concentration during the stimulus train was sufficient to activate presynaptic mGluR-III autoreceptors that diminished the efficacy of the glutamatergic inputs to the DA neurons in the VTA (Bonci et al., 1997), and that antagonism of these receptors by UBP1112 could reverse this modulation.
eCBs inhibit VTA glutamate release
Because glutamatergic afferents to VTA DA neurons are reportedly sensitive to CB1R agonists (Melis et al., 2004), it was of interest to determine whether the eCB released after SK channel blockade could inhibit EPSCs mediated by these axons as well as the GABAB IPSCs. Thus, apamin (100 nm) was applied while train- evoked AMPA-kainate EPSCs were recorded from VTA DA neurons, and the effect of AM251 was assessed. The EPSCs were significantly reduced by apamin (60.3 ± 4.7% of control; n = 6; p < 0.05) (Figs. 5C,D), and this was completely reversed by the CB1R antagonist AM251 (1 μm) (94.7 ± 7.8% of control; n = 5; p > 0.05) (Figs. 5C,D). This suggests that the eCBs released from postsynaptic DA neurons after SK channel blockade activated CB1Rs located on both GABAergic and glutamatergic afferents targeting these cells in the VTA. Thus, eCBs could simultaneously inhibit afferents originating from at least two different sources that are recognized as important contributors to burst-type firing patterns in VTA DA neurons (Seutin et al., 1994; Kitai et al., 1999; Erhardt et al., 2002).
Discussion
The balance between inhibition and excitation shapes VTA DA neuron excitability and firing patterns (Cooper, 2002). Here we demonstrate that manipulation of intrinsic physiological mechanisms regulating DA neuron excitability triggers a pronounced depression of synaptic input to VTA DA neurons via retrograde eCB signaling. We propose that this modulation of synaptic input by CB1Rs alters DA neuron excitability in an ongoing manner and may also represent a substrate underlying the rewarding properties of Δ9-THC.
Because the absence of CB1R protein or mRNA in VTA DA neurons argues against a more direct role for these receptors regulating DA neuron activity (Herkenham et al., 1991; Matsuda et al., 1993; Hernandez et al., 2000), we hypothesized that CB1R activation regulated GABAergic inputs impinging on GABABRs found on VTA DA neurons (Fig. 6). These inputs originate in NAc (Walaas and Fonnum, 1980; Sugita et al., 1992), VP (Kalivas et al., 1993), and PdP (Charara et al., 1996) and through activation of GABABRs can attenuate VTA DA neuron activity in vivo (Erhardt et al., 2002). Our observation that exogenous and endogenous CBs inhibited this input in an antagonist-reversible manner establishes that functional CB1Rs exist on these terminals. Because the GABAergic input to VTA DA neuron GABABRs is inhibited by CB1R activation, it is conceivable that Δ9-THC effects in the VTA occur through the inhibition of these afferents, enhancing VTA neuron activity (French, 1997; Cheer et al., 2003). This is further supported by data demonstrating increased VTA DA neuron bursting in vivo produced by GABAB antagonists (Erhardt et al., 2002) or Δ9-THC (French, 1997) and by a concomitant increase in DA release in the VTA and NAc (Chen et al., 1993; Gardner, 2002). Despite the demonstrated effects of Δ9-THC, however, evidence supporting eCB-mediated regulation of mesolimbic DA neurons remains inconclusive. Thus, in vivo administration of CB1R antagonists produces little change in ongoing VTA DA neuron activity (French, 1997; Cheer et al., 2003) or in the release of DA in the NAc (Cheer et al., 2004); however, CB1R antagonists increase DA overflow at mesolimbic terminals in the PFC (Tzavara et al., 2003), and the increase in NAc DA by morphine or ethanol is greatly reduced in mice lacking the CB1R (Mascia et al., 1999; Hungund et al., 2003). Based on these data and our present results, we propose that eCBs influence mesolimbic DA neuron activity, but only under circumstances in which the eCB-sensitive pathways to the VTA are active.
Figure 6.
