

Abstract
DNA testing is available for a growing number of hereditary diseases in neurology and other specialties. In addition to guiding breeding decisions, DNA tests are important tools in the diagnosis of diseases, particularly in conditions for which clinical signs are relatively nonspecific. DNA testing also can provide valuable insight into the risk of hereditary disease when decisions about treating comorbidities are being made. Advances in technology and bioinformatics will make broad screening for potential disease‐causing mutations available soon. As DNA tests come into more common use, it is critical that clinicians understand the proper application and interpretation of these test results.
Keywords: Genetic mapping, Genetic markers, Genetics, Neurology
Abbreviations
- DM
degenerative myelopathy
- NCL
neuronal ceroid lipofuscinosis
Veterinary medicine has entered the age of genomics. Advances in technology and bioinformatics have made information found in the DNA of our patients available at a rapidly decreasing cost. The unique population structure of purebred animals makes them ideally suited for gene discovery strategies. Lesser concerns about privacy and insurance, as well as greater control over breeding decisions allow the direct application of DNA information to decision making in veterinary medicine much easier than in human medicine. This means, however, that the veterinary community must become well versed in DNA testing and utilize those test results wisely. Perhaps nowhere is this potential greater than in diseases of the nervous system. Even with advances in imaging, the relative inaccessibility of the nervous system increases the value of additional diagnostic approaches such as DNA testing, although no diagnostic test can substitute for a thorough history and neurologic examination. Although in the past, genetic diseases often were considered only after other etiologies were excluded, the increasing availability of DNA testing permits earlier consideration of these conditions in the diagnostic plan.
We will discuss the principles and value of DNA testing in the practice of veterinary neurology. Although we focus on neurologic disease, the principles involved could apply to any other organ system. We will review some characteristics of hereditary diseases, the basic principles and potential pitfalls of DNA testing, and finally discuss how DNA testing is changing clinical neurology practice.
Some Characteristics of Hereditary Diseases
To select appropriate DNA testing, the clinician must first recognize when the clinical signs suggest a hereditary disease. Each hereditary disease is unique, but some general features of a disease will raise suspicion of a hereditary cause. Table 1 shows some of the characteristic features of several categories of hereditary neurologic diseases.1
Table 1.
General characteristics of some hereditary neurologic diseases.1
| Congenital malformations | Neonatal or young onset of signs |
| Static or progressive signs | |
| Diagnosed on advanced imaging or necropsy | |
| Organic acidurias | Neonatal or young onset |
| Waxing and waning encephalopathy | |
| Dietary influences | |
| Ketonuria, acidosis or anion gap, hypoglycemia, hyperammonemia | |
| Diagnosed on urine organic acid screens | |
| May see symmetric signal changes on MRI | |
| Lysosomal storage diseases | Neonatal to middle age onset |
| Varying signs of ataxia, blindness, weakness, dementia, seizures | |
| Inexorably progressive course | |
| Diagnosed on necropsy or occasionally liver or leukocyte inclusions, urine, or CSF metabolites | |
| Channelopathies | Neonatal to adult onset |
| Altered excitability of muscle or nerves | |
| Signs of myopathy, collapse, ataxia, or seizures | |
| May be episodic | |
| Diagnosed on clinical signs, electrodiagnostics or both | |
| Neurodegenerative diseases | Young to old age onset and progressive course |
| Selective or diffuse degeneration of neurons, myelin, or muscle | |
| May see selective brain atrophy on MRI Degenerative changes on biopsy or necropsy |
A high incidence of a disease in 1 breed or inbred line suggests a hereditary basis of the disease. It does not, however, necessarily reflect poor breeding practices. Inbreeding does not cause genetic disease per se, but makes the expression of recessive traits more likely. This could be a desirable trait that the breeder is trying to select for or an undesirable trait that also is segregating in the breed. A popular sire inevitably will be a silent carrier of undesirable traits as well as the desirable ones that made the animal popular. Unfortunately, the animal can pass both on to many offspring by frequent natural breeding or artificial insemination, but it may take several generations before a recessive genetic disease is recognized. Also, other factors such as the use of the breed such as in agility or search and rescue, the husbandry practices of breeders, or other environmental factors also could cause a disease to be particularly prevalent within a breed. Thus, all differential diagnoses for a presenting complaint must be considered (Fig 1). Nonetheless, if a purebred dog presents with a possible hereditary disease, references for breed‐associated diseases should be consulted.2, 3, 4 If the disease has been associated with that breed, then further search of the literature would be needed to determine how strong the evidence is for a hereditary basis and whether or not a specific mutation has been identified.
Figure 1.
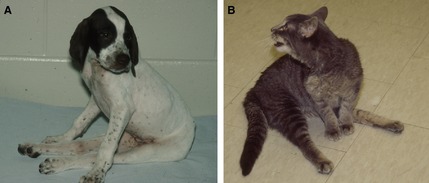
Two young animals presented for progressive, spastic paraplegia illustrate the importance of considering other differential diagnoses in suspected hereditary disease. (A) A purebred English Pointer with a history of affected littermates suggested a hereditary disease but was diagnosed with Neospora caninum infection. (B) A stray cat from the streets of St. Louis came from a random breeding population, but was diagnosed with a hereditary muscular dystrophy. From O'Brien 201275 used with permission.
Many hereditary diseases are congenital or have a young age of onset. Development of a complex organ like the nervous system requires the precise coordination of many processes which are regulated by different genes. If one of these processes is altered by a mutation, then development may not proceed normally. The effects often will be apparent in the neonate. Unfortunately, neonatal diseases have received relatively little attention in veterinary medicine. Many breeders are unwilling to invest in veterinary care for neonates and often view losses associated with “runts” or small litter size as something to be accepted. Neonatal diseases, however, can provide an ideal subject for gene mapping studies because the phenotype often is easily recognized and the entire family often is available for DNA sampling. Eliminating the disease not only prevents animal suffering, but also can decrease financial losses for the breeder.
Sometimes, developmental abnormalities have a delayed onset of signs. Different parts of the nervous system develop at different rates, and the genes controlling a process can change during different stages of development. For example, disorders of myelin development may not be apparent until the animal relies on adequate myelination to begin walking. Conversely, sometimes a neonatal form of a protein will be replaced by an adult form resulting in the animal “growing out of” the condition. Dachshunds with congenital myasthenia gravis show clinical signs and decreased acetylcholine receptor density as neonates, but signs resolve by 6 months of age. It is suspected that signs resolve as a defective embryonic subunit of the acetylcholine receptor (the λ subunit) is replaced by an adult form (the ε subunit).5 Early onset of a disease more commonly seen in adulthood, such as laryngeal paralysis, also would raise suspicion of a hereditary etiology.
