

Abstract
Somaclonal variation arises in plants and animals when differentiated somatic cells are induced into a pluripotent state, but the resulting clones differ from each other and from their parents. In agriculture, somaclonal variation has hindered micropropagation of elite hybrids and genetically modified crops, but the mechanism remains a mystery1. The oil palm fruit abnormality, mantled, is a somaclonal variant arising from tissue culture that drastically reduces yield, and has largely halted efforts to clone elite hybrids for oil production2–4. Widely regarded as epigenetic5, mantling has defied explanation, but here we identify the MANTLED gene using Epigenome Wide Association Studies. DNA hypomethylation of a LINE retrotransposon related to rice Karma, in the intron of the homeotic gene DEFICIENS, is common to all mantled clones and is associated with alternative splicing and premature termination. Dense methylation near the Karma splice site (the Good Karma epiallele) predicts normal fruit set, while hypomethylation (the Bad Karma epiallele) predicts homeotic transformation, parthenocarpy and dramatic loss of yield. Loss of Karma methylation and small RNA in tissue culture contributes to the origin of mantled, while restoration in spontaneous revertants accounts for non-Mendelian inheritance. The ability to predict and cull mantling at the plantlet stage will facilitate the introduction of higher performing clones and optimize environmentally sensitive land resources.
The African oil palm (Elaeis guineensis) is the most efficient oil bearing crop, but demand for edible oils and biofuels, combined with sustainability concerns over dwindling rainforest reserves, has led to intense pressure to improve oil palm yield. Introduction of the tenera hybrid (dura x pisifera) increased oil yield by up to 30%, leveraging the SHELL gene that confers single gene heterosis6,7. Clones (ramets) of individual high yielding tenera hybrid palms (ortets) provide a powerful shortcut to yield enhancement, with an additional 20 to 30% improvement6. Micropropagation through cell culture of immature apex leaf tissue (“the heart of palm”), and plantlet regeneration on hormone supplemented media (Methods), yields tens of thousands of genetically identical clonal palms. Unfortunately, shortly after the procedure was established, Tan Yap Pau of United Plantations, Malaysia, first noted a high frequency of homeotic floral phenotypes known as “mantling” among clonal ramets. Subsequently, Corley et al.2 documented the occurrence of mantled palms following prolonged periods in culture. In mantled palms, staminodes of pistillate flowers and stamens of staminate flowers develop as pseudocarpels8 often resulting in sterile parthenocarpic flowers with abortive fruit and very low oil yields (Fig. 1a–c, Extended Data Fig. 1). Pollination of mantled palms gave rise to variable numbers of mantled progeny, resembling rare naturally mantled variants known as poissoni, or diwakkawakka fruit forms3,4. The trait is non-Mendelian and sometimes reverts to normal9 and so has long been considered epigenetic5, with an overall decrease in DNA methylation found in mantled ramets5,10. The homeotic transformations observed in mantled palms resemble defects in B-function MADS box genes, suggesting strong candidates for epigenetic modification8. However, decades of research into candidate retroelements11,12 and candidate homeotic genes8,12,13 failed to identify epigenetic changes consistently found in somaclonal mantled palms.
Figure 1. Epigenome Wide Association Study (EWAS).

a, Normal, b, fertile mantled, c, parthenocarpic mantled fruit shown as whole fruit (top), longitudinal sectioned (middle) and cross sectioned (bottom). Black arrows, pseudocarpels. White arrows, kernel. d, Circos plot of Oil Palm Chromosomes. Track order: gene density (i); repeat density (ii); cytosine methylation density (whole genome bisulphite sequencing) in an ortet (iii); cytosine methylation densities (microarray) of ortet (iv), normal ramet (v) and mantled ramet (vi); differential cytosine methylation of normal minus mantled ramets (vii). Heatmaps represent average cytosine methylation densities in ~300 Kb windows independent of sequence context. e, Venn diagram of microarray features differentially methylated between leaves from mantled and normal ramets (p<0.05, two-sided Student t-test, Methods). Each set represents clonal lineages of given genotypes obtained from one source: Source A (red, 15 mantled, 15 normal), Source B (brown, 6 mantled, 14 normal), Source C (blue, 14 mantled, 15 normal), and Source D (green, 8 mantled, 10 normal). Red numbers indicate subsets including one of the four microarray features mapping to the Karma LINE element.
We performed a genome-wide, unbiased, DNA methylation analysis (an epigenome-wide association study, or EWAS) in search of loci epigenetically associated with the mantled phenotype, using a DNA microarray based on the E. guineensis (pisifera) reference genome14 (Methods). DNA methylation density was measured in 1–2 kb intervals surrounding each feature by DNA methylation-dependent comparative microarray hybridization15 and statistical analyses (Methods). Genome-wide DNA methylation maps were constructed from parthenocarpic mantled (n = 43) or normal (n = 54) ramets, as well as ortets from which these ramets were derived (n = 10). These maps strongly resembled those constructed by whole genome bisulphite sequencing in sample palms (Fig. 1d), demonstrating reproducibility.
At genome-wide resolution, the landscape of DNA methylation was remarkably consistent between ortets and ramets (Fig. 1d) with highest methylation within repetitive sequences14. However, thousands of loci were differentially methylated (Fig. 1d), most of which (~90%) were hypomethylated in mantled, consistent with previously reported reduced 5mC content5,10. Most hypomethylated loci (~75%) were transposons and repeats, while less frequent hypermethylated loci included genic sequences (Extended Data Fig. 2), resembling cell cultures of Arabidopsis16. 15 independent somaclonal lineages obtained from 4 independent sources were used to maximize genotypic diversity, and significant differentially methylated regions (DMRs) between normal and fully mantled samples were first identified within each source population (Methods). Results were then compared between populations (Fig. 1e). Although tens-of-thousands of DMRs were detected between mantled and normal clones in each population, 99.9% of these were exclusive to either one (94.4%) or 2 (5.5%) of the 4 populations, indicating significant genotypic variation in epigenetic response to tissue culture. A single microarray feature detected differential methylation between normal and mantled clones in all 4 populations (Fig. 1e). This feature lies within the ~35 kb intron 5 of EgDEF1 (Fig. 2a), the oil palm ortholog of the B class MADS box transcription factor genes, Antirrhinum majus DEFICIENS (DEF) and Arabidopsis APETALA3 (AP3)8,12,13.
Figure 2. Hypomethylation of Karma is associated with the mantled phenotype.