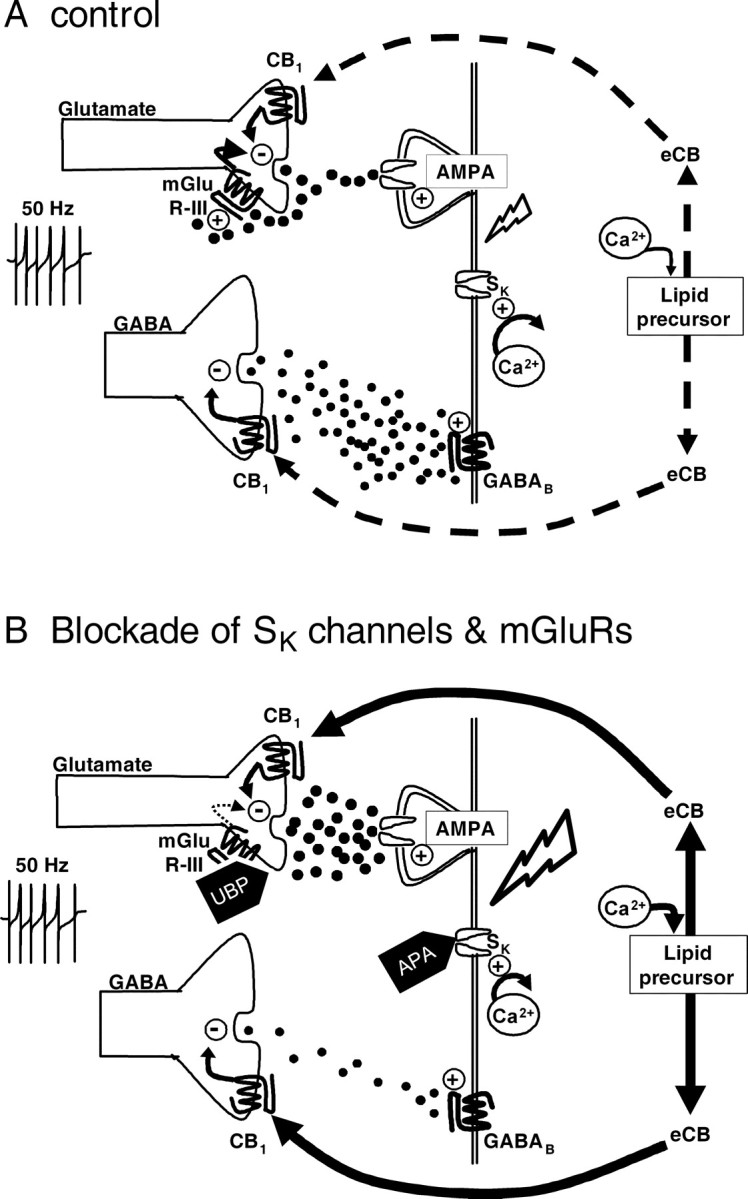
Proposed mechanism of CB1R activation by eCBs mediating the inhibition of GABA and glutamate release in the VTA. A, Under control conditions, electrical stimulation evokes the release of GABA from medium spiny terminals, which activates postsynaptic GABAB receptors on VTA DA neuron dendrites. Glutamate is also released by the stimulus. Glutamate then activates mGluR-IIIs located on glutamate terminals to limit further release and dampen DA neuron excitability. Under these conditions eCB release is absent, and cannabinoid agonists can activate CB1Rs on medium spiny neuron terminals to decrease GABA release onto DA neurons. B, Activation of glutamatergic afferents in the presence of apamin (which reduces AHPs and repolarization by blocking SK channels) increases the excitability of VTA DA neurons, leading to an increase in intracellular Ca2+ that then liberates eCBs from lipid precursors. The eCBs then bind to presynaptic CB1Rs on both GABAergic and glutamatergic axon terminals, decreasing the release of these neurotransmitters. Under these conditions, activation of CB1Rs with a synthetic agonist is occluded by the eCB; however, blockade of CB1Rs with AM251 blocks the eCB effect and increases GABAB IPSCs and AMPA EPSCs. Electrical stimulation in the presence of UBP, an mGluR-III antagonist, augments glutamate release, increasing activation of AMPA-kainate receptors on VTA DA neuron dendrites, which then increases excitability and intracellular Ca2+ levels. This leads to the production of eCBs, which then activate CB1Rs on axon terminals to decrease GABA and glutamate release. The combined application of UBP and apamin results in an additive increase in eCB production resulting in larger AM251 effects and the occlusion of CB1R agonist effects. Lightning bolt symbol represents membrane depolarization.
