

SUMMARY
Cocaine is a highly addictive drug that acts upon the brain’s reward circuitry via the inhibition of mono-amine uptake. Endogenous cannabinoids (eCB) are lipid molecules released from midbrain dopamine (DA) neurons that modulate cocaine’s effects through poorly understood mechanisms. We find that cocaine stimulates release of the eCB, 2-arach-idonoylglycerol (2-AG), in the rat ventral midbrain to suppress GABAergic inhibition of DA neurons, through activation of presynaptic cannabinoid CB1 receptors. Cocaine mobilizes 2-AG via inhibition of norepinephrine uptake and promotion of a cooperative interaction between Gq/11-coupled type-1 metabotropic glutamate and α1-adrenergic receptors to stimulate internal calcium stores and activate phospholipase C. The disinhibition of DA neurons by cocaine-mobilized 2-AG is also functionally relevant because it augments DA release in the nucleus accumbens in vivo. Our results identify a mechanism through which the eCB system can regulate the rewarding and addictive properties of cocaine.
Graphical Abstract
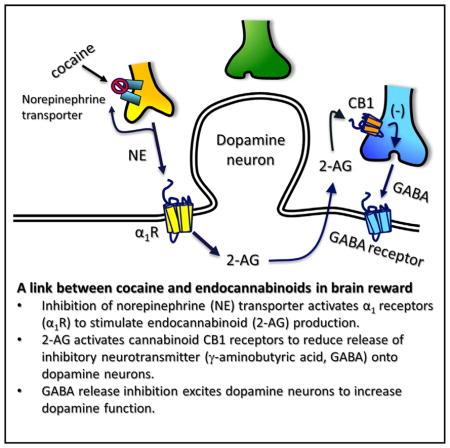
INTRODUCTION
The ventral tegmental area (VTA) is a central component of a mammalian brain reward circuit involved in processing environmental information to ensure survival. The VTA contains neurons that project extensively to the forebrain and release dopamine (DA). These neurons mediate the rewarding properties of all abused drugs (Di Chiara and Imperato, 1988; Wise and Rompre, 1989) and shift their activity from pacemaker to burst firing during exposure to drugs or stimuli predicting drug availability (Cooper, 2002; Covey et al., 2014). This enhancement of DA neuron activity results in increased DA in projection sites, such as the nucleus accumbens (NAc), which is critical to drug reinforcement (Di Chiara and Bassareo, 2007; Phillips et al., 2003).
Cannabinoid CB1 receptors (CB1R) are coupled to G-proteins, are extensively expressed on axon terminals in the CNS, and inhibit release of excitatory and inhibitory neurotransmitters to sculpt postsynaptic activity (Alger and Kim, 2011; Kano et al., 2009). CB1Rs are also activated by the primary psychoactive constituent of cannabis, Δ9-tetrahydrocannabinol (Δ9-THC), to mediate the physiological and psychotropic effects of this drug in humans (Hoffman and Lupica, 2013). Molecules, known as endogenous cannabinoids (eCBs), derived from membrane phospholipids, also activate CB1Rs to mediate short- and long-term forms of synaptic plasticity (Kano et al., 2009). Anandamide (Devane et al., 1992) and 2-arachidonoylglycerol (2-AG) (Mechoulam et al., 1995) are the best characterized eCBs, with 2-AG most closely associated with activity-dependent modulation of synapses (Kano et al., 2009). 2-AG can be mobilized during transient postsynaptic increases in Ca2+ through activation of voltage-dependent Ca2+ channels (Kreitzer and Regehr, 2001; Maejima et al., 2001; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). This type of 2-AG release mediates depolarization-induced suppression of inhibition (DSI) or excitation (DSE), in which 2-AG from postsynaptic cells acts to retrogradely activate CB1Rs on axon terminals to inhibit GABA or glutamate release, respectively. 2-AG synthesis can also be triggered through activation of Gαq/11-coupled metabotropic receptors via their coupling to phospholipase C-β (PLCβ). PLCβ hydrolyzes phosphatidylinositol 4,5-bisphosphate to form 1,2-dacylglycerol (DAG) and inositol triphosphate. DAG is then hydrolyzed by diacylglycerol lipase-α (DGLα) to form 2-AG (Hashimotodani et al., 2005; Kano et al., 2009; Maejima et al., 2005). Receptor-mediated 2-AG release can also be strongly augmented by coincident elevation of intracellular Ca2+ (Hashimotodani et al., 2007; Kano et al., 2009; Maejima et al., 2005).
eCBs are released from VTA DA neurons during depolarization (Melis et al., 2004) or conditions promoting burst activity (Riegel and Lupica, 2004). Furthermore, DGLα is strategically located on postsynaptic membranes adjacent to presynaptic CB1Rs in the VTA (Mátyás et al., 2008). All abused drugs, including Δ9-THC, increase DA release in forebrain areas such as the NAc. The cannabinoid-induced increase in DA likely occurs through an increase in VTA DA neuron firing rate and bursting (French et al., 1997; Gessa et al., 1998). Since cannabinoids act at CB1Rs in the VTA to inhibit the release of the inhibitory neurotransmitter GABA (Riegel and Lupica, 2004; Szabo et al., 2002), we hypothesize that disinhibition of VTA DA neurons leads to increases in their activity and phasic release of DA in the NAc (Cheer et al., 2007; Lupica et al., 2004; Riegel and Lupica, 2004).
The eCB system also regulates the rewarding actions of many abused drugs (Lupica and Riegel, 2005). Antagonism of CB1Rs reduces drug-induced reward-related behavior and the associated elevation of NAc DA (Cheer et al., 2007; Li et al., 2009). The rewarding property of the psychostimulant cocaine is primarily attributed to its ability to increase forebrain DA levels via inhibition of the DA transporter (DAT) (Ritz et al., 1987; Thomsen et al., 2009). However, because cocaine-induced elevations of DA release events depend on VTA DA neuron firing (Sombers et al., 2009) and eCB signaling (Cheer et al., 2007), eCB release in the VTA may be critical to cocaine reinforcement.
Here, we find that cocaine mobilizes 2-AG, which then activates CB1Rs to inhibit GABA release onto DA neurons, and that cocaine’s ability to elevate NAc DA in awake rats is attenuated by disruption of 2-AG signaling. Mobilization of 2-AG by cocaine depends on activation of Gq/11-coupled receptors, initiation of Ca2+ oscillations, and activation of PLC. We propose that cocaine increases DA in the NAc and reinforces behavior, in part, through disinhibition of DA neurons via suppression of GABA release by cocaine-induced 2-AG mobilization.
RESULTS
Inhibition of GABA Release onto VTA DA Neurons by 2-AG
IPSCs mediated by GABAB receptors on VTA DA neurons (S1a) are inhibited by presynaptic CB1Rs (Riegel and Lupica, 2004). Here, the synthetic CB1R agonist WIN55,212-2 (1 μM) inhibited GABAB IPSCs (77.9% ± 3.9% of control, n = 5; p < 0.05, paired t test) (Figures 1A and 1B). These responses were also increased by the CB1R antagonist/inverse agonist, AM251 (2 μM; 115.4% ± 1.6% of control, n = 6; p < 0.01, paired t test), suggesting that eCBs tonically inhibit GABAB IPSCs. Because many CB1R antagonists, such as AM251, are also inverse agonists (Landsman et al., 1997), their reversal of eCB effects may instead reflect allosteric modulation of CB1Rs. However, a “neutral” CB1R antagonist, 5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-[(E)-piperidinoiminomethyl]-1H-pyrazole (PIMSR1; 2 μM) (Hurst et al., 2006) also increased GABAB IPSCs in putative DA neurons (121.3% ± 5.7% of control, n = 6; p < 0.05, paired t test). Moreover, this did not differ significantly from that produced by AM251 (p = 0.35, unpaired t test, Figures 1A and 1B). This suggests that AM251 and PIMSR1 increased GABAB IPSC amplitude through antagonism of tonically released eCB acting at CB1Rs and not through allosteric modulation. As the effect of AM251 did not differ from PIMSR1, it was used in subsequent experiments.
Figure 1. Tonic Inhibition of GABA Signaling in VTA DA Neurons Is Mediated by 2-AG.

(A) Sample traces (left) and time-courses of GABAB IPSCs (right) during CB1R ligand treatment; CB1R agonist WIN55,212-2 (WIN, 1 μM), CB1R antagonist/inverse agonist, AM251 (2 μM), and neutral antagonist PIMSR1 (2 μM). Gray traces in all figures indicate those during treatment. Gray area in time course indicates period of drug application. Scale bar, 100 ms, 10 pA.
(B) Summary of CB1R ligand effects on GABAB IPSCs (F(2,14) = 30.75, p < 0.0001, one-way ANOVA, *p < 0.01, **p < 0.001, Tukey’s post hoc).
(C) The AM251-mediated increase in GABAB IPSCs is prevented by inclusion of the DGL inhibitor, THL (5 μM), in the pipette (*p < 0.05, unpaired t test). Scale bar, 100 ms, 10 pA.
(D) The AM251-induced increase of GABAB IPSCs is enhanced by the MAG lipase inhibitor JZL184 (10 μM; **p < 0.01, unpaired t test). Scale bar, 100 ms, 10 pA.