Other hereditary diseases may have a more delayed onset of signs. Animals with lysosomal storage diseases are deficient in the necessary enzyme from birth. Because the lysosome is primarily involved in recycling cellular components, development may proceed normally, but storage product accumulates in the lysosome over time, ultimately affecting neuronal function. If the mutation only partially affects enzyme activity, then clinical signs may be even further delayed. For example, American Bulldogs with the lysosomal storage disease neuronal ceroid lipofuscinosis (NCL) have 36% of normal activity of the affected enzyme cathepsin D.6 This residual activity appears to be enough to delay onset of signs until 1–3 years of age and the signs progress slowly. In contrast, sheep with <5% cathepsin D activity shows signs as neonates.7
Some mutations predispose the animal to a neurodegenerative process. In these cases, the nervous system develops normally but then undergoes premature degeneration. Some of these mutations can be very selective, such as the cerebellar ataxias where the Purkinje cells are the only cell type affected.8, 9 In other diseases, such as multiple system degeneration in Kerry Blue Terriers and Chinese Crested Dogs, multiple cell types are vulnerable to degeneration.10 In these cases, symmetrical signs and a fairly stereotypical onset and progression of signs are the hallmarks of hereditary disease.
Degenerative myelopathy (DM) appears to be a condition in which the mutation increases the animal's risk for neurodegeneration much later in life. In the study by Awano et al11 virtually all dogs with necropsy‐confirmed DM were homozygous for a mutation in the SOD1 gene, suggesting that the mutant allele was necessary to develop the disease. However, not all animals that were homozygous for the mutation developed the disease; a phenomenon called variable penetrance discussed further below.
Advances in genetic research have now made it possible to identify the mutations responsible for many of these diseases, and the numbers are growing rapidly. To utilize DNA testing effectively, however, the clinician must be able to select the test and interpret results appropriately. This requires an understanding of the terminology used, the types of tests available, and potential pitfalls of DNA testing.
Nomenclature: Speaking the Language of Genetics and Genomics
The nomenclature used in genetics and genomics is something new to many veterinarians. It is important, however, for the clinician to understand the language of DNA testing, and thus we will briefly review how genes are named, some of the types of mutations that occur, and how a mutation would be defined. Box 1 defines some of the commonly used terms. The names of genes are written as acronyms that are italicized to differentiate them from the names of proteins or other acronyms. The convention is to use all capital letters for human genes and lower case after the first letter for rodents. Domestic animal genes have followed the convention used for human genes. Like many things in biology and medicine, the names applied to genes evolve over time as we learn more about their biology, which can lead to confusion when the names vary in the literature.
Box 1.
Definitions of some terms used in genetics
Phenotype: the physical manifestations of a trait which can be influenced by genetics, epigenetics, and environment.
Genotype: the genetic makeup of an individual with respect to the gene being considered (the alleles).
Allele: one of alternate forms of a gene or locus.
Segregation: the separation of alleles during meiosis which determines which allele is passed to an offspring.
Recombination: the 2 copies of a chromosome may cross over during meiosis which results in exchange of genetic material and a new combination of alleles.
Locus: the location of a gene or genes on a chromosome; often used as a vague proxy for gene in gene mapping studies before the specific gene is identified.
Codon: the 3 nucleotide code for an amino acid or the initiation or stop of protein translation.
Variant: variation in the DNA sequence of an individual from the reference sequence. This could be a disease‐causing mutation or it could be neutral.
Single nucleotide polymorphisms (SNPs); variants where only a single nucleotide is substituted (eg, A>G); SNPs are the most common type of variant and are for example used to identify differences among individual chromosomes for association studies.
Frame shift: deletion or insertion of any number of nucleotides not divisible by 3 shifting the reading frame of the codons which follow. This in turn changes the subsequent amino acid sequence and the location of the stop codon.
Many genes are named based on the disease phenotype. For example, the NCLs are a group of lysosomal storage diseases characterized by accumulation in neurons of autofluorescent proteinaceous material that resembles ceroid and lipofuscin. The genes involved are named the CLN genes (Ceroid Lipofuscinosis, Neuronal). The genes often are numbered consecutively as different phenotypes or different modes of inheritance were recognized in humans. Thus, CLN1, CLN2, CLN3, and CLN4 are the first 4 phenotypes described and are differentiated by age of onset: infantile, late‐infantile, juvenile, and adult, respectively. CLN4 is further subdivided into CLN4A and CLN4B depending on whether the inheritance is autosomal recessive or dominant. If phenotypes are mapped to different loci in independent families, the underlying genes are given separate numbers. For example, CLN6 mutations cause late‐infantile onset NCL but the CLN6 gene mapped to a different chromosome than CLN2. Thus, this form must be associated with a different gene and was given a different number.
Alternatively, the name of a gene may reflect a biochemical or structural component of the nervous system such as an enzyme or ion channel. In this instance, the acronym will abbreviate the molecule and often will be followed by a number or letter referring to a subunit or subtype. For example, GRM1 refers to the gene that codes for the glutamate receptor, metabotropic, type 1. Often, as more is learned about a disease and gene, the molecular name replaces the phenotype‐based name. For example, the original name for GRM1 was SCAR13, the 13th form of autosomal recessive spinocerebellar ataxia to be recognized in humans.
Specific mutations in a gene are named based on the amino acid or nucleotide affected and the change the mutation produces. Because the term mutation was used in various contexts, geneticists today prefer the term variant to describe any difference in nucleotide or amino acid sequence, regardless of whether it is common or rare and regardless whether it is functionally neutral or causally involved in a specific phenotype. For the purposes of this manuscript, we use the term mutation to refer to a disease‐causing variant. The term polymorphism is used to refer to a nondisease‐causing variant or a variant with a high frequency in the population.
A wide variety of changes in the DNA sequence can occur. Missense mutations change a codon so that a different amino acid is specified. This alters the primary protein structure which may affect function. A nonsense mutation changes an amino acid‐specifying codon to a premature stop codon, which results in a truncated and usually nonfunctional protein. A variable number of nucleotides may be inserted or deleted by a mutation and produce a frame shift in the codons following that change. In addition to altering the subsequent amino acid sequence, frame shifts often lead to a premature stop codon. Retrotransposons, retroviral like sequences that can move within the genome, are 1 cause of insertions. Most disease‐causing variants are in the coding region (exons) of the gene, but some can affect splice sites. These can result in abnormal splicing of exons during transcription. Many other types of variants, such as noncoding regulatory variants also can be functionally relevant, but are difficult to identify with current technology.
The Human Genome Variation Society provides recommendations for the nomenclature describing variants at their website http://www.hgvs.org/mutnomen/recs.html,12 and some examples are given in Table 2. Although the specifics of the nucleotide change may not be of interest to clinicians, they must recognize that SOD1:c.118G>A and SOD1:c.52A>T refer to different variants just as ALT and ALP refer to different liver function tests.
Table 2.