a, Microarray feature data plotted below a map of the EgDEF1/MANTLED gene (vertical ticks, exons; horizontal line, introns; arrow, direction of transcription) including locations of Rider, Karma (dashed box) and Koala retrotransposons. CG and CHG sites are shown at the top. Log10 p values (54 normal vs. 43 parthenocarpic mantled ramets) are plotted (two-sided Student’s t-test). Arrow in p value plot, feature detected as hypomethylated in mantled ramets from all 4 sources (Fig. 1e). b, Genome-wide bisulphite sequencing of leaf samples from ortet (O, black, n=5), normal ramets (N, green, n=5) and parthenocarpic mantled ramets (M, red, n=5). Mean methylation density per cytosine is plotted on a 0 to 100% scale for each cytosine context and sample type. CHG DMR, differentially CHG methylated region corresponding to Karma. c, CHG methylation monitored in 86 additional ortets, mantled and normal ramet leaf samples by restriction enzyme digestion and qPCR (Methods). LDA was performed between normal (n=21) and mantled (n=28) samples with BbvI and RsaI restriction sites. FN1 and FN2, two false negative mantled samples. Green and red arrows, normal and mantled control samples, respectively. A similar analysis was performed on remaining normal (n=14) and mantled (n=23) samples with ScrFI restriction sites (Extended Data Fig. 4d). d–g, Karma bisulphite sequencing maps (antisense strand) of (d) normal control, (e) mantled control, (f) FN1 and (g) FN2. 13 CHG sites are shown to scale above. “S”, CHG at the Karma splice acceptor site (CAG/CTG). “B”, BbvI site. Bar, CHGs within the common microarray feature (Fig. 1e). Methylated and unmethylated CHG sites are indicated by green and red boxes, respectively. Open boxes, low quality base calls. Each row represents an individual Sanger DNA sequencing read.
EgDEF1 spans ~40 kb on chromosome 12 (Fig. 2a). A Ty1/copia Rider retrotransposon lies upstream, while a Ty3/gypsy retrotransposon, Koala, is located within intron 5. Consistent with important earlier work12, no DNA methylation difference within these retrotransposons was found in mantled clones across multiple populations (Fig. 2a, Extended Data Fig. 3). However, a third previously unreported repetitive element lies within intron 5, and has homology to rice Karma LINE elements. Karma is activated in rice embryogenic tissue culture, but only transposes in regenerated plants as transgenerational DNA hypomethylation of the element persists17. The 3.2 kb oil palm Karma element is flanked by a 13 bp target site duplication (TTCAAAATGATGA) and includes a defective reverse transcriptase open reading frame (ORF2) preceded by a splice acceptor (‘^’) and followed by a polyadenylation signal, resembling truncated Karma elements in rice17,18 (Supplementary Fig. 1). The unique microarray feature, which consistently detected hypomethylation in mantled clones, serendipitously includes the predicted splice acceptor site (GAACAG^ATGC). All three additional microarray features mapping within the Karma element also detected significant hypomethylation in mantled clones (Fig. 2a, Extended Data Figs. 3 and 4a–c).
To verify Karma hypomethylation, sample trios comprising genetically identical ortet, parthenocarpic mantled and normal ramets from 5 independent clonal lineages were subjected to whole genome bisulphite sequencing (Methods). CG methylation was unchanged across the EgDEF1 locus, but Karma CHG methylation (H=A, C or T) was dramatically reduced in mantled clones, revealing a DMR covering ~70 CHG sites. CHH methylation was much lower and only subtly reduced (Fig. 2b). To validate differential CHG methylation in unrelated clonal palms, quantitative Polymerase Chain Reaction (qPCR) assays were used to quantify CHG methylation at BbvI and RsaI restriction sites within the DMR (Methods, Fig. 2c, Extended Data Fig. 4b) in a panel of 49 palms from 21 clonal lineages and 4 independent sources: 8 ortets and 13 normal clones, 19 parthenocarpic mantled clones, 2 fertile mantled clones and 7 partially revertant clones yielding bunches with both mantled and normal fruit. Linear discriminant analysis provided 93% sensitivity and 100% specificity for detection of mantling (Fig. 2c). Fronds from all 7 of the revertant palms were scored as mantled, consistent with the observation that normal bunches arose late in development2. Similar results were obtained in 37 polymorphic palms using alternative restriction sites (Extended Data Fig. 4d). The two false negative mantled palms (Fig. 2c), were further analyzed by bisulphite sequencing of a region spanning the Karma splice acceptor site. While normal clones had dense CHG methylation, and mantled controls had lost all CHG methylation (Fig. 2d–e), the false negative mantled samples lost CHG methylation near the splice acceptor site (Fig. 2f–g, Extended Data Fig. 4e), which was therefore sufficient to predict the mantled phenotype. Because of their strong predictive properties, we named the mantled hyper- and hypo-methylated epialleles Good Karma and Bad Karma, respectively.
Two lineages of revertant palms had mixed bunches with both normal and mantled fruit9, resembling epialleles in maize regulated by transposons19. The first lineage included two revertant ramets with 99% and 95% normal fruit per bunch, respectively, in which abnormal fruit had only one or two small pseudocarpels (Fig. 3a–c). A second lineage included a mosaic ramet with only 7% normal fruit. In all 3 ramets, CHG methylation at the Bbv I site was low in fronds (Fig. 3d), consistent with other revertants (Fig. 2c). However, methylation was restored in fruit from the two revertant ramets, but not from the mantled mosaic ramet (Fig. 3d–f). As with similar epialleles in maize, Linnaria, and other plants19–21, reversion of the abnormal phenotype accompanied by restoration of DNA methylation is strong evidence that Karma hypomethylation is the cause of the mantled phenotype. Differential methylation between individual mantled and normal fruit was not observed, however, likely reflecting non-cell autonomy of the B class homeotic phenotype (Fig. 3a–d), also observed in Antirrhinum and Arabidopsis22. Bisulphite sequencing from normal and mantled fruit (Extended Data Fig. 5) revealed hyper-and hypomethylated reads at the splice acceptor site (Fig. 3e–f), suggesting that these fruit were indeed mosaic for hyper- and hypo-methylated cells. In one mosaic palm, direct evidence for mosaicism was obtained from different samples of the same vegetative frond (Extended Data Fig. 6).
Figure 3. Karma methylation in revertant palms.

a–c, Spikelet (a) from a revertant ramet including normal (b) and fertile mantled (c) fruit with one or two pseudocarpels (arrows). d, density of CHG methylation (% mCHG) at the BbvI site (Methods) in ramets yielding 100% normal fruit (n.f.) (green), revertant ramets yielding 99% (yellow) or 95% (orange) normal fruit and a mosaic ramet yielding 7% (red) normal fruit per bunch. Error bars, standard deviation (biological replicates of fronds (n=4), rachis sections (n=8) or fruit (n=2)). e–f, % mCHG for the 3 CHG sites found in the unique common microarray feature (Fig. 3b) in normal (green) and subtly mantled (red) fruit from revertant ramets yielding 99% (e) or 95% (f) normal fruit per bunch (two-tailed Fisher’s exact test, n.s., not significant). Alleles were analyzed separately based on a heterozygous SNP within the bisulphite sequencing amplicon.