With this in mind, our results revealed two independent manipulations that increased neuronal excitation and were sufficient to trigger eCB release. One of these involved the antagonism of presynaptic mGluR-IIIs regulating glutamate release onto VTA DA neurons (Bonci et al., 1997). Antagonism of mGluR-IIIs enhanced eCB release that then occluded the effect of WIN55,212-2 at CB1Rs. We conclude that this increase in eCB production resulted from enhanced glutamate release onto AMPA-kainate receptors located on the DA neurons, leading to an increase in intracellular Ca2+ levels. This is supported by our data demonstrating that mGluR antagonists increased EPSC amplitudes and by the blockade of the eCB-dependent inhibition of GABAB IPSCs by the AMPA-kainate antagonist NBQX. The role of intracellular Ca2+ was confirmed by blocking the eCB effect with BAPTA, consistent with studies in other brain areas and further revealing that the DA neurons are the likely source of the eCB in the VTA. These experiments demonstrate that presynaptic mGluR-IIIs are activated during mild levels of afferent activity to strongly regulate the efficacy of glutamatergic transmission and eCB production in the VTA. Although the origin of the activated glutamatergic afferents is unclear, our results are compatible with previous work demonstrating that depolarization of VTA neurons through somatic current injection also stimulated eCB production that activated CB1Rs located on glutamatergic afferents (Melis et al., 2004).
Consistent with previous reports, apamin increased DA neuron firing rates and membrane oscillations during electrical stimulation (Johnson et al., 1992; Cooper, 2002), whereas it also increased eCB production in the VTA. In addition, like mGluR-III antagonists, apamin occluded the effect of WIN55,212-2 at CB1Rs located on GABAergic afferents, revealing an AM251-induced increase in GABAB IPSCs. The blockade of SK channels by apamin also produced an AM251-reversible inhibition of AMPA-kainate EPSCs, presumably by initiating eCB release that activated CB1Rs on glutamatergic terminals. Thus, endogenous CB released from DA neurons simultaneously inhibited GABAergic and glutamatergic afferents in the VTA. By reducing the AHP-dependent repolarization that follows spike discharge, apamin increases DA neuron sensitivity to excitatory input, which in the presence of diminished inhibition can shift the balance of activity toward excitation and bursting (Overton and Clark, 1997; Cooper, 2002). Data obtained from in vitro and in vivo studies suggest conditions under which an “apamin-like” diminution of SK channel function may represent a physiologically relevant effect that might also increase eCB production in these neurons (Overton and Clark, 1997; Hyland et al., 2002). For instance, mechanisms that alter intracellular Ca2+ concentrations and thereby reduce the function of Ca2+-sensitive SK channels include mGluR-I stimulation of IP3 formation (Fiorillo and Williams, 1998), muscarinic receptor activation (Fiorillo and Williams, 2000; Scroggs et al., 2001), or α1 adrenergic receptor activation (Paladini et al., 2001). A previous study has shown that DA neuron bursting could be initiated by concomitant activation of NMDA receptors and SK channel blockade by apamin and that this effect was reversed by inhibitors of an electrogenic Na+ pump (Johnson et al., 1992); however, this explanation cannot be invoked to explain eCB release in our study because NMDA channel function was blocked by MK-801 throughout these experiments.
The shift from pacemaker-like firing to bursting is driven by patterned glutamatergic and GABAergic synaptic input to midbrain DA neurons (Grace and Bunney, 1985; Smith and Grace, 1992; Chergui et al., 1994; Tepper et al., 1995). Furthermore, bursting is normally lacking in DA neurons recorded in vitro, although it can be restored by the application of glutamate agonists and apamin (Johnson et al., 1992; Seutin et al., 1993; Kitai et al., 1999). GABAB antagonists can also initiate bursting (Erhardt et al., 2002), presumably by reducing GABAergic tone and “dis-inhibiting” the DA neurons (Seutin et al., 1994). In the present study, the increased release of glutamate and the enhanced sensitivity to glutamatergic input during mGluR antagonist and apamin application would be expected to increase DA neuron membrane oscillations and possibly induce bursting (Cooper, 2002); however, it was possible to observe eCB-dependent effects during each of these manipulations alone, suggesting that eCBs would be produced in greater amounts under conditions favoring bursting, but also at lower concentrations during milder forms of activity. Also, although both mGluR-III antagonism and SK channel blockade yielded an AM251-reversible inhibition of GABAB IPSCs and AMPA-kainate EPSCs, their additive effects demonstrated that eCB production was mediated through independent mechanisms. Collectively, these results imply an ongoing regulatory role for eCBs, the release of which is controlled by intrinsic physiological mechanisms in the VTA.