Examples of nomenclature to describe disease‐causing variants (mutations)
| Change Described | Variant | Convention | Example | ||
|---|---|---|---|---|---|
| Disease | Mutation | Explanation | |||
| Change in DNAa | DNA Substitution | Position of the nucleotide, wild‐type nucleotide, “>”, replacement nucleotide | Exercise‐induced collapse in Labrador Retrievers21 | DNM1:c.767G>T | Substitution of T for G at coding position 767 in the dynamin 1 gene |
| DNA Insertion or deletion | Underlined space between nucleotides, “ins” or “del”, nucleotide(s), or number for large ins/del | Polyneuropathy of juvenile Greyhounds25 | NDRG1:c.1080_1089 del TCGCCTGGAC | Deletion of TCGCCTGGAC between position 1080 and 1089 in the N‐myc downstream‐regulated gene 1 | |
| Bandera's neonatal ataxia in Coton de Tulears18 | GRM1:c.2316_2317ins62 | Insertion of 62 base pairs between position 2316 and 2317 in the glutamate receptor, metabotrophic 1 gene | |||
| Change in proteinb | Amino acid substitution (missense) | Position of the amino acid, wild‐type amino acid, substituted amino acid | Degenerative myelopathy in most breeds6 | SOD1:p.E40K or SOD1:p.Glu40Lys | Substitution of lysine (K) for glutamic acid (E) at position 40 in superoxide dismutase 1 protein |
| Premature stop (nonsense) | Position of the amino acid, wild‐type amino acid, “X” or “*” | Remitting focal epilepsy in Lagotto Romagnolo26 | LGI2:p.K518X or LGI2:p.Lys518* | Nonsense variant changes lysine at position 518 to a stop codon truncating the leucine‐rich glioma inactivated protein 2 | |
| Frame shift | Last normal amino acid, position, first variant amino acid, “fs*”, length of remaining altered amino acid sequence including the stop codon | Cerebellar cortical degeneration in Beagles8 | SPTBN2:p.G1952Rfs*27 | A deletion or insertion causes a frame shift changing glycine 1952 into arginine and an aberrant protein thereafter, which terminates after another 26 amino acids. The *27 means that a stop codon occurs at the 27th position after the first mutant amino acid. | |
In the mutation description, “:c.” indicates that the number refers to the DNA coding sequence (exons).
In the mutation description, “:p.” indicates that the number refers to the amino acid in the protein.
DNA Tests: Types of Test and Their Advantages and Disadvantages
Linkage mapping or association studies demonstrate a relationship among genetic markers at established chromosomal positions and the mutation responsible for a disease. This mapping establishes the locus, the chromosomal location where additional studies will be focused for identifying the specific causal mutation. Until a specific mutation is identified, the linked markers can be used to identify carriers of the mutation within a family. Linked marker DNA tests, however, need to be interpreted with caution because they can yield false‐positive and false‐negative results as illustrated in Figure 2.
Figure 2.
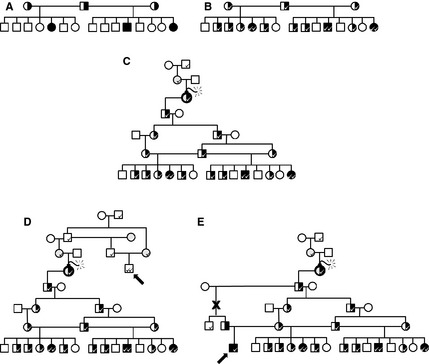
Linked marker DNA tests are useful in a family with known disease but can give false‐negative and false‐positive results as illustrated. (A) In this hypothetical pedigree of a recessive trait, males are squares and females are circles. Affected dogs are shown as filled symbols and carriers are half‐filled symbols. Parents of affected dogs are obligate carriers but genotype of normal offspring would be unknown. (B) A linked marker ( ) will segregate with the mutant allele and can be used to identify carriers within a family with known disease. (C) The marker allele could have been present within the family before the mutation occurred (
) will segregate with the mutant allele and can be used to identify carriers within a family with known disease. (C) The marker allele could have been present within the family before the mutation occurred ( ) which produced the disease‐causing allele. (D) Because the marker can segregate in the breed separate from the mutant allele, false‐positive results are possible (arrow: a dog that is normal but identified as affected by linkage). (E) Because the marker is only linked to the mutant allele, recombinations (X) can break that link leading to false results (arrow: a dog that is affected but identified as a carrier).
) which produced the disease‐causing allele. (D) Because the marker can segregate in the breed separate from the mutant allele, false‐positive results are possible (arrow: a dog that is normal but identified as affected by linkage). (E) Because the marker is only linked to the mutant allele, recombinations (X) can break that link leading to false results (arrow: a dog that is affected but identified as a carrier).
Once a linkage is identified, candidate genes within the locus are prioritized for further investigation based on biologic significance. For example, if a peripheral neuropathy was being investigated, genes affecting myelin production or axonal transport would be prime candidates. These genes then would be sequenced and variants evaluated for their effect on function. Whole‐genome sequencing has become cost‐effective as a tool to sequence all of the genes within a locus. When combined with bioinformatic techniques that permit comparison between cases and the whole‐genome sequences of controls, variants unique to the particular disease can be identified without first mapping a locus.13
The variants likely to affect protein function, such as a premature stop codon, a frame shift, or the substitution of an evolutionarily conserved amino acid then would be evaluated further. The first step would be to determine if the variant is significantly concordant with the phenotype in the families. For a recessive trait, all affected dogs and no normal dogs should be homozygous for the variant whereas the parents of affected dogs should all be heterozygous. A disease‐causing variant also should be absent or very rare in breeds that do not segregate the disease being investigated, and often a random sampling of DNA from other breeds is evaluated to test this hypothesis. Screening a broader population of the affected breed for the variant can give an estimate of the frequency of the allele in the breed, but often the DNA samples available constitute a highly biased sample. Further validation is required to show that the variant indeed is causing the disease and not just a functionally neutral, but tightly linked, marker. For example, immunohistochemistry may demonstrate that a candidate protein is not expressed or enzyme function assays may show a lack of activity. Similarities between the phenotype of the disease under study and either human diseases associated with that gene or transgenic mouse models would further support causation. Some discoveries will be novel genes about which limited information is available. Definitive proof for the causality of a variant in a novel gene is difficult to obtain and often beyond the scope of funding available for most veterinary diseases. Definitive proof of causality may be obtained by an in‐depth functional analysis of relevant biomarkers or by genetic approaches, which exploit special family structures occasionally seen in purebred domestic animals. DNA tests may become available at various stages of validation, and the clinician should evaluate the published evidence supporting the validity.
Once the causative variant in a gene has been identified, a DNA test which will be very sensitive and specific for the variant can be devised. Such tests frequently are termed “direct tests.” Even with such a specific DNA test, however, false results can occur because of phenocopy or incomplete penetrance.
The term phenocopy refers to a phenotype that is very similar to the trait under investigation but has a different cause. Sometimes it results from an acquired disease that mimics the signs of the hereditary disease. For example, canine distemper encephalitis could affect the cerebellum and produce cerebellar ataxia that appears identical to a hereditary cerebellar ataxia. This situation highlights the need to consider nonhereditary differential diagnoses and additional diagnostic tests.
Other mutations that produce similar clinical signs also can produce a phenocopy. Two virtually identical forms of NCL have been identified in Dachshunds that are caused by deficiencies of 2 different lysosomal enzymes, tripeptidyl peptidase 1 and palmitoyl‐protein thioesterase 1, and mutations have been identified in the genes that code for these enzymes, the TPP1 and PPT1 genes, respectively.14, 15 As illustrated in Figure 3, these lysosomal enzymes cleave proteins at different sites. Regardless of which site fails to get cleaved, however, processing of the protein is blocked, the autofluorescent protein byproduct builds up within the lysosome, and disease results. A DNA test for 1 mutation would produce a “false‐negative” result if NCL was caused by the alternative mutation. Thus, a Dachshund presenting between 7 and 10 months of age with progressive behavior changes, blindness, ataxia, and myoclonus should be tested for both mutations.