We next determined which eCB exerted control over GABAB IPSCs in putative VTA DA neurons. 2-AG is derived from membrane phospholipids via the sequential activity of PLC and DGLα (Kano et al., 2009). 2-AG is then inactivated by the enzyme monoacylglycerol lipase (MGL) in presynaptic terminals. Inclusion of the DGLα inhibitor tetrahdydrolipstatin (THL, 5 μM) in the intra-pipette solution prevented the enhancement of GABAB IPSCs by AM251 (AM251 = 140.7% ± 12.3%, n = 5; AM251 + intra-THL = 104.9% ± 7.7%, n = 6; unpaired t test, p < 0.05; Figure 1C). In contrast, the increase in GABAB IPSCs produced by AM251 was significantly larger in the presence of the MGL inhibitor JZL184 (10 μM; AM251 alone = 115.4% ± 1.6% of control, n = 6; AM251 + JZL184 = 132.6% ± 3.9% of control, n = 5; unpaired t test, p < 0.01; Figure 1D). This demonstrates that 2-AG is tonically released from putative DA neurons to inhibit GABAB IPSCs via activation of CB1Rs.
Cocaine Increases DA Transients in the NAc via eCB Action in the VTA
Systemic cocaine administration increases DA transient signals measured by fast scan cyclic voltammetry (FSCV) in the NAc and this is attenuated by systemic administration of the CB1R antagonist, rimonabant (SR141716A) (Cheer et al., 2007). Since DA neurons do not express CB1Rs (Mátyás et al., 2008), the effects of cocaine are likely presynaptic to these cells. Moreover, as CB1Rs are expressed on GABA axon terminals in the VTA (Mátyás et al., 2008; Riegel and Lupica, 2004; Szabo et al., 2002), we hypothesize that cocaine releases eCBs in the VTA to inhibit GABA release onto DA neurons to disinhibit these cells. To determine whether the eCB-dependent effects of cocaine occur through actions in the VTA, NAc DA transients were measured during intravenous (i.v.) cocaine administration in vivo and the effect of intra-VTA rimonabant tested (Figure 2). A one-way ANOVA for all combinations of intra-VTA and i.v. injections revealed a significant treatment effect (F(4,31) = 29.14, p < 0.0001). Under baseline conditions, NAc DA transients occurred at 0.028 ± 0.007 Hz, and as reported previously, were significantly increased by i.v. cocaine during intra-VTA vehicle infusion (cocaine + vehicle = 0.176 ± 0.01 Hz; p < 0.0001, Bonferroni post hoc comparison; Cheer et al., 2007). Injection of rimonabant into the VTA prior to i.v. cocaine significantly reduced this increase in DA transients (cocaine + rimonabant = 0.082 ± 0.10 Hz, n = 6 rats; p < 0.0001, Bonferroni post hoc comparison; Figure 2C). Moreover, intra-VTA rimonabant alone did not alter the frequency of NAc DA transients (0.032 ± 0.003 Hz, p > 0.05, Bonferroni post hoc comparison, Figure 2C).
Figure 2. Cocaine-Induced Increase in NAc DA Transients Require CB1R Activation in VTA.
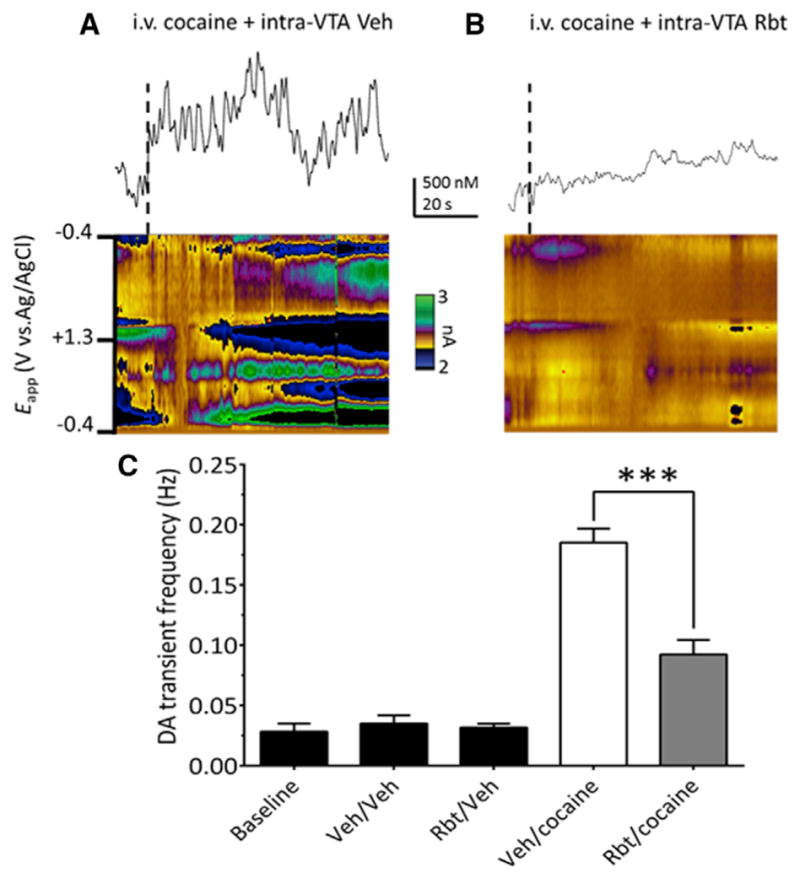
(A) Voltammetric current (encoded in color) plotted against the applied carbon-fiber electrode potential (ordinate) and the acquisition time (abscissa). Traces above color plots represent current (normalized to concentration) from the potential where DA is oxidized (~+0.6 V in green), when animals are given an i.v. bolus of cocaine (1 mg/kg), preceded by intra-VTA vehicle infusion.
(B) Systemic effects of cocaine are attenuated following CB1R blockade in VTA (250 ng rimonabant [Rbt] in 500 μl, unilateral and ipsilateral to the recording electrode).
(C) Effects of all treatments on NAc DA transient frequency (F(4,31) = 29.14, p < 0.0001, one-way ANOVA; n = 12 rats). Baseline indicates the frequency of DA transients without intracranial or systemic injections. In other groups the intra-VTA injection is indicated first, followed by the i.v. injection (e.g., intracranial Veh\intra-VTA Veh). Note that Rbt alone (Rbt/Veh) did not alter DA transient frequency, and Rbt significantly reduced the effect of i.v. cocaine on DA transients (Rbt/cocaine, ***p < 0.0001, Bonferroni post hoc test).
2-AG-Mediated Inhibition of GABAB IPSCs by Cocaine
We next determined whether cocaine enhanced eCB release in VTA in vitro and whether this reduced GABAergic input to DA neurons. Cocaine (10 μM) significantly inhibited GABAB IPSCs (67.5% ± 3.0% of control; n = 19; p < 0.0001, paired t test; Figures 3A and 3C) and this was significantly reduced by AM251 (2 μM; 87.2% ± 4.8% of control, n = 7; p < 0.01, one-way ANOVA, Bonferroni post hoc analysis; Figures 3A and 3C). Furthermore, the effects of cocaine on GABAB IPSCs reversed when its application was stopped (Figures S1B and S1C). To determine whether 2-AG was the eCB whose actions were increased by cocaine, we examined the effects of the DGLα inhibitor, THL on the inhibition of GABAB IPSCs produced by cocaine. Cocaine’s inhibition of GABAB IPSCs was not significantly reduced by THL (5 μM) when included in the intra-pipette solution (intra-THL: 75.0% ± 2.7% of control; p > 0.05, one-way ANOVA, Bonferroni post hoc analysis, n = 9; Figures 3B and 3C). However, extracellular application of THL (10 μM) significantly reduced the inhibitory effect of cocaine on GABAB IPSCs (extra-THL = 85.6% ± 4.7% of control, n = 5; p = 0.009, paired t test; p < 0.05, one-way ANOVA, Bonferroni post hoc analysis; Figures 3B and 3C). This suggests that cocaine releases 2-AG from sites that are more widely distributed than those involved in tonic inhibition of GABAB IPSCs under control conditions (Figure 1C).
Figure 3. Cocaine-Mobilized 2-AG Inhibits GABA Release and Increases DA Release in NAc via Actions in VTA.

(A) Acute cocaine (10 μM) perfusion inhibited GABAB IPSCs and this was partially blocked by AM251 (2 μM). Scale bar, 100 ms, 10 pA.
(B) Cocaine inhibition of GABAB IPSCs was reduced by inhibition of DGL with THL, applied either intracellularly via the pipette (intra-THL) or extracellularly, by addition to the aCSF (extra-THL). Scale bar, 100 ms, 10 pA.
(C) Summary of the experiments shows significant reduction of the cocaine inhibition of GABAB IPSCs by AM251 and THL (n = 5; F(3,36) = 6.35, p = 0.0014, one-way ANOVA; *p < 0.05 and **p < 0.01, Bonferroni post hoc analysis).
(D) Cocaine did not affect postsynaptic GABAB currents evoked by laser (indicated by vertical bar on sweeps inset) uncaging of O-CNB-Caged-GABA (100–200 μM in bath; n = 7, p > 0.05, paired t test. Scale bar, 100 ms, 10 pA.
(E) In vivo NAc FSCV current (encoded in color) is plotted against the applied potential (ordinate) and the acquisition time (abscissa). Traces above color plots are extracted currents (normalized to concentration) from the potential where DA is oxidized (~+0.6 V shown in green), when animals are given intravenous cocaine (1 mg/kg), preceded by an i.c.v. vehicle injection.
(F) Effects of cocaine are reduced in vivo by inhibition of 2-AG synthesis by THL in the VTA (750 ng THL in 500 μl, unilateral and ipsilateral to the recording electrode).