DNA methylation near splice acceptor sites impacts alternative splicing, although the mechanism remains unclear23. To assess alternative splicing, EgDEF1 transcript models were built based on transcriptome sequencing (data not shown) and validated by RT-PCR (Methods). As previously reported12, two forms of EgDEF1 transcripts were found in both normal and mantled inflorescences: the full-length EgDEF1 transcript (cDEF1), and a prematurely terminated transcript including exons 1 through 5 and 221 bp of intron 5 (tDEF1)12 (Fig. 4a; Extended Data Fig. 4c). However, we identified a third alternative transcript in mantled female inflorescences (Fig. 4a). This novel transcript (kDEF1) was spliced from the donor site of intron 5 to the proximal Karma acceptor site and is predicted to encode a truncated EgDEF1 peptide that terminates within the K domain of the MADS box protein, and has a unique C-terminal sequence (Extended Data Fig. 7a).
Figure 4. Alternative splicing and loss of 24nt small RNA.

a, EgDEF1/MANTLED transcripts assembled from transcriptome sequencing (data not shown) and RT-PCR (Methods). Black boxes, exons. Blue box, intron 5 sequence included in the tDEF1 transcript. Coordinates relative to the reference pisifera oil palm genome14. b, Quantitative RT-PCR of cDEF1, tDEF1 and kDEF1 transcripts in shoot apices (stage 0) and in early (stage 2) to late (stage 5) female inflorescences from normal and parthenocarpic mantled ramets. Error bars, standard deviations between 3 replicate assays of 3 replicate tissue samples per phenotype, per stage. Expression relative to an endogenous reference gene is shown (Methods). c, 24nt siRNA accumulation in shoot apices (stage 0) from normal (n=5) and parthenocarpic mantled (n=7) ramets, and from second passage apical leaf tissue cultures re-cloned from normal (n=2) or mantled (n=1) ramets (Methods). Values expressed as fragments per kilobase per million mapped reads (FPKM). Bars above (sense) and below (antisense) the line indicate mapped normalized 24nt siRNAs that are not significantly different in abundance in normal and mantled (grey) or significantly differentially expressed in normal (green) relative to mantled (red) (p <0.05, Student’s t-test, two tailed, assuming equal variance).
Expression of the three transcripts (Fig. 4b) was assessed in shoot apical meristem (stage 0) and 4 stages of immature female inflorescence development (Methods, Extended Data Fig. 7b–f). kDEF1 expression was strikingly restricted to stages 3 to 5 of mantled (but not normal) female inflorescence, strongly suggestive of a role in the mantled phenotype. In contrast, cDEF1 was detected at only slightly (though significantly) lower levels in mantled female inflorescences12, while tDEF1 was unchanged (Fig. 4b). Previously reported differences in timing of tDEF1 and cDEF1 transcription within each stage12 may be the consequences of kDEF1 transcription (Methods).
In plants, 24nt small interfering (si) RNAs guide non-CG methylation, and we identified a cluster of antisense 24nt Karma siRNAs in shoot apical meristem (stage 0), which were reduced or absent in mantled (Fig. 4c), and in later stages of normal inflorescence (Extended Data Fig. 8, Methods). In polyembryogenic tissue cultures derived from normal and abnormal clonal palms (Methods), small RNA underwent a switch from 24 to 21nt (Extended Data Fig. 9), resembling cell cultures and callus of Arabidopsis16. Karma methylation (Extended Data Fig. 10) and 24nt siRNA (Fig. 4c, Extended Data Fig. 8) were reduced in normal cultures between two and seven passages, and lost in abnormal, cultures, suggesting a model for the origin of mantled: if meristems are the source of 24nt siRNA24 (Fig. 4c), then leaf cells detached from the meristem would lose 24nt siRNA and non-CG DNA methylation over time in culture. Antisense small RNA might influence exon trapping, as it does in humans25,26, or else splicing might be associated with changes in chromatin, for example histone H3 lysine 4 methylation18. Good Karma would only be restored during shoot generation if DNA methylation were not entirely lost, or potentially if siRNAs were applied during the tissue culture process. 24nt siRNA could also facilitate non-Mendelian segregation4,9 which resembles paramutation (the interconversion of heterozygous epialleles) in some respects 11,23,27.
Despite its importance, mantling has been the elusive target of molecular genetic investigation for the past 3 decades. We have demonstrated that the mantled trait is a consequence of epigenetic modification of the Karma transposable element within the B class MADS box EgDEF1 gene, which we have named MANTLED. B class function in stamen identity is conserved in monocots, but similar to other monocot paleoAP3 genes, EgDEF1 overexpression fails to cause homeotic conversion in Arabidopsis8, because of a diverged C-terminal exon30. Nonetheless, like AP3 and DEF 28,29, the oil palm EgDEF1/MANTLED gene is expressed in inner perianth and stamen primordia as the perianth initiates (stage 2), followed by stamen and stamenoid primordia (stage 3)8. The appearance of kDEF1 transcripts during this transition (Fig. 4b) suggests that kDEF1 functions at this critical window to induce the mantled phenotype.
METHODS
Samples and tissue culture
Leaf tissue used in the EWAS discovery and validation panels was sampled from oil palm clones (known as ramets) derived from tenera mother palms (known as ortets) from various genetic backgrounds. The female parents of the ortets were predominantly of a Deli dura background whilst the pisifera male parents were derived from La Me, Yangambi, AVROS and Binga genotypes. These parental lines make up the major genetic backgrounds of the oil palm populations in Malaysia. These palms were collected from the Malaysian Palm Oil Board (MPOB), United Plantations Berhad, FELDA Global Ventures R&D Sdn Bhd and Applied Agricultural Resources Sdn Bhd (Extended Data Fig. 3). The ramets were derived from explants excised from non-chlorophyllous leaves of their respective ortets cultured on hormone-supplemented media31–34. The oil palm tissue culture process involves callus initiation and polyembryoid generation followed by shoot and root initiation35. In large scale production, the tissue culture process takes 48 to 52 months to complete. Once established, ramets undergo acclimatization in the nursery for 3 to 4 months, are moved to the field nursery for another 8 to 9 months, and finally are field planted. At every stage of the tissue culture process, off-types are culled. Flower and fruit bunch census are taken at the onset of flowering (2 to 3 years after field planting) and in subsequent years. Normal and mantled ramets were identified based on the census data collected.
Recloned tissue culture materials were generated using the standard protocol described above. Normal and abnormal ramets from identical genetic backgrounds were subjected to the tissue culture process and sub-sampling was carried out at the polyembryogenic stage, namely at subculture passage two (SC2) and seven (SC7), representing short and prolonged exposure in culture on hormone-supplemented media, respectively.
Female inflorescence samples used in staging the developmental phase were obtained from a total of 31 clonal palms with ages ranging from 3 to 10 years after field-planting. As a means to categorize the developmental phases, these inflorescence samples were histologically analyzed and classified as Stage 0, shoot apical meristem; Stage 2, initiation of perianth organs; Stage 3, development of perianth organs and initiation of reproductive organs; Stage 4, development of reproductive organs; Stage 5, fully formed reproductive organs according to Adam et al36.