The mesolimbic DA system plays an important role in modulating goal-directed behaviors by gating sensory inputs to the NAc (Wise, 2002). This is thought to arise from changes in DA neuron activity and the temporal characteristics of DA release (Floresco et al., 2003). For example, rapid transient increases in NAc DA release may signal prediction errors that encode novel environmental stimuli possessing reward value (Schultz, 2002). Interestingly, stimulation of glutamatergic afferents to the VTA can produce similar DA transients in the NAc coincident with approach behaviors (Roitman et al., 2004) that are correlated with DA neuron bursting (Floresco et al., 2003). In contrast, slower tonic increases in DA release may mediate movement and facilitate motivation by enhancing “extrasynaptic” DA levels in the NAc (Schultz, 2002; Floresco et al., 2003). These longer-lasting changes presumably reflect an increased number of active VTA DA neurons recruited by changes in synaptic inputs, including glutamate and GABA from the NAc-VP (Floresco et al., 2003). Cannabinoid agonists such as Δ9-THC also stimulate DA transients in the NAc (Cheer et al., 2004) and bursting in DA neurons (French, 1997). Our data demonstrating the inhibition of GABAergic inputs to the VTA by cannabinoids are consistent with reported increases in tonic DA release in the NAc (Gardner, 2002) and cell firing in the VTA after systemic administration of Δ9-THC (French, 1997). This disinhibitory effect of cannabinoids might also be amplified by the ability of CB1 agonists to diminish GABAA receptor-mediated IPSCs arising from axosomatic innervation of DA neurons by intrinsic GABAergic interneurons (Szabo et al., 2002).
In conclusion, by defining independent presynaptic and postsynaptic cellular mechanisms that control eCB release and regulate ongoing VTA DA neuron activity, we have shown that these neurons can regulate their own afferent input via the retrograde activation of presynaptic CB1Rs. Because the release of eCB molecules appears to depend on increased DA neuron activity, it seems likely that these cells may dynamically fine-tune their own synaptic inputs, thereby providing an additional mechanism to integrate the balance between excitation and inhibition. This regulation of afferent activity by eCBs acting at CB1Rs may also contribute to the shift from pacemaker to phasic DA neuron firing patterns in the presence of appropriate environmental stimuli. On the other hand, the rewarding properties of Δ9-THC may also reflect the activation of this eCB circuitry, which may have evolved as a regulatory mechanism within this central neural reward pathway.
Footnotes
This work was supported by the National Institute on Drug Abuse Intramural Research Program. We thank Drs. John Williams and Alex Hoffman for their helpful comments on this manuscript.
Correspondence should be addressed to Carl R. Lupica, Cellular Neurobiology Branch, Electrophysiology Unit, National Institute on Drug Abuse Intramural Research Program, National Institutes of Health, United States Department of Health and Human Services, Baltimore, MD 21224. E-mail: clupica@intra.nida.nih.gov.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2411070-09$15.00/0
References
- Alger BE (2002) Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol 68: 247-286. [DOI] [PubMed] [Google Scholar]
- Bonci A, Grillner P, Siniscalchi A, Mercuri NB, Bernardi G (1997) Glutamate metabotropic receptor agonists depress excitatory and inhibitory transmission on rat mesencephalic principal neurons. Eur J Neurosci 9: 2359-2369. [DOI] [PubMed] [Google Scholar]
- Cameron DL, Wessendorf MW, Williams JT (1997) A subset of ventral tegmental area neurons is inhibited by dopamine, 5-hydroxytryptamine and opioids. Neuroscience 77: 155-166. [DOI] [PubMed] [Google Scholar]
- Charara A, Smith Y, Parent A (1996) Glutamatergic inputs from the pedunculopontine nucleus to midbrain dopaminergic neurons in primates: Phaseolus vulgaris-leucoagglutinin anterograde labeling combined with postembedding glutamate and GABA immunohistochemistry. J Comp Neurol 364: 254-266. [DOI] [PubMed] [Google Scholar]