(G) Intra-VTA THL reduces DA transient frequency elicited by i.v. cocaine (F (4,10) = 9.395, p = 0.002, one-way ANOVA; n = 3 rats, **p < 0.001, Bonferroni post hoc analysis). Baseline indicates DA transient frequency without intracranial or systemic injections. In other groups, the intra-VTA injection is indicated first, followed by the i.v. injection condition (e.g., intracranial Veh\intra-VTA Veh). Note that THL alone (THL/Veh) did not alter DA transient frequency, and THL significantly reduced the effect of i.v. cocaine on DA transients (THL/cocaine, p < 0.001, Bonferroni post hoc test).
See also Figure S1.
The main site eCBs act in the CNS is at CB1Rs on axon terminals to suppress neurotransmitter release (Kreitzer and Regehr, 2001; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). To determine whether eCB-dependent effects of cocaine on GABAB IPSCs were presynaptic we measured GABA-mediated postsynaptic currents elicited by laser (355 nm) photolysis of O-(CNB-Caged) GABA (100–200 μM). Light-evoked GABAB currents were not altered by cocaine (control: 23.79 ± 4.85 pA; cocaine: 22.38 ± 4.74 pA; n = 7; p > 0.05, paired t test; Figure 3D), suggesting that inhibition of electrically evoked GABAB IPSCs by cocaine occurred presynaptically.
We next determined whether 2-AG in the VTA mediates the increase in NAc DA transients during cocaine application in vivo using THL injection in VTA. A one-way ANOVA for all combinations of intra-VTA and i.v. injections revealed a significant treatment effect (F(4,10) = 9.395, p = 0.002). Under baseline conditions, DA transients occurred at 0.044 ± 0.003 Hz and this was significantly increased by i.v. injection of cocaine and intra-VTA vehicle infusion (cocaine + vehicle = 0.094 ± 0.007 Hz; p < 0.001, Bonferroni post hoc comparison, Figure 3G). Intra-VTA THL significantly reduced the increase in DA transients by cocaine (cocaine + THL = 0.05 ± 0.011 Hz, n = 3 rats; p < 0.001, Bonferroni post hoc comparison; Figure 3G). Moreover, intra-VTA THL alone did not alter the frequency of DA transients, compared to pre-cocaine baseline (0.045 ± 0.006 Hz, p > 0.05, Bonferroni post hoc comparison, Figure 3G). Consistent with the in vitro experiments, these data suggest that cocaine enhances 2-AG levels in the VTA to disinhibit DA neurons, contributing to increased DA release in the NAc.
Cocaine-Induced 2-AG Signaling Is Independent of 5-HT and DA
Our study shows that AM251 and THL do not completely prevent inhibition of GABA release produced by cocaine (Figure 3), and another study shows that cocaine inhibits GABA release onto DA neurons in VTA by inhibiting serotonin (5-HT) reuptake and activation of 5-HT1B/D receptors (Cameron and Williams, 1994). Therefore, we determined whether the effects of cocaine on 2-AG involved 5-HT. The 5-HT1 receptor agonist sumatriptan (300 nM) decreased GABAB IPSCs (71.42% ± 4.50% of control, n = 8) and largely occluded the inhibition produced by cocaine (cocaine alone = 67.53% ± 3.01% of control, n = 19; sumatriptan + cocaine = 91.51% ± 3.52% of control, n = 8; p < 0.001, one-way ANOVA, Bonferroni post hoc analysis; Figures 4A, 4D, and 4G). The 5-HT1B/D antagonist, GR55562 (1 or 10 μM) had no effect alone on GABAB IPSCs but decreased the inhibition of this response by cocaine (cocaine alone = 67.53% ± 3.01% of control, n = 19; GR55562 + cocaine = 83.94% ± 3.91% of control, n = 10; p < 0.05, one-way ANOVA, Bonferroni post hoc analysis; Figures 4B and 4G). Additionally, the 5-HT uptake inhibitor citalopram (10 μM) and the 5-HT releaser fenfluramine (10 μM) both depressed GABAB IPSCs (citalopram: 77.77% ± 5.6% of control, n = 7; fenfluramine: 67.06% ± 3.42% of control, n = 11; Figure 4D).
Figure 4. Inhibition of GABAB IPSCs by Cocaine Is Independently Mediated by 5HT1B and CB1 Receptors.

(A) Inhibition of GABAB IPSC by cocaine was largely occluded by the 5HT1B receptor agonist sumatriptan (300 nM).
(B) The selective 5HT1B receptor antagonist GR55562 (1 or 10 μM) suppressed the inhibition by cocaine.
(C) The inhibition by cocaine was completely abolished when CB1 and 5HT1B receptors were blocked simultaneously.
(D) Serotonin-induced depression of GABAB IPSC is independent of CB1Rs. Application of the 5HT1B agonist sumatriptan, the selective serotonin reuptake inhibitor citalopram (10 μM), or the serotonin releaser fenfluramine (10 μM) all depressed GABAB IPSCs to a similar level. However, this is unaffected by the CB1R antagonist AM251 (4 μM; F(2,46) = 0.25, p = 0.77, two-way ANOVA). Scale bar, 100 ms, 10 pA.
(E) Pre-treatment with the selective DA transport inhibitor, GBR12935 (1 μM), does not occlude the inhibition of GABAB IPSCs by cocaine.
(F) The DA-D2 receptor antagonist sulpiride (5 μM) does not prevent the inhibition by cocaine.
(G) Summary the effect of cocaine on GABAB IPSCs with different drug treatments (F(5,51) = 12.83, p < 0.0001, one-way ANOVA; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 versus cocaine; #p < 0.05 versus GR55562; Bonferroni post hoc analysis). The complete elimination of the cocaine-induced inhibition of GABAB IPSCs by CB1R and 5-HT1B antagonism indicates that these effects are independent.
To determine whether the inhibition of GABAB IPSCs by the 5-HT system involves eCBs, we tested the effects of citalopram and fenfluramine during AM251 application (Figure 4D). We found that inhibition of GABAB IPSCs produced by these drugs was unaffected by CB1R antagonism (citalopram + AM251 = 75.03% ± 6.10% of control, n = 6; fenfluramine + AM251 = 70.26% ± 3.74% of control, n = 8; p > 0.05, unpaired t test; Figure 4D). Similarly, the effect of the 5-HT1 agonist sumatriptan was not altered by AM251 (sumatriptan + AM251 = 74.07% ± 3.41% of control, n = 12; Figure 4D). Therefore, we conclude that whereas cocaine enhances extracellular 5-HT levels to activate 5-HT1 receptors and inhibit GABA release onto DA neurons, this is independent of the inhibition produced by cocaine via 2-AG. Therefore, the inhibition of GABA release by cocaine in the VTA occurs via 2 independent mechanisms. Consistent with this, co-application of the 5-HT1B/D antagonist GR55562 and AM251 completely blocked the inhibition of GABAB IPSCs produced by cocaine (AM251 + GR55562 + cocaine = 101.09% ± 1.88% of control, n = 8; p < 0.0001 versus cocaine, one-way ANOVA; Figures 4C and 4G). Therefore, all subsequent experiments examining cocaine mobilization of 2-AG, were performed during blockade of 5-HT1B/1D receptors with GR55562 (1 μM).
Previous studies show that DA-D2 receptors stimulate 2-AG release (Melis et al., 2004; Pan et al., 2008). Therefore, we examined the involvement of D2 receptors and DA in 2-AG-mediated cocaine effects. In contrast to cocaine, the selective DAT inhibitor GBR12935 (1 μM) increased GABAB IPSCs and surprisingly did not occlude their inhibition by cocaine (GBR12935 + cocaine = 69.93% ± 4.67% of control, n = 7; Figures 4E and 4G). Similarly, a DA-D2 antagonist, sulpiride (5 μM), did not change the inhibition of GABAB IPSCs produced by cocaine (73.09% ± 5.45% of control, n = 5; Figures 4F and 4G). Therefore, the cocaine-induced inhibition of GABAB IPSCs did not result from increased extracellular DA levels or activation of DA-D2 receptors.
Gq/11-Coupled Receptor Involvement in the Cocaine-Induced Release of 2-AG
Several studies show that Gαq/11-coupled receptors promote the production of 2-AG in the brain (Kano et al., 2009; Kim et al., 2002; Kortleven et al., 2011; Maejima et al., 2001). Therefore, we examined their involvement in cocaine-induced 2-AG mobilization. Antagonists for Gq/11-coupled mGluR5, DA-D1, and neurotensin receptors did not alter the cocaine-induced inhibition of GABAB IPSCs in VTA DA neurons (Figure S2). However, antagonism of Gq/11-coupled α1-adrenergic receptors (α1R) with 2-[[β-(-4-hydroxyphenyl)ethyl]aminomethyl]-1-tetralone) (HEAT, 1 μM) or the selective, non-competitive, mGluR1 receptor antagonist, (3,4-dihydro-2H-pyrano[2,3-b]quinolin-7-yl)-(cis-4-methoxycy-clohexyl)-methanone (JNJ16259685 [JNJ]; 500 nM) completely blocked the 2-AG-dependent inhibition of GABAB IPSCs promoted by cocaine (cocaine = 83.94% ± 3.91% of control, n = 10; cocaine + HEAT = 99.68% ± 4.73% of control, n = 7; cocaine + JNJ = 99.39% ± 4.49% of control, n = 8; p < 0.01, one-way ANOVA, Bonferroni post hoc analysis; Figure 5A). This indicates that activation of both mGluR1 and α1Rs during cocaine application is required for the 2-AG-dependent inhibition of GABA release. We next examined the involvement of these receptors using selective agonists.
Figure 5. Mobilization of 2-AG by Cocaine Requires Synergistic Activation of mGluR1 and α1 Adrenergic Receptors and Activation of Phospholipase C.