Figure 3.
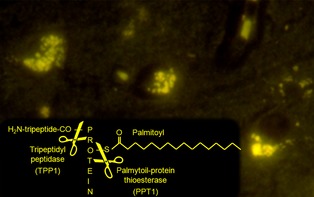
Within the lysosome, tripeptidyl peptidase 1 (TPP1) and palmatoyl‐protein thioesterase 1 (PPT1) contribute to protein degradation by cleaving off different portions of a protein, the N‐terminal tripeptide chain, and a palmitoyl fatty acid, respectively. A deficiency of either enzyme blocks degradation of the protein. Thus, mutations in either gene that codes for these enzymes (TPP1 and PPT1 respectively) lead to an identical lysosomal storage disease characterized by autofluorescent inclusions in neurons of the undegraded protein. Both diseases have been reported in Dachshunds.14, 15 A DNA test would detect the mutation in 1 gene, but not the other potentially leading to a “false‐negative” result.
Different mutations within the same gene also can produce phenocopy. In most breeds (including Bernese Mountain Dogs), a mutation in the gene that codes for superoxide dismutase 1 (SOD1:c.118G>A) is associated with DM.11 A few Bernese Mountain Dogs, however, were necropsy‐confirmed to have DM but were clear of the SOD1:c.118G>A mutation. Sequencing of the SOD1 gene from these dogs identified a different mutation, SOD1:c.52A>T, in the same gene.16 Because the DNA test specifically identifies the SOD1:c.118A allele, it would not detect the SOD1:c.52T allele. Thus, tests for both alleles are necessary in Bernese Mountain Dogs. Multiple mutations in a gene are common in human genetic diseases. This situation can lead to disease produced by compound heterozygosity. Here, the animal would have 1 mutant allele interfering with the gene function on the chromosome from 1 parent (eg, SOD1:c.52T) whereas the chromosome from the other parent contained a different mutant allele that also interferes with the function of that gene (eg, SOD1:c.118A). Thus, the animal is at risk for developing DM but would be identified as a carrier in either DNA test alone. NCL in Dachshunds and DM in Bernese Mountain Dogs represent examples of 2 different types of genetic heterogeneity. In NCL, there are 2 different mutations in 2 different genes (loci) leading to virtually the same clinical phenotype, this is termed “locus heterogeneity”. In DM, there are 2 different mutations affecting the same gene and leading to the same phenotype. This situation is termed as “allelic heterogeneity”.
Degenerative myelopathy also illustrates a cause of false‐positive DNA test results: incomplete penetrance. Although almost all dogs with DM are homozygous for the SOD1 mutation, not all dogs with the mutation develop disease.11 Because the average age of onset of DM is 9–11 years of age, many dogs with the SOD1 mutation die of other causes before they reach that age. However, there are well‐documented cases of dogs that are homozygous for the mutant allele living to 14–15 years of age without developing signs of DM. In these dogs, the mutation has not “penetrated” and produced disease. There are a number of possible explanations for incomplete penetrance. The SOD1 mutation may make the dog more susceptible to cumulative environmental stress that leads to neurodegenerative changes once a threshold has been exceeded, or an environmental trigger of the disease may be needed and the surviving dog may not have been exposed to that trigger. Alternatively, modifying genes may exist that affect the expression of the SOD1 mutation in some dogs.
Although incomplete penetrance is best documented in DM, it can occur in other genetic diseases as well. DNA test results typically report animals that are homozygous for a recessive disease‐associated allele as “at risk.” This recognizes the potential for incomplete penetrance and further emphasizes that a diagnosis can only be made based on appropriate clinical signs and other diagnostic tests. An animal that is heterozygous for a mutant allele would be reported as “carrier.” An animal that test homozygous for the normal (wild‐type or ancestral) allele may be reported as “clear.”
Using DNA Tests in Breeding Decisions
The most straightforward application of DNA testing is to direct breeding decisions. Once a DNA test based on a specific mutation in a breed has been developed, breeders will be able to detect carriers of that particular mutation with great accuracy. The first inclination is to recommend eliminating carriers of a mutant allele from the breeding population. Although this would be the fastest way to decrease the frequency of the allele in the population, doing so can have unwanted consequences. Some breeds have a relatively small gene pool, and with some diseases, the mutant allele frequency in the population can be high. Eliminating all of the carriers in these situations can create a detrimental bottle neck in the gene pool of the breed. Every dog will be carrying some potentially deleterious alleles, and restricting the gene pool could lead to the emergence of a different hereditary disease. Often a hereditary disease becomes widespread in a breed because of the “popular sire” effect discussed above. Eliminating those lines from the gene pool will decrease the frequency of the alleles that contribute to the desirable traits that made that sire popular as well as the undesirable one being targeted.
A more rational breeding strategy attempts to retain these desirable traits and the equally important genetic diversity in the breed. Dogs that are identified as carriers still can be bred, but they must always be bred to a dog that has tested clear of the mutant allele so that no affected dogs are produced. The offspring of such breeding then should be tested because 50% will carry the mutant allele. Genotype then should be one of the factors that determine which of those offspring are used for future breeding stock. If a carrier of the mutant allele has other desirable traits, they still can be bred as long as they are not bred to another carrier. If all other things are equal, a dog that is clear of the mutant allele would be the better choice for future breeding stock. Thus, no affected dogs are produced, but genetic diversity and other desirable traits are not compromised in the process. Over time, the frequency of the mutant allele in the population will decrease as more clear dogs are selected for future breeding.
Using DNA Tests in Clinical Decisions
DNA Tests as Diagnostic Tools
A number of neurologic diseases have now been associated with specific mutations in dogs and cats (Tables 3 and 4), and the number undoubtedly will increase as gene discovery becomes more efficient. DNA tests can be used like other diagnostic tests to help establish or eliminate differential diagnoses for a particular presenting complaint, such as cerebellar ataxia or episodic weakness.
Table 3.