- Cheer JF, Kendall DA, Mason R, Marsden CA (2003) Differential cannabinoid-induced electrophysiological effects in rat ventral tegmentum. Neuropharmacology 44: 633-641. [DOI] [PubMed] [Google Scholar]
- Cheer JF, Wassum KM, Heien MLAV, Phillips PEM, Wightman RM (2004) Cannabinoids enhance subsecond dopamine release in the nucleus accumbens of awake rats. J Neurosci 24: 4393-4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Marmur R, Pulles A, Paredes W, Gardner EL (1993) Ventral tegmental microinjection of delta 9-tetrahydrocannabinol enhances ventral tegmental somatodendritic dopamine levels but not forebrain dopamine levels: evidence for local neural action by marijuana's psychoactive ingredient. Brain Res 621: 65-70. [DOI] [PubMed] [Google Scholar]
- Chergui K, Suaud-Chagny MF, Gonon F (1994) Nonlinear relationship between impulse flow, dopamine release and dopamine elimination in the rat brain in vivo. Neuroscience 62: 641-645. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP (1997) Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol 37: 205-237. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Miller JC, Howson PA, Clark BP, Jane DE (2001) Synthesis of phenylglycine derivatives as potent and selective antagonists of group III metabotropic glutamate receptors. Bioorg Med Chem Lett 11: 777-780. [DOI] [PubMed] [Google Scholar]
- Cooper DC (2002) The significance of action potential bursting in the brain reward circuit. Neurochem Int 41: 333-340. [DOI] [PubMed] [Google Scholar]
- Erhardt S, Mathe JM, Chergui K, Engberg G, Svensson TH (2002) GABA(B) receptor-mediated modulation of the firing pattern of ventral tegmental area dopamine neurons in vivo. Naunyn Schmiedebergs Arch Pharmacol 365: 173-180. [DOI] [PubMed] [Google Scholar]
- Fiorillo CD, Williams JT (1998) Glutamate mediates an inhibitory postsynaptic potential in dopamine neurons. Nature 394: 78-82. [DOI] [PubMed] [Google Scholar]
- Fiorillo CD, Williams JT (2000) Cholinergic inhibition of ventral midbrain dopamine neurons. J Neurosci 20: 7855-7860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floresco SB, West AR, Ash B, Moore H, Grace AA (2003) Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat Neurosci 6: 968-973. [DOI] [PubMed] [Google Scholar]
- French ED (1997) delta9-Tetrahydrocannabinol excites rat VTA dopamine neurons through activation of cannabinoid CB1 but not opioid receptors. Neurosci Lett 226: 159-162. [DOI] [PubMed] [Google Scholar]
- French ED, Dillon K, Wu X (1997) Cannabinoids excite dopamine neurons in the ventral tegmentum and substantia nigra. NeuroReport 8: 649-652. [DOI] [PubMed] [Google Scholar]
- Gardner EL (2002) Addictive potential of cannabinoids: the underlying neurobiology. Chem Phys Lipids 121: 267-290. [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS (1985) Low doses of apomorphine elicit two opposing influences on dopamine cell electrophysiology. Brain Res 333: 285-298. [DOI] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, de Costa BR, Richfield EK (1991) Neuronal localization of cannabinoid receptors in the basal ganglia of the rat. Brain Res 547: 267-274. [DOI] [PubMed] [Google Scholar]
- Hernandez M, Berrendero F, Suarez I, Garcia-Gil L, Cebeira M, Mackie K, Ramos JA, Fernandez-Ruiz J (2000) Cannabinoid CB1 receptors colocalize with tyrosine hydroxylase in cultured fetal mesencephalic neurons and their activation increases the levels of this enzyme. Brain Res 857: 56-65. [DOI] [PubMed] [Google Scholar]
- Hungund BL, Szakall I, Adam A, Basavarajappa BS, Vadasz C (2003) Cannabinoid CB1 receptor knockout mice exhibit markedly reduced voluntary alcohol consumption and lack alcohol-induced dopamine release in the nucleus accumbens. J Neurochem 84: 698-704. [DOI] [PubMed] [Google Scholar]
- Hyland BI, Reynolds JNJ, Hay J, Perk CG, Miller R (2002) Firing modes of midbrain dopamine cells in the freely moving rat. Neuroscience 114: 475-492. [DOI] [PubMed] [Google Scholar]
- Isaacson JS (2000) Spillover in the spotlight. Curr Biol 10: R475-R477. [DOI] [PubMed] [Google Scholar]