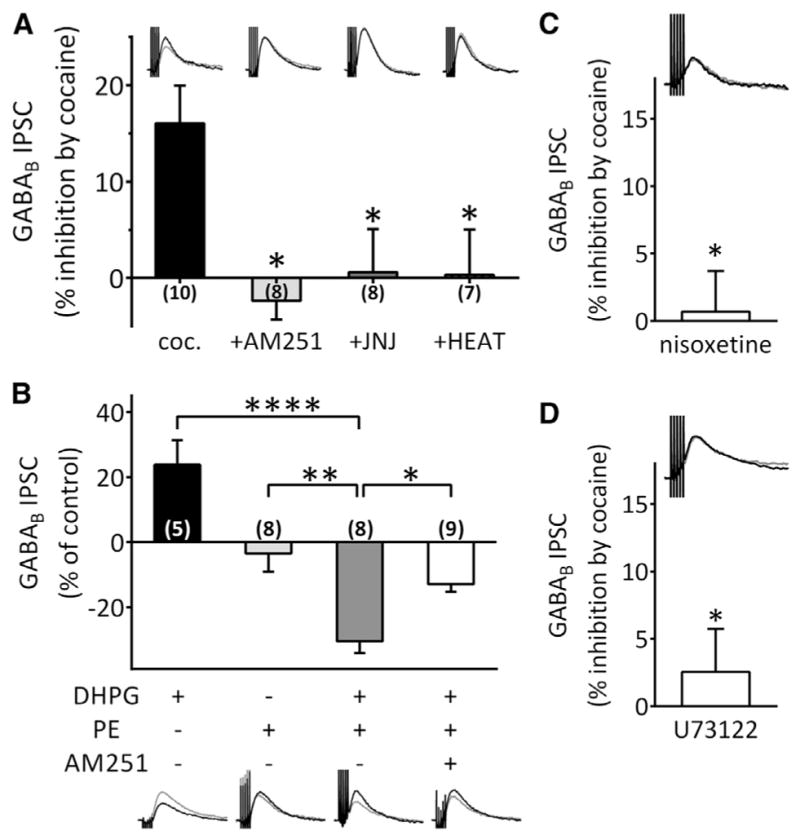
All experiments were performed in the presence of GR55562 to block 5-HT1B receptors.
(A) Inhibition of GABAB IPSCs by cocaine was blocked by AM251 (2 μM), the mGluR1 antagonist JNJ16259685 (JNJ, 500 nM) and the α1R antagonist HEAT (1 μM) (F(3,29) = 5.173, p = 0.0055, one-way ANOVA; *p < 0.05, Bonferroni post hoc analysis).
(B) Co-application of the mGluR1 agonist DHPG (1 μM) and the α1 agonist phenylephrine (PE, 100 μM) inhibited GABAB IPSCs, whereas alone these agonists did not exhibit such effects. Pre-incubation of slices with AM251 significantly blocked the synergistic effect of DHPG and PE (F(3,26) = 20.19, p < 0.0001, one-way ANOVA; *p < 0.05, **p < 0.01, ****p < 0.0001, Bonferroni post hoc analysis). Scale bar, 100 ms, 10 pA. Scale bar applies to (A).
(C) The NET inhibitor nisoxetine (10 μM) occluded the inhibition of GABAB IPSCs by cocaine (*p < 0.05, unpaired t test). Scale bar, 100 ms, 10 pA.
(D) An inhibitor of PLC, U73122 (5 μM), significantly blocked the effect of cocaine (*p < 0.05 versus cocaine, unpaired t test). Scale bar, 100 ms, 10 pA. See also Figures S2 and S3.
The mGluR1 agonist, DHPG (1 μM), alone, significantly increased the amplitude of GABAB IPSCs (123.80% ± 7.58% of control, n = 5; p < 0.05, paired t test; Figure 5B), whereas the α1R agonist, phenylephrine (PE, 100 μM) did not affect these currents (96.52% ± 5.60% of control, n = 8; p = 0.52, paired t test; Figure 5B). In contrast, co-application of DHPG and PE strongly inhibited GABAB IPSCs (69.53% ± 3.66% of control, n = 8; p < 0.001, paired t test; Figure 6B) and this was significantly reduced by AM251 pretreatment (87.09% ± 2.36% of control, n = 9; p < 0.05 versus HEAT + PE, unpaired t test; Figure 5B). This suggests that co-activation of Gq/11-coupled mGluR1 and α1Rs is necessary for cocaine to promote 2-AG mobilization and inhibit GABA release onto VTA DA neurons.
Figure 6. Cocaine-Induced Calcium Changes in VTA DA Neurons.

(A) Superimposed fluorescent and transmitted light images showing the expression of GCaMP6f in VTA DA neurons 2–3 weeks after injection of the AAV-GCaMP6f construct. Scale bar, 500 μm.
(B) Spontaneous slow calcium oscillations in VTA DA neurons. Left: confocal image of the GCaMP6f signal (200×). Scale bar, 50 μm. Right: example calcium oscillations (ΔF/F0) in the same cell indicated in the dashed square at left. Scale bar, 30 s, 20%.
(C) Sample traces of calcium signals (ΔF/F0) in response to cocaine application (10 μM, indicated in gray). Three types of changes were observed: no change, a decrease in spontaneous oscillations, and the initiation of slow oscillations (SO).
(D) Raster plot of oscillation events in 19 neurons imaged as in (B). Each vertical bar denotes the peak of an oscillation event. Each row exhibits the time course of oscillation change in single neurons.
(E) Cumulative frequencies of spontaneous, cocaine-decreased, and cocaine-initiated slow oscillations (cocaine SO) in calcium signal. The curve shifts left to slower frequencies in neurons whose calcium oscillations were decreased by cocaine (osc decrease versus spontaneous, p = 0.17, K-S test). The frequency of cocaine-initiated SO was significantly lower than that of spontaneous oscillations (cocaine-initiated versus spontaneous, p < 0.01, K-S test). Spontaneous oscillation, n = 43 neurons; oscillation decrease, n = 19 neurons; cocaine SO, n = 29 neurons recorded in eight brain slices from six rats.
(F) Cumulative distribution of half-widths of spontaneous, cocaine-decreased, and cocaine-initiated SO. Cocaine-initiated SO exhibit longer half-widths than spontaneous oscillations. (cocaine SO versus spontaneous, p < 0.0001, K-S test). Spontaneous oscillation, n = 43 neurons; oscillation decrease, n = 19 neurons; cocaine SO, n = 29 neurons recorded in eight brain slices from six rats.
(G) Increased correlation of calcium responses among neurons (same 19 neurons as D) during cocaine perfusion. Cross correlations among all neuron pairs were calculated, and the peak correlation coefficient (r = 0 to 1) is color-coded to form the heat map. The data indicate that VTA DA neuron calcium oscillations are more highly synchronized by cocaine.
(H) Cocaine-induced calcium oscillations depend on mGluR1, α1-adrenergic receptors, and internal calcium stores. The frequency of spontaneous calcium oscillations is reduced by JNJ16259685 (500 nM), but is not altered by HEAT (1 μM), U73122, or thapsigargin (thaps, 2 μM), respectively (F(4,265) = 3.806, p = 0.005, one-way ANOVA; control versus JNJ: p < 0.05, Bonferroni post hoc analysis). Each point represents the frequency of spontaneous Ca2+ oscillation from one neuron; the box and vertical lines indicate quartiles, minimal, and maximal frequencies. The percentage of neurons exhibiting spontaneous oscillations is shown for each treatment group.
(I) Cumulative distributions of the frequencies of spontaneous oscillations are only affected by JNJ (p = 0.0037, Kolmogorov–Smirnov test).
(J) Summary of the percentage of DA neurons responding to cocaine under control, JNJ16259685, HEAT, U73122, and thapsigargin treatments. Numbers denote the percentage of neurons demonstrating indicated changes upon cocaine treatment. All treatments decreased the cocaine-induced Ca2+ oscillation.
As cocaine is a potent inhibitor of the norepinephrine transporter (NET), we next examined whether the α1R-dependent effect of cocaine on GABAB IPSCs occurred via this mechanism. The selective NET inhibitor, nisoxetine (10 μM), completely blocked the inhibition of IPSCs produced by cocaine (99.31% ± 3.00% of control, n = 7; p < 0.05 versus cocaine, unpaired t test, Figure 5C), suggesting that cocaine promotes activation of α1Rs and the mobilization of 2-AG through the inhibition of NET.
Contribution of PLC-β and Intracellular Ca2+ to the Cocaine Mobilization of 2-AG
The lipolytic enzyme, PLC-β, promotes conversion of phosphoinositides to DAG, which can then be converted to 2-AG via DGLα (Alger and Kim, 2011; Hashimotodani et al., 2005; Kano et al., 2009). As PLCβ is activated by a rise in internal Ca2+, or activation of Gq/11-coupled receptors, we determined whether PLC-β inhibition alters the 2-AG inhibition of GABA release by cocaine. The cocaine inhibition of GABAB IPSCs was absent in DA neurons incubated in the PLC inhibitor, U73122 (5 μM; cocaine + U73122 = 97.46% ± 3.20% of control, n = 7; p < 0.05, unpaired t test, Figure 5D), suggesting that cocaine-induced 2-AG mobilization requires PLC.