Neurologic diseases in dogs with their known underlying molecular defects
| Phenotype | Gene | Variant | Breed | OMIA | Reference |
|---|---|---|---|---|---|
| Alpha fucosidosis | FUCA1 | c.376_389del14 | English Springer Spaniel | 000396‐9615 | 27, 28 |
| Bandera's neonatal cerebellar ataxia | GRM1 | c.2316_2317ins62 | Coton de Tulear | 000078‐9615 | 18 |
| Cerebellar abiotrophy (spinocerebellar ataxia type 5) | SPTBN2 | c.5921_5928del8 | Beagle | 000175‐9615 | 8 |
| Cerebellar ataxia | SEL1L | c.1972T>C | Finnish Hound | 001692‐9615 | 9 |
| Congenital myasthenic syndrome | CHAT | c.622G>A | Old Danish Pointing Dog | 000685‐9615 | 29 |
| Degenerative myelopathy | SOD1 | c.118G>A | Many | 000263‐9615 | 11 |
| SOD` | c.51A>T | Bernese Mountain Dog | 000263‐9615 | 16 | |
| Encephalopathy | SLC19A3 | c.624 insTTGC | Alaskan Husky | 001097‐9615 | 30 |
| Episodic falling | BCAN | 16 kb deletion | Cavalier King Charles Spaniel | 001592‐9615 | 31 |
| Exercise‐induced collapse | DNM1 | c.767G>T | Labrador Retriever | 001466‐9615 | 21 |
| Globoid cell leukodystrophy (Krabbe disease) | GALC | c.473A>C | Cairn Terrier/West Highland White Terrier | 000578‐9615 | 32 |
| GALC | c.790_791ins78 | Setter | 000578‐9615 | 33 | |
| GM1 gangliosidosis | GLB1 | c.1688_1706dup19 | Alaskan Husky | 000402‐9615 | 34 |
| GLB1 | c.179G>A | Portugese Water Dog | 000402‐9615 | 35 | |
| GLB1 | c.1647delC | Shiba | 000402‐9615 | 35 | |
| GM2 gangliosidosis (Tay Sachs disease) | HEXA | c.967G>A | Japanese Chin Dog | 001461‐9615 | 36 |
| GM2 gangliosidosis (Sandhoff disease) | HEXB | c.283delG | Toy Poodle | 001462‐9615 | 37 |
| Juvenile benign epilepsy | LGI2 | c.1552A>T | Lagotto Romagnolo | 001596‐9615 | 26 |
| L‐2‐hydroxyglutaric aciduria | L2HGDH | c[1297T>C;1299C>T] | Staffordshire Bullterrier | 001371‐9615 | 38 |
| L2HGDH | c.1A>G | Yorkshire Terrier | 001371‐9615 | 39, 40 | |
| Mucopolysaccharidosis I | IDUA | c.155+1G>A | Plott Hound | 000664‐9615 | 41 |
| Mucopolysaccharidosis IIIA | SGSH | c.708_709insC | New Zealand Huntaway | 001309‐9615 | 42 |
| Mucopolysaccharidosis IIIB | NAGLU | Insertion | Schipperke | 001342‐9615 | 43 |
| Mucopolysaccharidosis VI | ARSB | Exon 5 G>A | Miniature Pinscher | 000666‐9615 | 1 |
| ARSB | c.103_124del22 | Miniature Poodle | 000666‐9615 | 44 | |
| Myoclonus epilepsy (Lafora disease) | NHLRC1 | 12 bp repeat expansion | Dachshund | 000690‐9615 | 45 |
| Myotonia | CLCN1 | c.803C>T | Miniature Schnauzer | 000698‐9615 | 46 |
| CLCN1 | c.2665_2666insA | Australian Cattle Dog | 000698‐9615 | 47 | |
| Narcolepsy | HCRTR2 | SINE insertion intron 3 | Doberman Pinscher | 000703‐9615 | 48 |
| HCRTR2 | c.1105+5G>A | Labrador Retriever | 000703‐9615 | 48 | |
| HCRTR2 | c.160G>A | Dachshund | 000703‐9615 | 49 | |
| NCL, adult onset | ATP13A2 | c.1623delG | Tibetan Terrier | 001552‐9615 | 50, 51 |
| NCL 1 | PPT1 | c.736_737insC | Dachshund | 001504‐9615 | 15 |
| NCL 2 | TPP1 | c.325delC | Dachshund | 001472‐9615 | 14 |
| NCL 4a | ARSG | c.296G>A | Am. Staffordshire Terrier | 001503‐9615 | 52 |
| NCL 5 | CLN5 | c.619C>T | Border Collie | 001482‐9615 | 53 |
| NCL 6 | CLN6 | c.829T>C | Australian Shepherd | 001443‐9615 | 54 |
| NCL 8 | CLN8 | c.491C>T | English Setter | 001506‐9615 | 55 |
| NCL 10 | CTSD | c.597G>A | American Bulldog | 001505‐9615 | 6 |
| Neonatal encephalopathy with seizures | ATF2 | c.152T>G | Poodle | 001471‐9615 | 56 |
| Neuroaxonal dystrophy | MFN2 | c.1617_1619delGGA | Giant Schnauzer | 000715‐9615 | 57 |
| Polyneuropathy | NDRG1 | c.1080_1089del10 | Greyhound | 001292‐9615 | 25 |
| NDRG1 | c.293G>T | Alaskan Malamute | 001292‐9615 | 58 | |
| Polyneuropathy, LPN1 | ARHGEF10 | c.1955_1958+6del10 | Leonberger & St. Bernard | 001917‐9615 | –2 |
| Sensory ataxic neuropathy | MT‐TY (tRNA‐Tyr) | mtDNA:g.5304delT | Golden Retriever | 001467‐9615 | 59 |
| Shaking pup, tremor X‐linked | PLP1 | c.110A>C | Springer Spaniel | 000770‐9615 | 60 |
| Spinocerebellar ataxia (late onset) | CAPN1 | c.344C>T | Parson Russell Terrier & Jack Russell Terrier | 001820‐9615 | 19 |
| Spinocerebellar ataxia with myokymia, seizures, or both | KCNJ10 | c.627C>G | Jack Russell Terrier, Parson Russell Terrier & Russell Terrier | – | 13 |
| Spongiform leukoencephalomyelopathy | CYTB | mtDNA:g.14474G>A | Australian Cattle Dog/Shetland Sheepdog | 001130‐9615 | 61 |
| Startle disease (hyperekplexia) | SLC6A5 | 4.2 kb deletion | Irish setter | 001594‐9615 | 62 |
OMIA, Online Mendelian Inheritance in Animals.2
Table 4.
Neurologic diseases in cats with their known underlying molecular defects
| Phenotype | Gene | Variant | Breed | OMIA | Reference |
|---|---|---|---|---|---|
| GM1 gangliosidosis | GLB1 | c.1448G>C | Siamese, Korat, South‐East Asian native cats | 000402‐9685 | 63 |
| GM2 Gangliosidosis, GM2A deficiency | GM2A | c.516_519delGGTC | Domestic Shorthair | 001427‐9685 | 64 |
| GM2 Gangliosidosis, type II (Sandhoff disease) | HEXB | c.39delC | Korat | 001462‐9685 | 65 |
| HEXB | c.1467_1491inv25 | Domestic Shorthair | 001462‐9685 | 66 | |
| HEXB | c.667C>T | Japanese Domestic Cat | 001462‐9685 | 67 | |
| HEXB | c.1244‐8_1250del15 | Burmese | 001462‐9685 | 68 | |
| Mannosidosis, alpha | MAN2B1 | c.1749_1752delCCAG | Persian | 000625‐9685 | 69 |
| Mucolipidosis II | GNPTA | c.2655C>T | Domestic Shorthair | 001248‐9685 | 3 |
| Mucopolysaccharidosis I | IDUA | 3 bp deletion4 | Domestic Shorthair | 000664‐9685 | 70 |
| Mucopolysaccharidosis VI | ARSB | c.1427T>C | Siamese | 000666‐9685 | 71 |
| Mucopolysaccharidosis VII | GUSB | c.1051G>A | Domestic Shorthair | 000667‐9685 | 72 |
| Niemann‐Pick C disease | NPC1 | c.2864G>C | Domestic Shorthair | 000725‐9685 | 73 |
| Spinal muscular atrophy | LIX1 (& LNPEP) | 140 kb deletion | Maine Coon | 000939‐9685 | 74 |
OMIA, Online Mendelian Inheritance in Animals.2
In some cases, the phenotype is straightforward. For example, 2 young Coton de Tulears that have never been able to walk because of severe cerebellar ataxia in a litter with 3 normal pups is characteristic of Bandera's neonatal cerebellar ataxia.17, 18 A DNA test of a pup showing typical signs would confirm the diagnosis and ensure that testing of the normal offspring for the mutation in GRM1 is carried out if they are to be used for breeding. Only if the DNA test results were negative for the mutation would other differential diagnoses need to be considered. Conversely, because the neurotransmitter receptor deficit does not produce any readily identifiable structural changes,17 no other routine diagnostic test would be able to confirm the diagnosis.