- Johnson SW, North RA (1992) Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol (Lond) 450: 455-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, Seutin V, North RA (1992) Burst firing in dopamine neurons induced by N-methyl-d-aspartate: role of electrogenic sodium pump. Science 258: 665-667. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Churchill L, Klitenick MA (1993) GABA and enkephalin projection from the nucleus accumbens and ventral pallidum to the ventral tegmental area. Neuroscience 57: 1047-1060. [DOI] [PubMed] [Google Scholar]
- Kita H, Kitai ST (1987) Efferent projections of the subthalamic nucleus in the rat: light and electron microscopic analysis with the PHA-L method. J Comp Neurol 260: 435-452. [DOI] [PubMed] [Google Scholar]
- Kitai ST, Shepard PD, Callaway JC, Scroggs R (1999) Afferent modulation of dopamine neuron firing patterns. Curr Opin Neurobiol 9: 690-697. [DOI] [PubMed] [Google Scholar]
- Kosinski CM, Standaert DG, Testa CM, Penney J, Young AB (1998) Expression of metabotropic glutamate receptor 1 isoforms in the substantia nigra pars compacta of the rat. Neuroscience 86: 783-798. [DOI] [PubMed] [Google Scholar]
- Lan R, Liu Q, Fan P, Lin S, Fernando SR, McCallion D, Pertwee R, Makriyannis A (1999) Structure-activity relationships of pyrazole derivatives as cannabinoid receptor antagonists. J Med Chem 42: 769-776. [DOI] [PubMed] [Google Scholar]
- Mascia MS, Obinu MC, Ledent C, Parmentier M, Bohme GA, Imperato A, Fratta W (1999) Lack of morphine-induced dopamine release in the nucleus accumbens of cannabinoid CB1 receptor knockout mice. Eur J Pharmacol 383: R1-R2. [DOI] [PubMed] [Google Scholar]
- Matsuda LA, Bonner TI, Lolait SJ (1993) Localization of cannabinoid receptor mRNA in rat brain. J Comp Neurol 327: 535-550. [DOI] [PubMed] [Google Scholar]
- Melis M, Pistis M, Perra S, Muntoni AL, Pillolla G, Gessa GL (2004) Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci 24: 53-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC, Howson PA, Conway SJ, Williams RV, Clark BP, Jane DE (2003) Phenylglycine derivatives as antagonists of group III metabotropic glutamate receptors expressed on neonatal rat primary afferent terminals. Br J Pharmacol 139: 1523-1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olpe HR, Karlsson G, Pozza MF, Brugger F, Steinmann M, Van Riezen H, Fagg G, Hall RG, Froestl W, Bittiger H (1990) CGP 35348: a centrally active blocker of GABAB receptors. Eur J Pharmacol 187: 27-38. [DOI] [PubMed] [Google Scholar]
- Overton PG, Clark D (1997) Burst firing in midbrain dopaminergic neurons. Brain Res Rev 25: 312-334. [DOI] [PubMed] [Google Scholar]
- Paladini CA, Fiorillo CD, Morikawa H, Williams JT (2001) Amphetamine selectively blocks inhibitory glutamate transmission in dopamine neurons. Nat Neurosci 4: 275-281. [DOI] [PubMed] [Google Scholar]
- Roitman MF, Stuber GD, Phillips PEM, Wightman RM, Carelli RM (2004) Dopamine operates as a subsecond modulator of food seeking. J Neurosci 24: 1265-1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoepp DD (2001) Unveiling the functions of presynaptic metabotropic glutamate receptors in the central nervous system. J Pharmacol Exp Ther 299: 12-20. [PubMed] [Google Scholar]
- Schultz W (1998) Predictive reward signal of dopamine neurons. J Neurophysiol 80: 1-27. [DOI] [PubMed] [Google Scholar]
- Schultz W (2002) Getting formal with dopamine and reward. Neuron 36: 241-263. [DOI] [PubMed] [Google Scholar]
- Scroggs RS, Cardenas CG, Whittaker JA, Kitai ST (2001) Muscarine reduces calcium-dependent electrical activity in substantia nigra dopaminergic neurons. J Neurophysiol 86: 2966-2972. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Pickel VM (1992) Prefrontal cortical efferents in the rat synapse on unlabeled neuronal targets of catecholamine terminals in the nucleus accumbens septi and on dopamine neurons in the ventral tegmental area. J Comp Neurol 320: 145-160. [DOI] [PubMed] [Google Scholar]