To assess the role of Ca2+ in the release of 2-AG by cocaine, in vitro calcium imaging experiments were performed in VTA DA neurons expressing the genetically-encoded calcium indicator, GCaMP6f, in TH-Cre rats. Robust expression of GCaMP6f was confirmed (Figure 6A), and DA neurons exhibiting stable Ca2+-associated fluorescence and spontaneous Ca2+ oscillations were observed (Figures 6A and 6B; Movie S1). The mean frequency of spontaneous calcium oscillations in a sub-group of neurons (29.1%, 168 of 578 neurons) (Figure 6B; Movie S1) under resting conditions was 0.042 ± 0.002 Hz (range 0.0026–0.167 Hz, Figures 6H and 6I). With bath application of cocaine (10 μM), the Ca2+ signal in 21.2% (32 of 151) of the DA neurons was unchanged, whereas it was reduced by cocaine in 59.6% (90 of 151) of DA neurons (Figures 6C, 6D, and 6J; Movie S1). Among this latter population, 47.8% (43 of 90) exhibited slow calcium oscillations during baseline whose frequency was reduced by cocaine (control = 0.041 ± 0.004 Hz; cocaine = 0.012 ± 0.003 Hz; p < 0.0001, paired t test; Figures 6C–6E). The remaining neurons (19.2%, 29 of 151) exhibited no Ca2+ oscillations during baseline but demonstrated oscillations during cocaine application (0.023 ± 0.004 Hz; p < 0.01 versus spontaneous oscillation, Kolmogorov-Smirnov [K-S] test; Figures 6C–6E and 6J). These cocaine-induced slow Ca2+ oscillations (SO) exhibited lower frequencies and broader half-widths than oscillations observed during baseline (spontaneous baseline oscillation half-width = 11.21 ± 2.48 s; cocaine-induced oscillation half-width = 24.17 ± 3.19 s; p < 0.0001, K-S test; Figure 6F), suggesting that cocaine SOs reflect distinct Ca2+ sources and/or release mechanisms. The cocaine SOs were also more highly synchronized among VTA DA neurons than the spontaneous Ca2+ oscillations under baseline conditions, as indicated by cross correlation analysis (Figure 6G).
In electrophysiological experiments, the cocaine-induced depression of GABA release was mediated by 2-AG and abolished by antagonism of mGluR1 or α1Rs (Figure 5A). Therefore, we examined effects of these antagonists on cocaine-induced changes in intracellular Ca2+. The mGluR1 antagonist, JNJ16259685 (500 nM), prevented the cocaine-induced increase in the Ca2+ SO, as only 4.1% (4 of 97 cells) of DA neurons showed this response (Figure 6J, **p < 0.0001, chi-square test). Moreover, the proportion of DA neurons showing no change in intracellular Ca2+ with cocaine increased to 32.0% (31 of 97 neurons, Figure 6J), and the proportion of VTA DA neurons exhibiting a decrease in Ca2+ signal upon cocaine application was not altered by JNJ162959685 (63.9%, 62 of 97 neurons, Figure 6J). The α1R antagonist, HEAT (1 μM), also prevented the increased the cocaine-induced Ca2+ SO, as only 5.8% (5 of 86 neurons) showed this response (Figure 6J, p < 0.0001, chi-square test). However, unlike the mGluR1 antagonist, HEAT also reduced the proportion of neurons exhibiting a decrease in Ca2+ oscillations (3.5%, 3 of 86, Figure 6J). The PLCβ inhibitor, U73122, also prevented the cocaine-induced SO, as only 5.0% (4 of 80 cells, Figure 6J, p < 0.0001, chi-square test) of the DA neurons showed this effect.
The number of neurons showing an increase in Ca2+ SO with cocaine was also reduced when intracellular stores were depleted with the sarco/endoplasmic reticulum Ca2+ ATPase inhibitor, thapsigargin (2 μM; oscillation increase in 2.6%, 2 of 77 neurons, Figure 6J, p < 0.0001, chi-square test). Additionally, the proportion of neurons showing inhibition of the Ca2+ signal by cocaine was increased following thapsigargin (81.8%, 63 of 77 neurons; Figure 6J). The frequency of baseline Ca2+ oscillations was significantly reduced by JNJ162959685 (p = 0.0037 versus cocaine, K-S Test, Figures 6H and 6I), but unchanged by HEAT, U73122, or thapsigargin (Figures 6H and 6I). However, the percentage of neurons showing these oscillations in the absence of cocaine was lower in U73122-treated slices and higher in thapsigargin-incubated slices (control: 29.1%; HEAT: 22.0%; JNJ: 20.8%; U73122: 11.3%; thapsigargin: 43.6%; Figure 6H).
Depletion of Internal Ca2+ Stores Blocks the 2-AG-Dependent Effects of Cocaine
To determine whether elimination of the cocaine-induced Ca2+ SOs also blocks the cocaine-mediated release of 2-AG, we measured GABAB IPSCs after treatment with thapsigargin (2 μM; Figure 6K). We found that thapsigargin prevented the cocaine inhibition of GABAB IPSCs in DA neurons (95.63% ± 1.67% of control, n = 7; p < 0.05 versus cocaine alone, unpaired t test; Figure 6K), confirming that internal Ca2+ is necessary for cocaine-induced 2-AG mobilization.
DISCUSSION
Here, we find that cocaine mobilizes 2-AG in the VTA to inhibit GABAergic inputs to GABAB receptors on DA neurons via pre-synaptic CB1R activation. We also show that cocaine’s ability to increase the frequency of DA transients in the NAc is dependent on 2-AG synthesis and CB1R signaling in the VTA, collectively implicating an eCB-dependent disinhibition of VTA DA neurons. Our results thereby provide a mechanism to explain the disruption of cocaine-motivated behavior that is observed during antagonism of the eCB system in animal models of addiction and relapse to drug seeking (De Vries et al., 2001; Li et al., 2009; Soria et al., 2005; Xi et al., 2006, 2008). Based on these findings, we hypothesize that cocaine-induced 2-AG mobilization in the VTA facilitates DA release in the NAc to support cocaine reinforcement.
The cocaine-induced inhibition of GABA release is disrupted by several manipulations that target the eCB system in the VTA, including (1) blockade of CB1Rs, (2) inhibition of PLCβ, and (3) inhibition of DGLα. We also find that 2-AG mobilization by cocaine requires activation of both mGluR1 and α1Rs, since antagonism of either receptor prevents inhibition of GABAB IPSCs in DA neurons. As the inhibition of GABA release by cocaine was blocked by a NET inhibitor, our data further suggest that cocaine increases extracellular NE and activates α1Rs through this mechanism. Prior studies show that cocaine increases NE via inhibition of NET (Reith et al., 1997), and NE neurons project to the VTA to form connections with DA neurons (Jones et al., 1977; Mejías-Aponte et al., 2009). Moreover, genetic elimination of α1Rs is associated with lower baseline levels of DA and prevents psychostimulant-mediated increases in NAc DA (Auclair et al., 2002). A more recent study shows that cocaine increases extracellular NE in VTA to activate α1Rs, contributing to excitation of DA neurons and DA release in the NAc (Goertz et al., 2014). This, together with our data, suggests that modulation of NE function in the VTA likely plays a critical role in the behavioral effects of cocaine.
We also find that mGluR1s are critical for the 2-AG-dependent effects of cocaine in the VTA. However, electrically-evoked EPSCs in VTA DA neurons are unaffected by cocaine under our recording conditions (Figure S3), suggesting that synaptic glutamate release is not involved. Despite this, prior work shows that cocaine increases glutamate concentrations measured via microdialysis (Kalivas and Duffy, 1995), and mGluR1s are activated by glutamate in DA neurons using electrical stimulation similar to ours (Fiorillo and Williams, 1998). Therefore, we suggest that sufficient extracellular glutamate is present under baseline conditions to activate mGluR1s and this cooperatively interacts with α1Rs to stimulate 2-AG production.
Imaging of Ca2+ dynamics in TH-Cre rats with GCaMP6f reveals that cocaine initiates Ca2+ SOs in ~20% of VTA DA neurons, whereas baseline spontaneous Ca2+ oscillations are suppressed in most cells. Antagonism of either mGluR1s or α1Rs with JNJ16259685 or HEAT, respectively, largely eliminates the cocaine SOs; an effect also seen with blockade of PLCβ by U73122 or depletion of intracellular Ca2+ by thapsigargin. Corresponding blockade of both cocaine-induced SOs and inhibition of GABAB IPSCs by each of these manipulations supports the idea that the 2-AG-dependent actions of cocaine are related to an increase in Ca2+ activity and dependent on cooperative mGluR1 and α1AR signaling.
The activity of PLCβ is enhanced in neurons by intracellular Ca2+ and this is synergistically increased by co-activation of Gq/11-coupled receptors to greatly augment 2-AG mobilization (Alger and Kim, 2011; Kano et al., 2009). Here, we show that cocaine causes activation of Gq/11-coupled α1Rs in DA neurons through an increase in extracellular NE via inhibition of NET. The requirement for co-activation of mGluR1 and α1Rs suggests that there is an interaction at the Gq/11-coupled receptor level that increases the activity of PLCβ to synergistically augment 2-AG release. This cooperativity between mGluR1 and α1Rs in the inhibition of GABA release onto VTA DA neurons may serve as a coincidence detector, such that when sufficient NE is present, together with glutamate activation of mGluR1s, 2-AG is mobilized to reduce GABAergic input, thereby enhancing DA neuron activity via disinhibition. Consequently, this will amplify forebrain DA release, as shown here, and potentially enhance the reinforcing properties of cocaine.
Our data also suggest that the cocaine-induced 2-AG mobilization occurs in the ~20% of VTA DA neurons demonstrating cocaine-induced Ca2+ oscillations. This, together with our observation that extracellular application of THL is more effective at blocking the cocaine-induced inhibition of GABA release than intracellular application, supports the idea that 2-AG is released from this subset of neurons to more widely affect synaptic inputs. Moreover, since GCaMP6f was expressed only in DA neurons in our study, the possibility that cocaine releases 2-AG from non-DA neurons was not evaluated and therefore remains a possibility.