In an older animal presenting with signs of cerebellar ataxia, a much wider range of differential diagnoses would need to be considered. To fine‐tune movement in real time, the cerebellum depends upon fast conduction of proprioceptive information during movement. Because anything that affects myelination will affect conduction velocity and the timing of the coordination efforts, demyelinating disease often will produce cerebellar signs. Although only a small portion of the volume, the cerebellum contains over half of the neurons in the brain. This complex network that ensures proper coordination of movement all converges on the Purkinje cells, the sole efferent from the cerebellar cortex (Fig 4). A variety of neurotransmitters and their receptors mediate the communication of information to the Purkinje cells, and a variety of voltage‐gated ion channels are necessary to maintain normal membrane potential and transmit the signals. Supporting the extensive dendritic arborization needed to conduct this function places tremendous metabolic demands on the Purkinje cells. Thus, a wide variety of hereditary as well as toxic, metabolic, demyelinating, neoplastic, infectious, or inflammatory processes can affect the cerebellum but produce a fairly uniform clinical sign: cerebellar ataxia.
Figure 4.
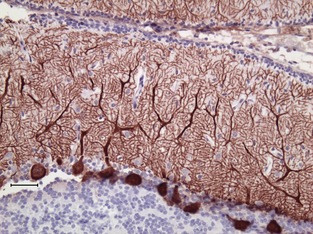
The Purkinje cells (brown in this histologic section labeled with antibodies against the calcium buffering protein calbindin) are the sole output from the complex information process that fine‐tunes movement by the cerebellar cortex. A diverse cadre of mutations can affect the function or structure of the Purkinje cell, but produce a very similar phenotype of cerebellar ataxia. (Courtesy Gayle Johnson).
Particularly if the animal presents with an acute onset of signs, imaging, cerebrospinal fluid analysis, or other tests would be indicated to eliminate common, potentially treatable diseases such as Neospora caninum infection, infarction, or neoplasia before considering hereditary diseases. If these are eliminate, or if a purebred dog is presented for ataxia, particularly if it is slowly progressive, hereditary disease becomes a more likely differential. The cost‐benefit analysis needed to decide whether to run a DNA test then clearly is weighted toward performing a DNA test if the mutation has been reported in the breed and the clinical signs are consistent with the suspected disease.
If a mutation has not been associated with the presenting signs in the breed in question, or if the dog is a mixed breed, then the decision of whether to run DNA tests is more difficult. If the breed being evaluated is closely related to a breed with a known mutation, there is a reasonable chance that the same mutation is found in both populations. For example, Parson Russell Terriers were derived from Jack Russell Terriers, which in turn were derived from Smooth‐haired Fox Terriers. Thus, the same disease‐causing mutation such as the CAPN1 or the KCNJ10 mutations causing spinocerebellar ataxia13, 19 may well be segregating in all 3 breeds. If a mutation has been described in Labrador Retrievers and the patient is a Yorkshire Terrier, the odds are higher that even if the same gene is involved, different mutations occurred independently. Nonetheless, it may well be worth the relatively low cost of a DNA test developed for a different breed. Identifying the same mutation in a new breed not only would support the diagnosis, but it would also inform Yorkshire Terrier breeders that the mutation is segregating in their breed as well. The key is recognizing that a negative test does not eliminate the possibility of a different mutation in the same gene as the etiology in the new breed. In these difficult situations, it also may be valuable to consult with a veterinary geneticist, ideally the one who was involved in the identification of the mutation.
In addition, conducting DNA tests on phenotypes with diverse pathogeneses allows us to refine our categorization of the disease. A Labrador Retriever with exercise intolerance will not be able to perform physically at a level expected of a working dog. Thus, accurately diagnosing the cause is important for the overall genetic health of the breed as well as the welfare of the patient. Once cardiovascular or respiratory causes are excluded, neuromuscular diseases must be considered. Before the advent of DNA testing, muscle biopsy was used to diagnose centronuclear myopathy, but now a DNA test for the PTPLA insertion that causes the disease can readily identify these cases.20 This advance, however, clearly identified a subgroup of dogs with exercise intolerance that was not homozygous for this mutation. Further research identified a mutation in the DNM1 gene responsible for another type of exercise‐induced collapse in Labradors.21 This research identified a new subgroup of dogs with exercise intolerance that are normal for both previously identified mutations. Further research may identify a genetic etiology for the disease in this group.
Identifying Risk of Comorbidity
In the case of a hereditary neurodegenerative disease such as DM, knowing the genotype of an animal can help the client and clinician make informed decisions when comorbidities are possible. An older Pembroke Welsh Corgi or German Shepherd Dog may well develop intervertebral disk disease which could be readily diagnosed by spinal imaging. In the past, when deciding whether or not to proceed with surgery, the clinician could only advise the client that concurrent DM was a possibility in these breeds. By genotyping the patient for the SOD1 mutation, it is now possible to give the client more accurate information on the relative risk of concurrent DM. If the dog tests homozygous for the SOD1:c.118A allele (“at risk”), the decision may well be to move forward with surgery to treat the compressive myelopathy, but it would be done with the knowledge that the patient is at risk for concurrent DM.
Future Directions
Whole‐Genome Sequencing
As new mutations are identified, the number of DNA tests needed to make clinical and breeding decisions will increase dramatically. On the clinical side, the ability to broadly screen for mutations will never replace the need for a thorough history, physical examination, and other diagnostic tests to direct DNA testing appropriately. For signs such as cerebellar ataxia or exercise intolerance, a large number of potential disease‐causing mutations may need to be considered, and the cost of running a large number of individual DNA tests could become prohibitive. One solution to this problem may be the advent of whole‐genome sequencing. Once only available to laboratories with larger research grants, the cost of sequencing the entire genome of an individual has been decreasing to the point where it will soon be feasible to perform a whole‐genome sequence as a routine diagnostic procedure. Advances in bioinformatics will permit the rapid analysis of these large datasets to identify variants that are relevant to the disease process in question. If intellectual property concerns can be addressed, such broad genomic screening someday may become as commonplace as the serum biochemistry profile is today.
Treatment for Genetic Diseases
The ultimate goal with hereditary diseases in animals should be prevention with wise breeding strategies, but it is unlikely that such endeavors will be completely successful. In the case of an old age‐onset disease such as DM, even if breeders eliminated the mutant allele today, a cohort of dogs that are homozygous for the mutation will be reaching the age of onset for the disease over the next 10–15 years. Thus, there is a need for effective treatments for these conditions. Furthermore, the ethical and practical considerations of human genetic diseases create a more pressing need for successful treatments, and the advances made in our patients could facilitate the application of similar interventions for human diseases. A number of therapeutic strategies currently are being investigated. Although treatments such as antisense oligonucleotides to silence mutant genes or virus vector‐delivered gene replacement may seem futuristic, advances in our knowledge and technology may make these treatments as commonplace tomorrow as chemotherapy for cancer is today.