E. guineensis genome microarray design
The microarray design was based on the E. guineensis genome14 and contains over 1 million 60 base probes. Probes were selected from non-overlapping 1.5 kb windows across all scaffolds of the P1 pisifera genome build, choosing the 60mer with the lowest composite 15mer frequency count within the genome. When compared to the publically available EG5 genome assembly14, 860,861 probes from the microarray matched at 100% stringency (81,194 probes falling on exons and the remaining covering intronic and intergenic regions of the genome). The microarrays were manufactured by Roche NimbleGen using the HX1 platform.
DNA methylation dependent fractionation
Genomic DNA (60 μg) was mechanically hydrosheared to 1–4 kb fragments. Sheared DNA was divided into four equal portions, two of which were digested with 10 units/μg McrBC (New England Biolabs) under manufacturer’s recommended conditions and the other two were mock treated without adding enzyme. Following digestion, DNAs were treated with proteinase K (50mg/ml) for 1 hour at 50°C and then precipitated with EtOH under standard conditions. Resuspended DNAs were resolved by agarose gel electrophoresis, and DNA in the 1–4 kb size range was excised from gels and extracted. McrBC requires that two methylated half sites (RmC, where R = A or G) lie within 40–3,000 bp of each other, and cutting occurs in the proximity of one of the half sites37. Because 1 to 4 kb fragments are treated with McrBC, and undigested fragments are isolated, hybridization of a microarray probe complimentary to sequence distant from the methylation site results in a ‘wingspan’ effect in which probes are able to detect DNA methylation from a distance up to ~1.5 kb38. For each fraction, 200 ng was used for cyanogen dye labeling (Cy3 or Cy5). For each sample, four microarrays were hybridized in a duplicated dye swap design to differentially labeled untreated and DNA methylation depleted fractions. Sample size was chosen to allow multiple clonal lineages including both normal and parthenocarpic samples from each of four independent sources.
Microarray data processing, normalization and statistical analysis
Among the ~860,000 microarray probes matching the oil palm genome with 100% sequence stringency, a subset of ~460,000 uniquely mapped probes was selected to reduce noise from non-specific hybridizations. For data processing, corrections on spatial non-uniformity of fluorescent signal intensities, done separately for the Cy3 and Cy5 dye channels, were made. Data were then normalized by background subtraction using negative control probes, followed by scaling. After normalization, median log signal of the control probes for each dye of each array was set to zero. MAD (median absolute deviation) log signal of all the probes on an array was used as a constant for a given treatment (untreated or DNA methylation depleted). DNA methylation was measured as the sample average (over the two pairs of dye-swapped technical replicates) of normalized log2 ratios of untreated over methylation depleted DNA. Statistical analysis was first conducted within samples derived from each source independently. Within each group, a two-sided t-test was performed between normal and mantled phenotypes. Using a cutoff of p=0.05, one probe that was significantly differentially methylated in all the 4 groups was identified. To confirm this finding with a different statistical approach, quantile normalization of methylation measurements (the sample average of normalized log ratios of untreated over methylation depleted DNA) was performed on all samples together, and then a t-test on all normal versus all mantled samples was conducted. This process identified the same probe which was found in the initial analysis, as well as an additional three immediately neighboring probes. Computer code is available upon request.
Quantitative PCR DNA methylation assays
Primer pairs were designed to amplify two Karma element regions as diagrammed in Extended Data Fig. 4a–b). A 633 bp amplicon included methylation sensitive restriction sites containing CHG positions 188 bp (BbvI) and 375 bp (ScrFI) downstream of the Karma splice site CHG. A 632 bp amplicon that amplified a region near the center of the Karma element included a RsaI methylation sensitive restriction site. Primer pairs were confirmed to amplify a single band of the correct size by agarose gel electrophoresis. For qPCR DNA methylation assays, 100 ng of genomic DNA was digested with 10 units/μg of the indicated restriction enzyme under standard conditions. An equal amount of genomic DNA was mock treated in a reaction lacking enzyme. Digestion reactions were incubated at 37°C for 16 hours. qPCR was carried out using 10 ng each of the mock treated and enzyme digested samples in 1X Roche SYBR Green Master Mix on a Roche LC480 instrument. qPCR amplifications were performed in duplicate. For each duplicate mock/digested amplification pair, the delta Ct value was calculated as the digested Ct minus the mock Ct and duplicated dCt values were averaged. DNA methylation density was calculated as % Dense Methylation = 2^(−dCt(digested-mock)). Samples were genotyped by restriction digestion of PCR amplicons with either BbvI or RsaI to confirm that all samples used for DNA methylation validation included intact restriction sites on both alleles. Enzyme digestions were quality controlled by performing qPCR assays monitoring three independent invariantly unmethylated endogenous genomic loci and one invariantly methylated endogenous genomic locus. All quality control passed digestions reported <5% methylation of the unmethylated controls and >95% methylation of the methylated control. Primer sequences are available upon request.
Whole genome bisulphite sequencing
1 μg genomic DNA from each of 15 mature leaf samples (5 lineage trios of ortet, normal ramet and mantled ramet) was used to construct TruSeq fragment libraries (Illumina), and up to 500 ng of adapted library molecules were bisulphite converted using the EZ DNA Methylation-Lightning Kit (Zymo Research). Each library was sequenced in one lane of a HiSeq 2000 flow cell to generate paired 100 bp reads. Reads were mapped to an in silico bisulphite converted reference E. guineensis (pisifera) genome. For each cytosine context (CG, CHG or CHH), the number of mapped reads corresponding to unconverted cytosines relative to the total number of reads including the particular base was used to calculate the percent methylation at each cytosine position.
Clone based bisulphite sequencing
Because all cytosines are potential sites for DNA methylation in plants, and because whole genome bisulphite sequencing demonstrated that CG methylation is maintained at high levels in both normal and mantled ramets, bisulphite sequencing amplicon primers were designed to include CG dinucleotides, but exclude CHG and CHH trinucleotides. Within primer sequences, CG dinucleotides were assumed to be methylated. The amplicon (amplified from the antisense strand) contained 13 CHG sites, including the CHG site at the Karma splice acceptor site. 2 μg of each sample was bisulphite converted as described for whole genome bisulphite sequencing. 30 ng converted DNA was used for PCR amplification in 1X HiFi Hotstart Uracil+ Ready Mix (Kappa). Amplicons were cloned using the TOPO TA Cloning Kit (Invitrogen) following A-tailing by Klenow treatment. For each sample, 48 white colonies were individually picked, propagated and plasmid DNA extracted. Plasmid inserts were PCR amplified and Sanger sequenced (ABI 3730) using vector-specific primers. Sequencing was performed on 48 clones per sample, and reads from plasmids not including the amplicon insert are not shown. Sequences were base called in CONSED and methylation densities at each CHG site were calculated. Where possible, heterozygous non-cytosine single nucleotide polymorphisms were scored so that each allele could be analyzed independently. In cases where a polymorphism changed a CHG site to either a CG or CHH site, the non-CHG variant was not included in calculations of CHG methylation. Because CHH methylation was determined to be consistently very low in both normal and mantled ramets, conversion of CHH sites within the amplicon was used to control for bisulphite conversion rates. All samples analyzed displayed <4% methylation of CHH sites, demonstrating that bisulphite conversion was >96% complete in all samples. Primer sequences are available upon request.