- Seutin V, Johnson SW, North RA (1993) Apamin increases NMDA-induced burst-firing of rat mesencephalic dopamine neurons. Brain Res 630: 341-344. [DOI] [PubMed] [Google Scholar]
- Seutin V, Johnson SW, North RA (1994) Effect of dopamine and baclofen on N-methyl-d-aspartate-induced burst firing in rat ventral tegmental neurons. Neuroscience 58: 201-206. [DOI] [PubMed] [Google Scholar]
- Smith ID, Grace AA (1992) Role of the subthalamic nucleus in the regulation of nigral dopamine neuron activity. Synapse 12: 287-303. [DOI] [PubMed] [Google Scholar]
- Sugita S, Johnson SW, North RA (1992) Synaptic inputs to GABAA and GABAB receptors originate from discrete afferent neurons. Neurosci Lett 134: 207-211. [DOI] [PubMed] [Google Scholar]
- Swanson LW (1982) The projections of the ventral tegmental area and adjacent regions: a combined fluorescent retrograde tracer and immunofluorescence study in the rat. Brain Res Bull 9: 321-353. [DOI] [PubMed] [Google Scholar]
- Szabo B, Siemes S, Wallmichrath I (2002) Inhibition of GABAergic neurotransmission in the ventral tegmental area by cannabinoids. Eur J Neurosci 15: 2057-2061. [DOI] [PubMed] [Google Scholar]
- Tanda G, Goldberg SR (2003) Cannabinoids: reward, dependence, and underlying neurochemical mechanisms—a review of recent preclinical data. Psychopharmacology (Berl) 169: 115-134. [DOI] [PubMed] [Google Scholar]
- Tepper JM, Martin LP, Anderson DR (1995) GABAA receptor-mediated inhibition of rat substantia nigra dopaminergic neurons by pars reticulata projection neurons. J Neurosci 15: 3092-3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa CM, Standaert DG, Young AB, Penney Jr JB (1994) Metabotropic glutamate receptor mRNA expression in the basal ganglia of the rat. J Neurosci 14: 3005-3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa CM, Friberg IK, Weiss SW, Standaert DG (1998) Immunohistochemical localization of metabotropic glutamate receptors mGluR1a and mGluR2/3 in the rat basal ganglia. J Comp Neurol 390: 5-19. [PubMed] [Google Scholar]
- Tokuno H, Moriizumi T, Kudo M, Nakamura Y (1988) A morphological evidence for monosynaptic projections from the nucleus tegmenti pedunculopontinus pars compacta (TPC) to nigrostriatal projection neurons. Neurosci Lett 85: 1-4. [DOI] [PubMed] [Google Scholar]
- Tzavara ET, Davis RJ, Perry KW, Li X, Salhoff C, Bymaster FP, Witkin JM, Nomikos GG (2003) The CB1 receptor antagonist SR141716A selectively increases monoaminergic neurotransmission in the medial prefrontal cortex: implications for therapeutic actions. Br J Pharmacol 138: 544-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walaas I, Fonnum F (1980) Biochemical evidence for gamma-aminobutyrate containing fibres from the nucleus accumbens to the substantia nigra and ventral tegmental area in the rat. Neuroscience 5: 63-72. [DOI] [PubMed] [Google Scholar]
- Westerink BH, Kwint HF, deVries JB (1996) The pharmacology of mesolimbic dopamine neurons: a dual-probe microdialysis study in the ventral tegmental area and nucleus accumbens of the rat brain. J Neurosci 16: 2605-2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, North RA, Shefner SA, Nishi S, Egan TM (1984) Membrane properties of rat locus coeruleus neurones. Neuroscience 13: 137-156. [DOI] [PubMed] [Google Scholar]
- Wise RA (2002) Brain reward circuitry: insights from unsensed incentives. Neuron 36: 229-240. [DOI] [PubMed] [Google Scholar]
- Wolfart J, Roeper J (2002) Selective coupling of T-type calcium channels to SK potassium channels prevents intrinsic bursting in dopaminergic midbrain neurons. J Neurosci 22: 3404-3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Yokoo H, Tanaka T, Emoto H, Tanaka M (1994) Opposite changes in the mesolimbic dopamine metabolism in the nerve terminal and cell body sites induced by locally infused baclofen in the rat. Brain Res 636: 111-114. [DOI] [PubMed] [Google Scholar]
- Zheng F, Johnson SW (2003) Metabotropic glutamate and muscarinic cholinergic receptor-mediated preferential inhibition of N-methyl-d-aspartate component of transmissions in rat ventral tegmental area. Neuroscience 116: 1013-1020. [DOI] [PubMed] [Google Scholar]