Relevance to Acquired Disease
Although an individual genetic disease may be rare, and can be expected to become more uncommon as DNA testing is accepted by breeders, identifying the mutation causing a hereditary diseases also can shed light on the pathogenesis of more common, acquired diseases. The recognition of the gene responsible for a hereditary disease identifies a molecular pathway that is important to the normal function of the nervous system. Thus, if we can identify, for example, the gene responsible for a hereditary form of epilepsy, it may direct studies into the function of that ion channel or metabolic pathway in seizures secondary to brain tumors or head trauma. Bandera's neonatal ataxia highlights the importance of the metabotropic glutamate receptor in cerebellar function in dogs.18 Idiopathic cerebellitis in dogs is thought to be an immune‐mediated disease.22, 23 This raises the question of whether idiopathic cerebellitis may be caused by antibodies directed against the mGluR1 receptor as they are in human paraneoplastic cerebellar ataxia24 or whether these receptors are the target of metronidazole toxicity.
Conclusions
Advances in genomic research undoubtedly will change the way veterinary medicine is practiced in the future. As with any advance, there will be difficulties in achieving the promise that genomics holds for the future. Understanding the basic principles of genomics as they apply to DNA testing will be essential for the practicing veterinarian to be able to fully capitalize on these advances and avoid potential pitfalls.
Acknowledgments
We thank all the veterinarians who contribute the phenotype information and DNA samples that make gene discovery possible. We also thank Drs Gary Johnson and Joan Coates for critically reviewing the manuscript.
Conflict of Interest Declaration: Authors disclose no conflict of interest.
Biographies
D.P. O'Brien is the Chancellor's Chair in Comparative Neurology in the Department of Veterinary Medicine & Surgery at the University of Missouri, College of Veterinary Medicine. He received his DVM degree from the University of Illinois. After 3 years in general practice, he returned to the University of Illinois to complete a residency in neurology and a PhD in neuroscience. He is board certified in neurology by the American College of Veterinary Internal Medicine and the recipient of the 2011 Kirk Award for Professional Excellence from the ACVIM. His research focuses on hereditary and acquired diseases of the nervous system including epilepsy, movement disorders, developmental disorders, and neurodegenerative diseases.

T. Leeb studied chemistry and received a PhD in biochemistry from the University of Munich. He did his postdoctoral training at the University of Texas Health Science Center at San Antonio and the University of Göttingen before he became a Professor of Molecular Veterinary Genetics at the University of Veterinary Medicine Hannover. Since 2005, he is Professor for Veterinary Genetics and Animal Breeding, Director of the Institute of Genetics, and Head of the Department for Clinical Research and Veterinary Public Health at the University of Bern. Tosso's main research interest is the identification of genetic variants causing heritable phenotypes with a special emphasis on traits and diseases of the skin and the nervous system. He has developed more than 20 genetic test assays and published more than 200 research articles. He is a curator of the Online Mendelian Inheritance in Animals (OMIA) database and serves on the editorial board of several scientific journals including PLoS Genetics. He is a member of the German Academy of Sciences Leopoldina and received a Humboldt Research Award for his contributions to Veterinary Genetics.

Footnotes
Foureman P, Berman L, Stieger K, et al. Mucopolysaccharidosis type VI in Miniature Pinschers: Screening for the mutation. J Vet Intern Med 2004;18:408–409 (abstract)
Ekenstedt KJ, Becker D, Minor KM, et al. An ARHGEF10 deletion is highly associated with a juvenile‐onset inherited polyneuropathy in Leonberger and Saint Bernard dogs. PLoS Genet, submitted
Giger U, Tcherneva E, Caverly J, et al. A missense point mutation in N‐acetylglucosamine‐1‐phospotrans‐ferase causes mucolipidosis II in domestic shorthair cats. J Vet Intern Med 2006;20:781 (abstract)
This variant was determined from a partial IDUA cDNA sequence. It is therefore not possible to give an accurate variant designation.
References
- 1. O'Brien DP. Basic Genomics. Denver, CO: Proceedings ACVIM Forum, 2011. [Google Scholar]
- 2. Faculty of Veterinary Science, University of Sydney . 2014. Available at: http://omia.angis.org.au/. Accessed April 2, 2014.
- 3. Lorenz MD, Coates JR, Kent M. Handbook of Veterinary Neurology, 5th ed St. Louis, MO: Elsevier; 2011. [Google Scholar]
- 4. Platt SR, Olby NJ. BSAVA Manual of Canine and Feline Neurology. Gloucester, MA: British Small Animal Veterinary Association; 2012. [Google Scholar]
- 5. Dickinson PJ, Sturges BK, Shelton GD, LeCouteur RA. Congenital myasthenia gravis in Smooth‐Haired Miniature Dachshund dogs. J Vet Intern Med 2005;19:920–923. [DOI] [PubMed] [Google Scholar]
- 6. Awano T, Katz ML, O'Brien DP, et al. A mutation in the cathepsin D gene (CTSD) in American Bulldogs with neuronal ceroid lipofuscinosis. Mol Genet Metabol 2006;87:341–348. [DOI] [PubMed] [Google Scholar]
- 7. Tyynela J, Sohar I, Sleat DE, et al. A mutation in the ovine cathepsin D gene causes a congenital lysosomal storage disease with profound neurodegeneration. EMBO J 2000;19:2786–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Forman OP, De RL, Stewart J, et al. Genome‐wide mRNA sequencing of a single canine cerebellar cortical degeneration case leads to the identification of a disease associated SPTBN2 mutation. BMC Genet 2012;13:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kyostila K, Cizinauskas S, Seppala EH, et al. A SEL1L mutation links a canine progressive early‐onset cerebellar ataxia to the endoplasmic reticulum‐associated protein degradation (ERAD) machinery. PLoS Genet 2012;8:e1002759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. O'Brien DP, Johnson GS, Schnabel RD, et al. Genetic mapping of canine multiple system degeneration and ectodermal dysplasia Loci. J Hered 2005;96:727–734. [DOI] [PubMed] [Google Scholar]
- 11. Awano T, Johnson GS, Wade CM, et al. Genome‐wide association analysis reveals a SOD1 mutation in canine degenerative myelopathy that resembles amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 2009;106:2794–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. den Dunnen JT, Antonarakis SE. Nomenclature for the description of human sequence variations. Hum Genet 2001;109:121–124. [DOI] [PubMed] [Google Scholar]