The rewarding and abuse-related properties of cocaine are thought to primarily result from inhibition of DA uptake in fore-brain areas such as the NAc (Phillips et al., 2003; Ritz et al., 1987; Sulzer, 2011; Thomsen et al., 2009). However, cocaine also acts in the VTA to increase DA neuron firing rates and DA release events in the NAc (Sombers et al., 2009), and these events are decreased by systemic CB1R antagonists (Cheer et al., 2007). DA transients result from burst firing that occurs upon presentation of natural reward, cues that predict their availability or during administration of abused drugs (Aragona et al., 2008; Cheer et al., 2007; Oleson et al., 2012; Phillips et al., 2003; Roitman et al., 2004; Sombers et al., 2009) and function to reinforce behavior (Steinberg et al., 2014). Here, we demonstrate that the eCB influence on DA transients is likely mediated by 2-AG acting at CB1Rs in the VTA, since intra-VTA administration of a CB1R antagonist, or an inhibitor of DGLα blocked this effect of cocaine. Although inhibition of VTA DA neurons is the most frequent response to cocaine in anesthetized rodents, an increase in the bursting activity of a proportion of these cells is consistently reported (Einhorn et al., 1988; Mejias-Aponte et al., 2015). Furthermore, a much larger proportion of VTA DA neurons is excited by cocaine in awake-freely moving rats, compared to anaesthetized animals (Koulchitsky et al., 2012). Our lab has shown that that conditions promoting burst firing of DA neurons causes activity- and extracellular-Ca2+-dependent eCB release that presynaptically inhibits GABA release onto these cells (Riegel and Lupica, 2004). In accord with the present observations that cocaine mobilizes 2-AG via Gq11-dependent mechanisms to enhance the inhibition of GABA inputs to these cells, we suggest that the eCB system in the VTA is an important component of a positive feedback mechanism that is engaged during exposure to cocaine and contributes to its rewarding properties. In this scheme (Figure 7), we propose that blockade of NET by cocaine promotes activation of Gq/11-coupled α1Rs and this, together with mGluR1 activation, initiates 2-AG release, which then suppresses GABA release to disinhibit (K) Pre-treatment of slices with thapsigargin (2 μM) significantly attenuates the inhibition of GABAB IPSCs by cocaine (n = 7; *p < 0.05, unpaired t test). Inset at right shows averaged electrically evoked GABAB IPSC traces (n = 5) under control conditions (top) and after treatment with thapsigargin. Control sweeps are shown in black; those obtained during cocaine application are shown in gray. Scale bar, 100 ms, 10 pA. See also Movie S1. VTA DA neurons. This disinhibition then promotes burst firing of DA neurons, mobilizing additional 2-AG through an activity-dependent mechanism. This then supports further activation of these cells, via disinhibition, to augment DA neuron activity and DA release in downstream projections. In this way, the eCB system promotes strong activation of DA neurons to contribute to the rewarding actions of cocaine. Therefore, the mechanisms that we have identified in the present study may be critical in the development of pharmacotherapies for psychostimulant addiction that target the eCB system.
Figure 7. Putative Mechanism of Cocaine Mobilization of 2-AG and Inhibition of GABA Neurotransmission in the VTA.
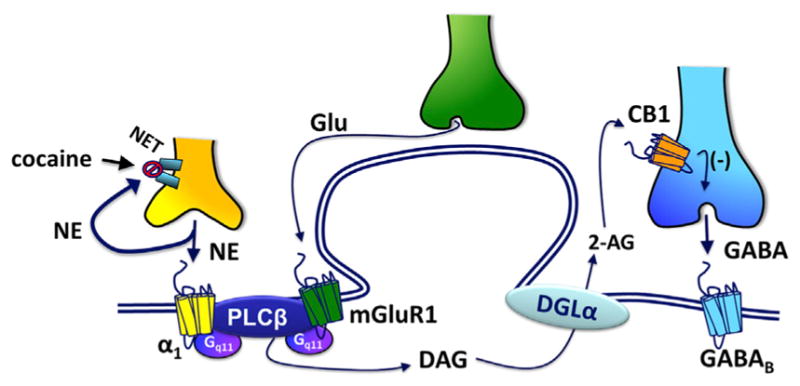
Glutamate spillover under baseline conditions activates mGluR1s, and α1-adrenergic receptors located on VTA DA neurons are activated by NE whose clearance is prevented by cocaine blockade of the NET. This co-activation of Gq11-coupled mGluR1 and α1Rs cooperatively stimulates PLC-β leading to DAG formation. DAG is then converted to 2-AG, via DGL-α, then crosses the extracellular space through an unknown mechanism to activate CB1Rs located on GABA axon terminals. Activation of these presynaptic CB1Rs inhibits GABA release onto postsynaptic GABAB receptors, leading to 2-AG-mediated disinhibition of DA neurons. This increases DA transients in the NAc to enhance the rewarding properties of cocaine.
EXPERIMENTAL PROCEDURES
Animal protocols were conducted under NIH guidelines and were approved by the NIDA Intramural Research Program Animal Care and Use Committee.
Slice Preparation
Male Sprague-Dawley rats, 15 to 21 days old (Charles Rivers) were decapitated, their brains rapidly removed and transferred to an aerated (95% O2/5% CO2), ice-cold solution (in mM: sucrose 220, KCl 2.5, MgCl2 7, CaCl2 0.5, NaH2PO4 1.25, NaHCO3 26, glucose 20). Horizontal slices containing VTA (250 μM) were cut using a tissue slicer (VT1000, Leica), then transferred to a holding chamber filled with oxygenated artificial CSF (aCSF; in mM: NaCl 126, KCl 3, MgCl2 1.5, CaCl2 2.4, NaH2PO4 1.2, NaHCO3 26, glucose 11) at room temperature.
Confocal experiments used 2- to-3-month-old female TH-Cre rats. Two to three weeks after bilateral injection of a GCaMP6f construct into the VTA, rats were anesthetized by isoflurane and decapitated. Horizontal VTA slices (230 μM) were cut in a solution containing (in mM) NMDG 93, KCl 2.5, MgSO4 10, CaCl2 0.5, NaH2PO4 1.2, NaHCO3 30, glucose 25, HEPES 20, sodium ascorbate 5, sodium pyruvate 3, thiourea 2; PH 7.2–7.4. Slices were first incubated in this solution at 34°C for 10–15 min and then transferred to room temperature aCSF until imaging.
Electrophysiology
A slice was transferred to a recording chamber and immersed in heated (32°C), flowing (2 ml/min), oxygenated aCSF. Slices were visualized with an upright microscope equipped with differential interference contrast (DIC) optics (BX51WI, Olympus). Recorded neurons were in lateral VTA, medial to the terminal nucleus of the accessory optic tract and anterior to the third cranial nerve. Whole-cell voltage-clamp recordings were made with an Axopatch 200B (Molecular Devices) amplifier. Pipettes (3–5 MΩ) were filled with (in mM): K-gluconate 140, NaCl 2, MgCl2 1.5, HEPES 10, EGTA 0.1, Mg-ATP 4, Na2-GTP 0.3, Tris-phosphocreatine 10 (pH 7.2), 290 mOSM. Cell-attached recordings were performed with the same electrodes immediately after patch formation to identify DA neurons by electrophysiological properties; spontaneous, pacemaker firing (1–4 Hz), and action potential widths >1.5 ms (Chieng et al., 2011). DNQX (10 μM), D-AP5 (25 μM), picrotoxin (100 μM), and strychnine (1 μM) were present to block AMPA, NMDA, GABAA, and glycine receptors, respectively. After rupture of the membrane patch, 5 stimuli at 40 Hz (200–600 μA, 200 μs) were applied every 60 s, via a bipolar electrode placed in anterior VTA, to evoke GABAB receptor currents. Stimulation protocols were generated and signals acquired using the WinLTP program (WinLTP, The University of Bristol, Bristol, UK) on a personal computer.
Expression of Genetically Encoded GCaMP6f and Imaging
TH-Cre rats were anesthetized with isoflurane, and AAV1.Syn.Flex.GCaMP6f. WPRE.SV40 virus (titer: ~1012 genomes/ml, 500 nl each site, UPENN Vector Core) was injected bilaterally into VTA (from bregma, A/P: −5.4 mm; M/L: ± 0.7 mm; D/V: −8.0 mm), using a micro pump (UltraMicroPump, WPI, 100 nl/min) and Hamilton syringe (1701RN) with hypodermic tubing (30 gauge). The tubing was left in place for 5 min after completion of the injection.
Two to three weeks after surgery, GCaMP6f fluorescence was observed using a confocal microscope (LSM710, Zeiss, W Plan-Apochromat 20× objective) and 488 nm laser in slices in aCSF. Images were collected at 0.5 Hz. After 5–10 min of baseline, slices were exposed to drug for 20 min. Image stacks were processed and analyzed offline with ImageJ (NIH). When observed, a StackReg plug-in was used to correct x-y drift. Regions of interests were defined over cell bodies. The time-lapse signal was expressed as ΔF/F0, where F0 is the baseline fluorescence and ΔF = F − F0. The frequency of calcium oscillations was determined by fast Fourier transformation (FFT) of the signal. Half-width was measured using Event Detection in Clampfit 9 (Molecular Devices). Cross correlation of the calcium signals was performed in Clampfit 9, and the peak correlation coefficient was chosen within a ±10-s window.