mRNA and siRNA sequencing
Transcriptome sequencing was performed on shoot apex, early inflorescence (<2cm) and late stage inflorescence (3 normal and 3 mantled female inflorescence biological replicates each). 2–3 μg total RNA was used to construct individually barcoded Illumina TruSeq stranded libraries. Libraries were pooled in sets of 4, and each pool was sequenced in one lane of a HiSeq 2000 flow cell to generate paired 100 bp reads. Small RNA fractions of female shoot apex tissue at stage 0 and female inflorescence tissue at stages 2, 3, 4 and 5 (7 mantled and 5 normal biological replicates at stage 0, 6 mantled and 8 normal biological replicates each at stages 2 and 3, 7 mantled and 5 normal biological replicates at stage 4, and 5 mantled and 4 normal biological replicates at stage 5), as well as second-passage tissue cultures recloned from mantled (n=1) or normal (n=2) ramets, were used to construct Illumina TruSeq small RNA libraries and sequenced following the same strategy as mRNA sequencing. mRNA sequencing data was used to construct gene models for all observed EgDEF1 alternative transcripts. siRNA reads mapping to the genomic scaffold including EgDEF1 were identified and normalized as fragments per 1,000 reads mapped to the genome (FPKM). FPKM values for each 24mer were compared between biological replicates of normal and mantled samples by a two-tailed Student t-test, assuming equal variance.
Quantitative RT-PCR Assays
To specifically quantify cDEF1 expression, a forward primer spanning the junction of exons 1 and 2 was utilized with a reverse primer within exon 7 (Extended Data Fig. 7b–d). The same forward primer was utilized with a reverse primer including intron 5 sequence to specifically quantify tDEF1 expression. Finally, a forward primer spanning the junction of exons 4 and 5 was utilized with a reverse primer within Karma, downstream of the exon 5/Karma splice junction, to specifically quantify kDEF1 expression. Assays were optimized using normal and mantled late stage inflorescence total RNA, and cDNAs were Sanger sequenced to confirm the identity of the amplicons. Standard curves generated from serially diluted cDNAs were generated for each primer pair, as well as for two internal oil palm reference gene assays39 (Extended Data Fig. 7e–f). Gene expression was quantified in developing inflorescence stages 0, 2, 3, 4 and 5. All first strand cDNA reverse transcription reactions were performed from 1 μg total RNA using a cocktail of reverse primers specific to EgDEF1 exons 6 and 7, as well as 3′ regions of Karma. For each stage, three technical replicates were performed for each of the three biological replicates per phenotype, per stage. qRT-PCR reactions were performed using 1 μL first strand cDNA in 1X Roche SYBR Master Mix on a Roche LC480 instrument. Cycle thresholds above 33 cycles were not included in calculations, and detectable expression was calculated only for samples in which expression was detected in at least 2 of 3 technical replicates. Expression levels were quantified by extrapolation from the standard curve for each assay, and expression levels relative to the reference gene were calculated. Primer sequences are provided in Extended Data Fig 7d.
We found only a subtle decrease in expression of cDEF1 in female mantled relative to normal inflorescence at stage 3 (Fig. 4b). A more significant decrease was previously reported by Adam et al.8 but was not detected consistently40. More recently, an increase in the ratio of tDEF1 to tDEF1 + cDEF1 was reported by Jaligot et al12 who used samples from early and late time points at each stage of inflorescence development12. However, when absolute values of (tDEF1/tDEF1+cDEF1) from early and late samples from any given stage are pooled (Fig 6B and Fig S8B in Jaligot et al., 2014), then only modest increases in expression are observed in mantled samples in agreement with our results (Fig. 4b).
Extended Data
Extended Data Figure 1. Spikelets from clonal palms of different fruit form phenotypes.

a, Spikelets from a normal ramet. b, Spikelet from a fertile mantled ramet. c, Spikelet from a parthenocarpic mantled ramet. d, Spikelet from a revertant ramet displaying both normal (N) and mantled (M) fruits in the same spikelet.
Extended Data Figure 2. Annotation of genome-wide differentially methylated loci.
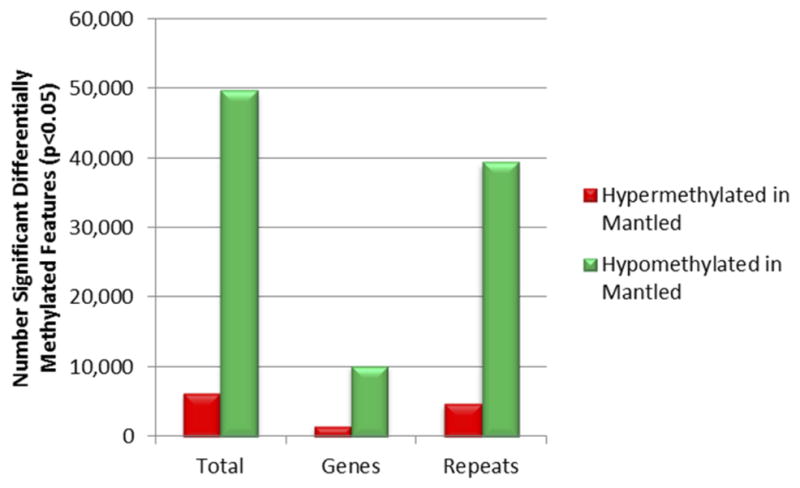
Sequences of microarray features reporting significant differential DNA methylation between fully normal and fully mantled leaf DNA samples of one or more clonal lineages were mapped to the reference E. guineensis pisifera genome14. Features were assigned to gene and repeat classes according to annotations of genomic elements mapped within 3 Kb of the microarray feature sequence, as this is the distance at which McrBC is capable of monitoring DNA methylation density. The Repeat class includes all repetitive sequences, including transposons and pisifera-specific repetitive sequences14. Features mapping within 3 Kb of both a gene and a repeat were assigned to both classes. The number of features reporting hypermethylation (red) and hypomethylation (green) are plotted.
Extended Data Figure 3. Summary of DNA methylation changes predicted by EWAS within clonal lineages.