Measurement of DA Transients In Vivo Using FSCV
Male Sprague-Dawley rats purchased with implanted jugular vein catheters (n = 9, 300–350 g; Charles Rivers) were anesthetized with isoflurane. A guide cannula (Bioanalytical Systems) was positioned above the NAc shell (from bregma, A/P: +1.7 mm, M/L: +0.8 mm, D/V: −2.5 mm), and a combination bipolar stimulating electrode/steel guide cannula (26 gauge; Plastics One) was implanted ipsilaterally targeting the VTA (from bregma, A/P: 5.4 mm, M/L: 1.2 mm, D/V: 8.2 mm). An Ag/AgCl reference electrode was placed in contralateral cortex. All components were permanently affixed with dental cement. Animals were allowed 3–5 days for recovery.
FSCV was used to monitor DA release using a triangular input waveform (0.4 V to 1.3 V, 400 V/s) at 10 Hz, applied to a glass-encased carbon fiber electrode lowered into the NAc. Endogenous and electrically evoked (biphasic; 2 ms/phase, 24 pulse, 60 Hz, 125 μA) DA release was recorded prior to the experimental session to verify electrode performance and create a training set for principal component regression (PCR) to extract the DA component from the raw voltammetric data. Rapid DA concentration changes (i.e., DA “transients”) were analyzed using Mini Analysis (Synaptosoft) and custom written MATLAB (The MathWorks) routines to determine drug effects on transient frequency and amplitude (Cheer et al., 2007).
Intra-VTA infusions of vehicle, rimonabant, or tetrahydrolipstatin (THL) occurred in the experimental chamber using a microprocessor-controlled infusion pump (Harvard Apparatus) at a flow rate of 0.5 μl/min. An infusion needle was inserted through a guide cannula ending 1 mm above the tip of the bipolar stimulating electrode (Plastics One); the needle was cut to extend 1 mm beyond the cannula tip. A within-subjects design was used where animals first received microinfusions of vehicle, followed by 3 mg/kg intravenous cocaine. A second infusion of rimonabant or THL occurred after the baseline frequency of DA transients was reestablished (~45–60 min) and preceded a second cocaine injection (3 mg/kg).
Drugs
For in vitro experiments, drugs were first dissolved in vehicle to make 1,000- or 10,000-fold concentrated stock solutions. The drugs were then applied to the slices by diluting the stock solution in the perfusion reservoir. DNQX, D-(−)-2-amino-5-phosphonopentanoic acid (D-AP5), GR55562, HEAT, JNJ16259685, MTEP, AM 251, THL, phenylephrine, sumatriptan, and U73122 were purchased from Tocris. Picrotoxin was purchased from Sigma. AAV1.Syn.Flex.GCaMP6f was purchased from the University of Pennsylvania Vector Core. 5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-[(E)-piperi-dinoiminomethyl]-1H-pyrazole (PIMSR1) was provided by Dr. Herbert H. Seltz-man at Research Triangle Institute (NIDA, Raleigh, NC; RTI International). For FSCV experiments, cocaine (Sigma-Aldrich) was dissolved in saline, rimonabant (SR141716A; RTI), and THL (Sigma-Aldrich) were freshly suspended in a 1:1:18 ratio of ethanol, emulphor (Alkamuls EL-620; Rhodia), and saline (0.9%).
Data Analysis
Group data are presented as the mean ± SEM in all cases. All statistical tests, including t tests, one-way ANOVAs, and K-S tests were performed using GraphPad Prism 5 (GraphPad Software). Post hoc analysis (Bonferroni test) was performed when an ANOVA yielded a significant (p < 0.05) main effect.
Supplementary Material
Highlights.
Endogenous cannabinoids modulate the behavioral effects of cocaine
Cocaine mobilizes 2-arachidonoylglycerol (2-AG) in the midbrain
Cocaine-mobilized 2-AG disinhibits dopamine (DA) neurons and increases DA release
Cocaine mobilizes 2-AG via Gq/11-coupled receptor coupling to internal calcium
Acknowledgments
The AAV1.Syn.Flex.GCaMP6f.WPRE.SV40 virus was generously made available by Vivek Jayaraman, PhD, Rex A. Kerr, PhD, Douglas S. Kim, PhD, Loren L. Looger, PhD, Karel Svoboda, PhD, and the GENIE Project, Janelia Farms, Howard Hughes Medical Institute. The TH-cre rat breeders were donated by Dr. Karl Deisseroth to the NIDA-IRP Optogenetic and Transgenic Core. We thank Drs. Antonello Bonci and Alexander Hoffman for critically reading the manuscript. The work was funded by the NIH/NIDA-Intramural Research Program (C.R.L., H.W., and T.T.), and NIDA extramural grants DA022340, DA033926 (J.F.C.).
Footnotes
Supplemental Information includes three figures and one movie, and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.08.041.
AUTHOR CONTRIBUTIONS
H.W., C.R.L., and J.F.C. conceived the study. H.W. collected and analyzed most of the electrophysiological and calcium imaging data. T.T. collected some of the electrophysiological data. D.P.C. collected and analyzed all of the in vivo data. All authors contributed to writing the manuscript.
References
- Alger BE, Kim J. Supply and demand for endocannabinoids. Trends Neurosci. 2011;34:304–315. doi: 10.1016/j.tins.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragona BJ, Cleaveland NA, Stuber GD, Day JJ, Carelli RM, Wightman RM. Preferential enhancement of dopamine transmission within the nucleus accumbens shell by cocaine is attributable to a direct increase in phasic dopamine release events. J Neurosci. 2008;28:8821–8831. doi: 10.1523/JNEUROSCI.2225-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auclair A, Cotecchia S, Glowinski J, Tassin JP. D-amphetamine fails to increase extracellular dopamine levels in mice lacking alpha 1b-adrenergic receptors: relationship between functional and nonfunctional dopamine release. J Neurosci. 2002;22:9150–9154. doi: 10.1523/JNEUROSCI.22-21-09150.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron DL, Williams JT. Cocaine inhibits GABA release in the VTA through endogenous 5-HT. J Neurosci. 1994;14:6763–6767. doi: 10.1523/JNEUROSCI.14-11-06763.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheer JF, Wassum KM, Sombers LA, Heien MLAV, Ariansen JL, Aragona BJ, Phillips PEM, Wightman RM. Phasic dopamine release evoked by abused substances requires cannabinoid receptor activation. J Neurosci. 2007;27:791–795. doi: 10.1523/JNEUROSCI.4152-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chieng B, Azriel Y, Mohammadi S, Christie MJ. Distinct cellular properties of identified dopaminergic and GABAergic neurons in the mouse ventral tegmental area. J Physiol. 2011;589:3775–3787. doi: 10.1113/jphysiol.2011.210807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DC. The significance of action potential bursting in the brain reward circuit. Neurochem Int. 2002;41:333–340. doi: 10.1016/s0197-0186(02)00068-2. [DOI] [PubMed] [Google Scholar]
- Covey DP, Roitman MF, Garris PA. Illicit dopamine transients: reconciling actions of abused drugs. Trends Neurosci. 2014;37:200–210. doi: 10.1016/j.tins.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vries TJ, Shaham Y, Homberg JR, Crombag H, Schuurman K, Dieben J, Vanderschuren LJ, Schoffelmeer AN. A cannabinoid mechanism in relapse to cocaine seeking. Nat Med. 2001;7:1151–1154. doi: 10.1038/nm1001-1151. [DOI] [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Bassareo V. Reward system and addiction: what dopamine does and doesn’t do. Curr Opin Pharmacol. 2007;7:69–76. doi: 10.1016/j.coph.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einhorn LC, Johansen PA, White FJ. Electrophysiological effects of cocaine in the mesoaccumbens dopamine system: studies in the ventral tegmental area. J Neurosci. 1988;8:100–112. doi: 10.1523/JNEUROSCI.08-01-00100.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorillo CD, Williams JT. Glutamate mediates an inhibitory post-synaptic potential in dopamine neurons. Nature. 1998;394:78–82. doi: 10.1038/27919. [DOI] [PubMed] [Google Scholar]
- French ED, Dillon K, Wu X. Cannabinoids excite dopamine neurons in the ventral tegmentum and substantia nigra. Neuroreport. 1997;8:649–652. doi: 10.1097/00001756-199702100-00014. [DOI] [PubMed] [Google Scholar]
- Gessa GL, Melis M, Muntoni AL, Diana M. Cannabinoids activate mesolimbic dopamine neurons by an action on cannabinoid CB1 receptors. Eur J Pharmacol. 1998;341:39–44. doi: 10.1016/s0014-2999(97)01442-8. [DOI] [PubMed] [Google Scholar]
- Goertz RB, Wanat MJ, Gomez JA, Brown ZJ, Phillips PE, Paladini CA. Cocaine increases dopaminergic neuron and motor activity via midbrain α1 adrenergic signaling. Neuropsychopharmacology. 2014 doi: 10.1038/npp.2014.296. Published online November 6, 2014 http://dx.doi.org/10.1038/npp.2014.296. [DOI] [PMC free article] [PubMed]
- Hashimotodani Y, Ohno-Shosaku T, Tsubokawa H, Ogata H, Emoto K, Maejima T, Araishi K, Shin HS, Kano M. Phospholipase Cbeta serves as a coincidence detector through its Ca2+ dependency for triggering retrograde endocannabinoid signal. Neuron. 2005;45:257–268. doi: 10.1016/j.neuron.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Kano M. Ca(2+)-assisted receptor-driven endocannabinoid release: mechanisms that associate pre-synaptic and postsynaptic activities. Curr Opin Neurobiol. 2007;17:360–365. doi: 10.1016/j.conb.2007.03.012. [DOI] [PubMed] [Google Scholar]
- Hoffman AF, Lupica CR. Synaptic targets of Δ9-tetrahydrocan-nabinol in the central nervous system. Cold Spring Harb Perspect Med. 2013;3:a012203. doi: 10.1101/cshperspect.a012237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst D, Umejiego U, Lynch D, Seltzman H, Hyatt S, Roche M, McAllister S, Fleischer D, Kapur A, Abood M, et al. Biarylpyrazole inverse agonists at the cannabinoid CB1 receptor: importance of the C-3 carboxamide oxygen/lysine3.28(192) interaction. J Med Chem. 2006;49:5969–5987. doi: 10.1021/jm060446b. [DOI] [PubMed] [Google Scholar]
- Jones BE, Halaris AE, McIlhany M, Moore RY. Ascending projections of the locus coeruleus in the rat. I. Axonal transport in central noradrenaline neurons. Brain Res. 1977;127:1–21. doi: 10.1016/0006-8993(77)90377-8. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Duffy P. D1 receptors modulate glutamate transmission in the ventral tegmental area. J Neurosci. 1995;15:5379–5388. doi: 10.1523/JNEUROSCI.15-07-05379.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Kim J, Isokawa M, Ledent C, Alger BE. Activation of muscarinic acetylcholine receptors enhances the release of endogenous cannabinoids in the hippocampus. J Neurosci. 2002;22:10182–10191. doi: 10.1523/JNEUROSCI.22-23-10182.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortleven C, Fasano C, Thibault D, Lacaille JC, Trudeau LE. The endocannabinoid 2-arachidonoylglycerol inhibits long-term potentiation of glutamatergic synapses onto ventral tegmental area dopamine neurons in mice. Eur J Neurosci. 2011;33:1751–1760. doi: 10.1111/j.1460-9568.2011.07648.x. [DOI] [PubMed] [Google Scholar]
- Koulchitsky S, De Backer B, Quertemont E, Charlier C, Seutin V. Differential effects of cocaine on dopamine neuron firing in awake and anesthetized rats. Neuropsychopharmacology. 2012;37:1559–1571. doi: 10.1038/npp.2011.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001;29:717–727. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- Landsman RS, Burkey TH, Consroe P, Roeske WR, Yamamura HI. SR141716A is an inverse agonist at the human cannabinoid CB1 receptor. Eur J Pharmacol. 1997;334:R1–R2. doi: 10.1016/s0014-2999(97)01160-6. [DOI] [PubMed] [Google Scholar]
- Li X, Hoffman AF, Peng XQ, Lupica CR, Gardner EL, Xi ZX. Attenuation of basal and cocaine-enhanced locomotion and nucleus accumbens dopamine in cannabinoid CB1-receptor-knockout mice. Psychopharmacology (Berl) 2009;204:1–11. doi: 10.1007/s00213-008-1432-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupica CR, Riegel AC. Endocannabinoid release from midbrain dopamine neurons: a potential substrate for cannabinoid receptor antagonist treatment of addiction. Neuropharmacology. 2005;48:1105–1116. doi: 10.1016/j.neuropharm.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Lupica CR, Riegel AC, Hoffman AF. Marijuana and cannabinoid regulation of brain reward circuits. Br J Pharmacol. 2004;143:227–234. doi: 10.1038/sj.bjp.0705931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M. Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron. 2001;31:463–475. doi: 10.1016/s0896-6273(01)00375-0. [DOI] [PubMed] [Google Scholar]
- Maejima T, Oka S, Hashimotodani Y, Ohno-Shosaku T, Aiba A, Wu D, Waku K, Sugiura T, Kano M. Synaptically driven endocannabinoid release requires Ca2+-assisted metabotropic glutamate receptor subtype 1 to phospholipase Cbeta4 signaling cascade in the cerebellum. J Neurosci. 2005;25:6826–6835. doi: 10.1523/JNEUROSCI.0945-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mátyás F, Urbán GM, Watanabe M, Mackie K, Zimmer A, Freund TF, Katona I. Identification of the sites of 2-arachidonoylglycerol synthesis and action imply retrograde endocannabinoid signaling at both GABAergic and glutamatergic synapses in the ventral tegmental area. Neuropharmacology. 2008;54:95–107. doi: 10.1016/j.neuropharm.2007.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- Mejías-Aponte CA, Drouin C, Aston-Jones G. Adrenergic and noradrenergic innervation of the midbrain ventral tegmental area and retrorubral field: prominent inputs from medullary homeostatic centers. J Neurosci. 2009;29:3613–3626. doi: 10.1523/JNEUROSCI.4632-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejias-Aponte CA, Ye C, Bonci A, Kiyatkin EA, Morales M. A subpopulation of neurochemically-identified ventral tegmental area dopamine neurons is excited by intravenous cocaine. J Neurosci. 2015;35:1965–1978. doi: 10.1523/JNEUROSCI.3422-13.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis M, Pistis M, Perra S, Muntoni AL, Pillolla G, Gessa GL. Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci. 2004;24:53–62. doi: 10.1523/JNEUROSCI.4503-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron. 2001;29:729–738. doi: 10.1016/s0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Oleson EB, Beckert MV, Morra JT, Lansink CS, Cachope R, Abdullah RA, Loriaux AL, Schetters D, Pattij T, Roitman MF, et al. Endo-cannabinoids shape accumbal encoding of cue-motivated behavior via CB1 receptor activation in the ventral tegmentum. Neuron. 2012;73:360–373. doi: 10.1016/j.neuron.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Hillard CJ, Liu Q-S. Endocannabinoid signaling mediates cocaine-induced inhibitory synaptic plasticity in midbrain dopamine neurons. J Neurosci. 2008;28:1385–1397. doi: 10.1523/JNEUROSCI.4033-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips PEM, Stuber GD, Heien MLAV, Wightman RM, Carelli RM. Subsecond dopamine release promotes cocaine seeking. Nature. 2003;422:614–618. doi: 10.1038/nature01476. [DOI] [PubMed] [Google Scholar]
- Reith ME, Li MY, Yan QS. Extracellular dopamine, norepinephrine, and serotonin in the ventral tegmental area and nucleus accumbens of freely moving rats during intracerebral dialysis following systemic administration of cocaine and other uptake blockers. Psychopharmacology (Berl) 1997;134:309–317. doi: 10.1007/s002130050454. [DOI] [PubMed] [Google Scholar]
- Riegel AC, Lupica CR. Independent presynaptic and postsynaptic mechanisms regulate endocannabinoid signaling at multiple synapses in the ventral tegmental area. J Neurosci. 2004;24:11070–11078. doi: 10.1523/JNEUROSCI.3695-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science. 1987;237:1219–1223. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- Roitman MF, Stuber GD, Phillips PEM, Wightman RM, Carelli RM. Dopamine operates as a subsecond modulator of food seeking. J Neurosci. 2004;24:1265–1271. doi: 10.1523/JNEUROSCI.3823-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sombers LA, Beyene M, Carelli RM, Wightman RM. Synaptic overflow of dopamine in the nucleus accumbens arises from neuronal activity in the ventral tegmental area. J Neurosci. 2009;29:1735–1742. doi: 10.1523/JNEUROSCI.5562-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria G, Mendizábal V, Touriño C, Robledo P, Ledent C, Parmentier M, Maldonado R, Valverde O. Lack of CB1 cannabinoid receptor impairs cocaine self-administration. Neuropsychopharmacology. 2005;30:1670–1680. doi: 10.1038/sj.npp.1300707. [DOI] [PubMed] [Google Scholar]
- Steinberg EE, Boivin JR, Saunders BT, Witten IB, Deisseroth K, Janak PH. Positive reinforcement mediated by midbrain dopamine neurons requires D1 and D2 receptor activation in the nucleus accumbens. PLoS ONE. 2014;9:e94771. doi: 10.1371/journal.pone.0094771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D. How addictive drugs disrupt presynaptic dopamine neuro-transmission. Neuron. 2011;69:628–649. doi: 10.1016/j.neuron.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo B, Siemes S, Wallmichrath I. Inhibition of GABAergic neurotransmission in the ventral tegmental area by cannabinoids. Eur J Neurosci. 2002;15:2057–2061. doi: 10.1046/j.1460-9568.2002.02041.x. [DOI] [PubMed] [Google Scholar]
- Thomsen M, Han DD, Gu HH, Caine SB. Lack of cocaine self-administration in mice expressing a cocaine-insensitive dopamine transporter. J Pharmacol Exp Ther. 2009;331:204–211. doi: 10.1124/jpet.109.156265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Wise RA, Rompre PP. Brain dopamine and reward. Annu Rev Psychol. 1989;40:191–225. doi: 10.1146/annurev.ps.40.020189.001203. [DOI] [PubMed] [Google Scholar]
- Xi ZX, Gilbert JG, Peng XQ, Pak AC, Li X, Gardner EL. Cannabinoid CB1 receptor antagonist AM251 inhibits cocaine-primed relapse in rats: role of glutamate in the nucleus accumbens. J Neurosci. 2006;26:8531–8536. doi: 10.1523/JNEUROSCI.0726-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi ZX, Spiller K, Pak AC, Gilbert J, Dillon C, Li X, Peng XQ, Gardner EL. Cannabinoid CB1 receptor antagonists attenuate cocaine’s rewarding effects: experiments with self-administration and brain-stimulation reward in rats. Neuropsychopharmacology. 2008;33:1735–1745. doi: 10.1038/sj.npp.1301552. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.