Rows indicate independent clonal lineages from four oil palm industry sources (Source A–D, as indicated in Fig. 1e). The numbers of fully normal and fully mantled palms per lineage represented are indicated to the left. Columns represent each microarray feature mapping to the EgDEF1 (open box at top) and upstream region. The relative positions of Rider, Karma and Koala elements are indicated. The arrow indicates the direction of EgDEF1 transcription. Features reporting significant hypomethylation or hypermethylation in mantled relative to normal clones are indicated as black and gray boxes, respectively. White boxes indicate features reporting no significant DNA methylation difference. Only clonal lineages including more than 1 ramet per phenotype are shown in order to determine statistical significance within each clonal lineage (n=41 normal; n=37 parthenocarpic mantled). Ramets from four additional clonal lineages were included in the source-by-source analysis shown in Fig. 1e.
Extended Data Figure 4. DNA methylation assays and supporting DNA methylation data.

a, Diagram of the EgDEF1 gene including the Karma element within intron 5 (orange box). Black boxes represent exons and the horizontal line represents introns. Scale bar is in base pair units. b, Blue tick marks represent the relative positions of the four microarray features reporting significant hypomethylation of mantled clones in all source lineages. The left-most feature includes the Karma splice acceptor site. Horizontal lines labeled B (BbvI) and R (RsaI) indicate the relative positions of amplicons used for qPCR based CHG methylation assays. The BbvI amplicon also includes a ScrFI site (S) utilized in panel d. The relative position of the bisulfite sequencing amplicon used to determine Karma splice site CHG methylation is shown below the qPCR amplicons. c, Diagrams of the three alternatively spliced EgDEF1 transcripts. Black boxes represent exons included in each transcript. The dotted lines represent intronic sequences spliced out of the mature mRNA transcripts. The red box represents Karma element sequence spliced to EgDEF exon 5 in the kDEF1 transcript. The blue box represents EgDEF1 intron 5 sequence included in the tDEF1 transcript that does not utilize the exon 5 splice donor site. d, In addition to adult leaf samples analyzed by BbvI and RsaI qPCR assays (Fig. 2c), 37 samples were found to have a SNP in the BbvI site and were therefore analyzed by ScrFI and RsaI qPCR assays (Methods, Extended Data Fig. 4b). LDA was performed between normal (n=14) and mantled (n=22 parthenocarpic mantled; n=1 fertile mantled) samples. Combining these results with those shown in Figure 2c, sensitivity and specificity for detection of mantling are each 94%. e, Bisulphite sequencing of controls, FN1 and FN2 (Fig. 2c). mCHG density was calculated for the 3 CHG sites covered by the unique common microarray feature (Fig. 1e, Fig. 2d–g). FN1, FN2 and the mantled control were significantly hypomethylated relative to the normal control (* p<0.0001, two-tailed Fisher’s exact test).
Extended Data Figure 5. Clone based bisulfite sequencing maps of normal and mantled phenotype fruits from epigenetic mosaics.

The heatmap format is as described in Figure 2d–g. Gray boxes indicate a site in which a SNP on allele a resulting in a CHG to CHH site conversion. Mosaic Clone #1 represents a revertant clone yielding 95% normal fruit. Mosaic Clone #2 represents a revertant clone yielding 99% normal fruit. Alleles were analyzed independently based on a SNP not affecting a potentially methylated base. Statistical analyses of methylation at the three CHG sites spanning the Karma splice site are shown in Fig. 3e–f.
Extended Data Figure 6. CHG methylation in rachis sectors of an oil palm yielding 7% normal fruit (Clone lineage 2 in Fig. 3d).

Rachis of three successive fronds was dissected into 8 equal sectors. DNA methylation in each sector per frond was measured by BbvI and ScrFI assays, as described in Methods. Average DNA methylation density measurements of three technical replicates per frond, per sector, per assay are plotted on a radial graph representing the 8 rachis sections around the palm trunk (ScrFI assay, light blue; BbvI assay dark blue). Sector numbering was ratcheted for frond 2 vs. 1 and frond 3 vs. 2 based on the R2 best fit of CHG methylation density around the circumference of the palm to correct for out-of-register numbering of rachis sectors between successive fronds (data not shown). Consistent with the fact that this oil palm yields only 7% normal phenotype fruit, the majority of DNA methylation measurements are consistent with the mantled phenotype. However, sectors 8 and 2 display gains of CHG methylation in rachis sectors of all three fronds, and reach or approach normal levels in sectors 8 and 2 of frond 2, thus demonstrating mosaicism directly.
Extended Data Figure 7. Protein sequences and summary of qRT-PCR assay designs.

a, Residues highlighted in red are encoded by Karma sequence splice to exon 5 of EgDEF1. The alternate splicing event disrupts the transcription activation domain of EgDEF1. 12 variant amino acids are coded by Karma sequencing, followed by a stop codon. b, Diagram of EgDEF1 locus including positions of qRT-PCR primers. cDEF1 transcripts were detected using primer a (spanning the splice junction of exons 1 and 2) and primer c (internal to exon 7). kDEF1 transcripts were detected using primer b (spanning the splice junction of exons 4 and 5) and primer d (internal to Karma ORF2). tDEF1 transcripts were detected using primer a and primer e (spanning the 3′ end of exon 5 and including tDEF1-specific intron 5 sequence. c, All assays were confirmed to give a single band of the correct size by agarose gel electrophoresis. Amplicons were Sanger sequence verified. Note that no band is amplified using the kDEF1 primer pair in samples from normal inflorescence, consistent with lack of expression of kDEF1 in normal inflorescence. d, Sequences of primers diagrammed in panel b. e–f, PCR amplicons including each qRT-PCR amplified sequence were serially diluted and quantified in triplicate by qPCR using the indicated primer pairs. Dilutions (x-axis) were plotted against the measured cycle threshold (y-axis). e, Standard curves for cDEF1 (blue), kDEF1 (red) and tDEF1 (green). Line equations were used to calculate the efficiency of each primer pair. The efficiency of each primer pair was used in calculations for quantification of expression of each associated transcript. f, Standard curves for two endogenous oil palm control genes. The efficiency of each primer pair was used in calculations for quantification of expression of each associated transcript. Expression of each alternative transcript was calculated relative to the control PD00569 control. Control qRT-PCR primers are described in Chan et al.39.
Extended Data Figure 8. 24nt sRNA analysis of inflorescence development.

sRNA expression at inflorescence stages 0 (shoot apical meristem), 2, 3, 4 and 5, was analyzed by Illumina sRNA sequencing (Methods). FPKM normalized expression values for each measured 24nt sRNA are plotted in scale with the genomic elements diagrammed at the top of the figure. Gray bars indicate detected 24nt sRNA that are not significantly differentially expressed between normal relative to mantled tissues (p > 0.05, student’s t test, two tailed assuming equal variance). Differentially expressed 24nt sRNAs are plotted as green or red bars for normal or mantled tissues, respectively. Bars above and below the zero line represent sense and antisense sRNAs, respectively, and are plotted on the same scale in both directions.
Extended Data Figure 9. Relative abundance of 21nt and 24nt sRNA in normal and mantled reclones and stage 0 inflorescence.

a, Distribution of small RNA lengths derived from mantled reclone (blue) and stage 0 inflorescence (red). b, Distribution of small RNA lengths derived from normal reclone (blue) and stage 0 inflorescence (red). Read lengths of small RNA sequencing reads are plotted as the percentage of total reads for each incremental sRNA nucleotide length.
Extended Data Figure 10. CHG methylation in recloned tissue cultures.

Tissue cultures were reconstituted from normal and mantled ramets from two clonal lineages (“clones of clones”). Methylation at three CHG sites across the Karma DMR was quantified by qPCR assays at two (SC2) and seven (SC7) passages in tissue culture. Cultures derived from normal ramets displayed higher CHG methylation than those derived from mantled ramets. In both normal and mantled reclones, CHG methylation generally decreased with time in culture. At SC2, the time point at which 24nt sRNAs were measured (Figure 5c), the culture from normal ramet lineage 1 had lost methylation at the Bbv I (the site nearest the Karma splice acceptor site).
Supplementary Material
Acknowledgments
We acknowledge the contributions of staff members of the Breeding and Tissue Culture Unit at MPOB for creating the valuable clonal lines, and for their extensive data collection and sampling efforts. We thank Genomics Unit at MPOB for conducting DNA fingerprinting to verify clonal lines. We thank The McDonnell Genome Institute at Washington University for genomic bisulphite sequencing and transcriptome sequencing support, and MOgene for microarray hybridizations. At Orion Genomics, we thank Nathan Sander, Jerry Reed, Joel Brune, Khaing Soe, Jill McDonald, Clyde Brown and Barbara Dove for technical support, and Miin-Feng Wu and Mohit Sachdeva for assistance with the manuscript and additional informatics support. We would also like to thank Tamas Dalmay, University of East Anglia, UK for recommendations on small RNA library construction. We appreciate the constant support of the Director-General of MPOB, Datuk Dr. Yuen-May Choo, and the Ministry of Plantation Industries and Commodities, Malaysia.
Footnotes
Supplementary Information is available in the online version of the paper.
Data Deposition Statement Microarray data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE68410 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE68410). Small RNA sequence data from the region of interest have been deposited in NCBI’s SRA database under the accession numbers SAMN03569290 through SAMN03569351. Whole genome bisulphite sequence data have been deposited in NCBI’s SRA under accession numbers SAMN03569063 through SAMN03569077. The cDNA sequence of the kDEF1 transcript has been deposited in GenBank under accession number KR347486.
Author Contributions M.O.-A. led the work on the MANTLED marker/gene. M.O.-A. R.S., E.-T.L.L., and R.S. conceptualized the research programme. M.O.-A., R.S., E.-T.L.L., R.N., N.L., S.W.S., J.M.O., R.S. and R.A.M. developed the overall strategy, designed experiments and coordinated the project. A.T.H, Z.I. and S.K.R. performed tissue culture on selected ortets and field planted the ramets. Field data collection and fruit bunch census were conducted at various Research Stations by F.A.M., N.A.A.B., M.M., N.A., Z.Y. and M.D.A. M.O.-A., C.-C.L, X.A., C.-N.C., W.-C.W., S.S.R.S.A. and Y.-Y.K. identified samples for discovery and validation panels. M.O.-A., C.-C.L and X.A. identified materials used in the mosaic experiments. M.O.-A., S.-E.O., S.-Y.K., N.S. and N.A. conducted laboratory experiments, histological staging of inflorescences and data analyses. N.J. and S.W.S performed microarray analyses. B.B. and M.A.B., prepared fractions for microarray hybridizations. B.B. designed and analyzed qPCR experiments. B.B. and M.B. performed qPCR assays. A.V.B. designed and analyzed clone based bisulphite sequencing experiments, and A.V.B and M.B. performed bisulphite sequencing assays. M.B. designed and performed qRT-PCR experiments. C.W. and J.M.O. analyzed transcriptome data. K-L.C., N.A., S.W.S., M.H., C.W. and A.V.B. provided bioinformatics support. M.O.-A., R.S., E.-T.L.L., R.N., N.L., S.W.S., J.M.O., R.S. and R.A.M. prepared and revised the manuscript.
References
- 1.Stroud H, et al. Plants regenerated from tissue culture contain stable epigenome changes in rice. Elife. 2013;2:e00354. doi: 10.7554/eLife.00354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Corley RHV. Oil palm. In: Monselise SP, editor. CRC Handbook of fruit set and development. CRC Press; Boca Raton, Florida: 1986. pp. 253–259. [Google Scholar]
- 3.Zeven AC. The ‘mantled’ oil palm (Elaeis guineensis Jacq.) J W Afr Inst Oil Palm Res. 1973;5:31–33. [Google Scholar]
- 4.Mgbeze GC, Iserhienrhien A. Somaclonal variation associated with oil palm (Elaeis guineensis Jacq.) clonal propagation: A review. African Journal of Biotechnology. 2014;13:989–997. [Google Scholar]
- 5.Jaligot E, Rival A, Beule T, Dussert S, Verdeil JL. Somaclonal variation in oil palm (Elaeis guineensis Jacq.): the DNA methylation hypothesis. Pl Cell Rep. 2000;19:684–690. doi: 10.1007/s002999900177. [DOI] [PubMed] [Google Scholar]
- 6.Corley RHV, Law IH. The future for oil palm clones. In: Pushparajah E, editor. Plantation management for the 21st century. Incorp. Soc. Planters; Kuala Lumpur: 1997. pp. 279–289. [Google Scholar]
- 7.Singh R, et al. The oil palm SHELL gene controls oil yield and encodes a homologue of SEEDSTICK. Nature. 2013;500:340–344. doi: 10.1038/nature12356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adam H, et al. Functional characterization of MADS box genes involved in the determination of oil palm flower structure. J Exp Bot. 2007;58:1245–1259. doi: 10.1093/jxb/erl263. [DOI] [PubMed] [Google Scholar]
- 9.Rao V, Donough CR. Preliminary evidence of a genetic cause for the floral abnormalities in some oil palm ramets. Elaies. 1990;2:199–207. [Google Scholar]
- 10.Matthes M, Singh R, Cheah SC, Karp A. Variation in oil palm (Elaeis guineensis Jacq.) tissue culture-derived regenerants revealed by AFLPs with methylation-sensitive enzymes. Theoretical Applied Genetics. 2001;102:971–979. [Google Scholar]
- 11.Kubis SE, Castilho AM, Vershinin AV, Heslop-Harrison JS. Retroelements, transposons and methylation status in the genome of oil palm (Elaeis guineensis) and the relationship to somaclonal variation. Plant Mol Biol. 2003;52:69–79. doi: 10.1023/a:1023942309092. [DOI] [PubMed] [Google Scholar]
- 12.Jaligot E, et al. DNA methylation and expression of the EgDEF1 gene and neighboring retrotransposons in mantled somaclonal variants of oil palm. PLoS One. 2014;9:e91896. doi: 10.1371/journal.pone.0091896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Syed Alwee S, et al. Characterization of oil palm MADS box genes in relation to the mantled flower abnormality. Plant Cell, Tissue and Organ Culture. 2006;85:331–334. [Google Scholar]
- 14.Singh R, et al. Oil palm genome sequence reveals divergence of interfertile species in Old and New worlds. Nature. 2013;500:335–339. doi: 10.1038/nature12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lippman Z, et al. Role of transposable elements in heterochromatin and epigenetic control. Nature. 2004;430:471–476. doi: 10.1038/nature02651. [DOI] [PubMed] [Google Scholar]
- 16.Tanurdzic M, et al. Epigenomic consequences of immortalized plant cell suspension culture. PLoS Biol. 2008;6:2880–2895. doi: 10.1371/journal.pbio.0060302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Komatsu M, Shimamoto K, Kyozuka J. Two-step regulation and continuous retrotransposition of the rice LINE-type retrotransposon Karma. Plant Cell. 2003;15:1934–1944. doi: 10.1105/tpc.011809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cui X, et al. Control of transposon activity by a histone H3K4 demethylase in rice. Proc Natl Acad Sci U S A. 2013;110:1953–1958. doi: 10.1073/pnas.1217020110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martienssen R, Barkan A, Taylor WC, Freeling M. Somatically heritable switches in the DNA modification of Mu transposable elements monitored with a suppressible mutant in maize. Genes Dev. 1990;4:331–343. doi: 10.1101/gad.4.3.331. [DOI] [PubMed] [Google Scholar]
- 20.Cubas P, Vincent C, Coen E. An epigenetic mutation responsible for natural variation in floral symmetry. Nature. 1999;401:157–161. doi: 10.1038/43657. [DOI] [PubMed] [Google Scholar]
- 21.Saze H, Kakutani T. Heritable epigenetic mutation of a transposon-flanked Arabidopsis gene due to lack of the chromatin-remodeling factor DDM1. EMBO J. 2007;26:3641–3652. doi: 10.1038/sj.emboj.7601788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perbal MC, Haughn G, Saedler H, Schwarz-Sommer Z. Non-cell-autonomous function of the Antirrhinum floral homeotic proteins DEFICIENS and GLOBOSA is exerted by their polar cell-to-cell trafficking. Development. 1996;122:3433–3441. doi: 10.1242/dev.122.11.3433. [DOI] [PubMed] [Google Scholar]
- 23.Regulski M, et al. The maize methylome influences mRNA splice sites and reveals widespread paramutation-like switches guided by small RNA. Genome Res. 2013;23:1651–1662. doi: 10.1101/gr.153510.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baubec T, Finke A, Mittelsten Scheid O, Pecinka A. Meristem-specific expression of epigenetic regulators safeguards transposon silencing in Arabidopsis. EMBO Rep. 2014;15:446–452. doi: 10.1002/embr.201337915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taniguchi-Ikeda M, et al. Pathogenic exon-trapping by SVA retrotransposon and rescue in Fukuyama muscular dystrophy. Nature. 2011;478:127–131. doi: 10.1038/nature10456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alló M, et al. Control of alternative splicing through siRNA-mediated transcriptional gene silencing. Nat Struct Mol Biol. 2009;16:717–724. doi: 10.1038/nsmb.1620. [DOI] [PubMed] [Google Scholar]
- 27.Arteaga-Vazquez M, et al. RNA-mediated trans-communication can establish paramutation at the b1 locus in maize. Proc Natl Acad Sci U S A. 2010;107:12986–12991. doi: 10.1073/pnas.1007972107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sommer H, et al. Deficiens, a homeotic gene involved in the control of flower morphogenesis in Antirrhinum majus: the protein shows homology to transcription factors. EMBO J. 1990;9:605–613. doi: 10.1002/j.1460-2075.1990.tb08152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jack T, Brockman LL, Meyerowitz EM. The homeotic gene APETALA3 of Arabidopsis thaliana encodes a MADS box and is expressed in petals and stamens. Cell. 1992;68:683–697. doi: 10.1016/0092-8674(92)90144-2. [DOI] [PubMed] [Google Scholar]
- 30.Lamb RS, Irish VF. Functional divergence within the APETALA3/PISTILLATA floral homeotic gene lineages. Proc Natl Acad Sci U S A. 2003;100:6558–6563. doi: 10.1073/pnas.0631708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones LH. Propagation of clonal oil palms by tissue culture. Planter, Kuala Lumpur. 1974;50:374–381. [Google Scholar]
- 32.Rabechault H, Martin J-P. Multiplication vegetative du palmier a huile (Elaeis guineensis Jacq.) a l’aide de cultures de tissues foliares. C R Acad Sci Paris, Ser D. 1976;283:1735–1737. [Google Scholar]
- 33.Durand-Gasselin T, Le Guen V, Konan K, Duval Y. Oil palm (Elaeis guineensis Jacq.) plantations in Cote-d’Ivoire obtained through in vitro culture. First results. Oléagineux. 1990;45:10–11. [Google Scholar]
- 34.Kushairi A, et al. Current status of oil palm tissue culture in Malaysia. Proceedings of the Clonal and Quality Replanting Material Workshop. Towards Increasing the Annual National Productivity by One Tonne FFB/ha/year; 2006. pp. 3–14. [Google Scholar]
- 35.Rohani O, et al. Tissue culture of oil palm. In: Yusof \B, Jalani BS, Chan KW, editors. Advances in Oil Palm Research. Malaysian Palm Oil Board; 2000. pp. 238–283. [Google Scholar]
- 36.Adam H, et al. Determination of flower structure in Elaeis guineensis: do palms use the same homeotic genes as other species? Ann Bot. 2007;100:1–12. doi: 10.1093/aob/mcm027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stewart FJ, Raleigh EA. Dependence of McrBC cleavage on distance between recognition elements. Biol Chem. 1998;379:611–616. [PubMed] [Google Scholar]
- 38.Ordway JM, et al. Comprehensive DNA methylation profiling in a human cancer genome identifies novel epigenetic targets. Carcinogenesis. 2006;27:2409–2423. doi: 10.1093/carcin/bgl161. [DOI] [PubMed] [Google Scholar]
- 39.Chan PL, et al. Evaluation of reference genes for quantitative real-time PCR in oil palm elite planting materials propagated by tissue culture. PLoS ONE. 2014;9 doi: 10.1371/journal.pone.0099774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaligot E, et al. Epigenetic imbalance and the floral developmental abnormality of the in vitro-regenerated oil palm Elaeis guineensis. Ann Bot. 2011;108:1453–1462. doi: 10.1093/aob/mcq266. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.