- 13. Gilliam D, O'Brien DP, Coates JR et al. A homozygous KCNJ10 mutation in Jack Russell Terriers and related breeds with spinocerebellar ataxia with myokymia, seizures or both. J Vet Intern Med 2014;28:871–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Awano T, Katz ML, O'Brien DP, et al. A frame shift mutation in canine TPP1 (the ortholog of human CLN2) in a juvenile Dachshund with neuronal ceroid lipofuscinosis. Mol Genet Metabol 2006;89:254–260. [DOI] [PubMed] [Google Scholar]
- 15. Sanders DN, Farias FH, Johnson GS, et al. A mutation in canine PPT1 causes early onset neuronal ceroid lipofuscinosis in a Dachshund. Mol Genet Metabol 2010;100:349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wininger FA, Zeng R, Johnson GS, et al. Degenerative myelopathy in a Bernese Mountain Dog with a novel SOD1 missense mutation. J Vet Intern Med 2011;25:1166–1170. [DOI] [PubMed] [Google Scholar]
- 17. Coates JR, O'Brien DP, Kline KL, et al. Neonatal cerebellar ataxia in Coton de Tulear dogs. J Vet Intern Med 2002;16:680–689. [DOI] [PubMed] [Google Scholar]
- 18. Zeng R, Farias FH, Johnson GS, et al. , et al. A truncated retrotransposon disrupts the GRM1 coding sequence in Coton de Tulear dogs with Bandera's neonatal ataxia. J Vet Intern Med 2011;25:267–272. [DOI] [PubMed] [Google Scholar]
- 19. Forman OP, De RL, Mellersh CS. Missense mutation in CAPN1 is associated with spinocerebellar ataxia in the Parson Russell Terrier dog breed. PLoS ONE 2013;8:e64627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pele M, Tiret L, Kessler JL, et al. SINE exonic insertion in the PTPLA gene leads to multiple splicing defects and segregates with the autosomal recessive centronuclear myopathy in dogs.[erratum appears in Hum Mol Genet. 2005 Jul 1, 14(13), pp. 1905–6]. Hum Mol Genet 2005;14:1417–1427. [DOI] [PubMed] [Google Scholar]
- 21. Patterson EE, Minor KM, Tchernatynskaia AV, et al. A canine DNM1 mutation is highly associated with the syndrome of exercise‐induced collapse. Nat Genet 2008;40:1235–1239. [DOI] [PubMed] [Google Scholar]
- 22. Hazell KLA, Child G, Chin G. Clinical characteristics and outcome after treatment of shaker dog syndrome in 90 dogs. Aust Vet Pract 2011;41:167–171. [Google Scholar]
- 23. Tipold A, Fatzer R, Jaggy A, et al. Presumed immune‐mediated cerebellar granuloprival degeneration in the Coton de Tulear breed. J Neuroimmunol 2000;110:130–133. [DOI] [PubMed] [Google Scholar]
- 24. Sillevis SP, Kinoshita A, De LB, et al. Paraneoplastic cerebellar ataxia due to autoantibodies against a glutamate receptor. N Engl J Med 2000;342:21–27. [DOI] [PubMed] [Google Scholar]
- 25. Drogemuller C, Becker D, Kessler B, et al. A deletion in the N‐myc downstream regulated gene 1 (NDRG1) gene in Greyhounds with polyneuropathy. PLoS ONE 2010;5:e11258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Seppala EH, Jokinen TS, Fukata M, et al. LGI2 truncation causes a remitting focal epilepsy in dogs. PLoS Genet 2011;7:e1002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Occhiodoro T, Anson DS. Isolation of the canine alpha‐L‐fucosidase cDNA and definition of the fucosidosis mutation in English Springer Spaniels. Mamm Genome 1996;7:271–274. [DOI] [PubMed] [Google Scholar]
- 28. Skelly BJ, Sargan DR, Herrtage ME, Winchester BG. The molecular defect underlying canine fucosidosis. J Med Genet 1996;33:284–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Proschowsky HF, Flagstad A, Cirera S, et al. Identification of a mutation in the CHAT gene of Old Danish Pointing Dogs affected with congenital myasthenic syndrome. J Hered 2007;98:539–543. [DOI] [PubMed] [Google Scholar]
- 30. Vernau KM, Runstadler JA, Brown EA, et al. Genome‐wide association analysis identifies a mutation in the thiamine transporter 2 (SLC19A3) gene associated with Alaskan Husky encephalopathy. PLoS ONE 2013;8:e57195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Forman OP, Penderis J, Hartley C, et al. Parallel mapping and simultaneous sequencing reveals deletions in BCAN and FAM83H associated with discrete inherited disorders in a domestic dog breed. PLoS Genet 2012;8:e1002462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Victoria T, Rafi MA, Wenger DA. Cloning of the canine GALC cDNA and identification of the mutation causing globoid cell leukodystrophy in West Highland White and Cairn terriers. Genomics 1996;33:457–462. [DOI] [PubMed] [Google Scholar]
- 33. McGraw RA, Carmichael KP. Molecular basis of globoid cell leukodystrophy in Irish setters. Vet J 2006;171:370–372. [DOI] [PubMed] [Google Scholar]
- 34. Kreutzer R, Leeb T, Muller G, et al. A duplication in the canine beta‐galactosidase gene GLB1 causes exon skipping and GM1‐gangliosidosis in Alaskan Huskies. Genetics 2005;170:1857–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang ZH, Zeng B, Shibuya H, et al. Isolation and characterization of the normal canine beta‐galactosidase gene and its mutation in a dog model of GM1‐gangliosidosis. J Inherit Metab Dis 2000;23:593–606. [DOI] [PubMed] [Google Scholar]
- 36. Sanders DN, Zeng R, Wenger DA, et al. GM2 gangliosidosis associated with a HEXA missense mutation in Japanese Chin dogs: A potential model for Tay Sachs disease. Mol Genet Metabol 2013;108:70–75. [DOI] [PubMed] [Google Scholar]
- 37. Rahman MM, Chang HS, Mizukami K et al. A frameshift mutation in the canine HEXB gene in Toy Poodles with GM2 gangliosidosis variant 0 (Sandhoff disease). Vet J 2012; E‐pub ahead of print. doi: 10.1016/j.tvjl.2012.05.021. [DOI] [PubMed] [Google Scholar]
- 38. Penderis J, Calvin J, Abramson C, et al. L‐2‐hydroxyglutaric aciduria: Characterisation of the molecular defect in a spontaneous canine model. J Med Genet 2007;44:334–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Farias FH, Zeng R, Johnson GS, et al. A L2HGDH initiator methionine codon mutation in a Yorkshire Terrier with L‐2‐hydroxyglutaric aciduria. BMC Vet Res 2012;8:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sanchez‐Masian DF, Artuch R, Mascort J, et al. L‐2‐hydroxyglutaric aciduria in two female Yorkshire Terriers. J Am Anim Hosp Assoc 2012;48:366–371. [DOI] [PubMed] [Google Scholar]
- 41. Menon KP, Tieu PT, Neufeld EF. Architecture of the canine IDUA gene and mutation underlying canine mucopolysaccharidosis I. Genomics 1992;14:763–768. [DOI] [PubMed] [Google Scholar]
- 42. Yogalingam G, Pollard T, Gliddon B, et al. Identification of a mutation causing mucopolysaccharidosis type IIIA in New Zealand Huntaway dogs. Genomics 2002;79:150–153. [DOI] [PubMed] [Google Scholar]
- 43. Ellinwood NM, Wang P, Skeen T, et al. A model of mucopolysaccharidosis IIIB (Sanfilippo syndrome type IIIB): N‐acetyl‐alpha‐D‐glucosaminidase deficiency in Schipperke dogs. J Inherit Metab Dis 2003;26:489–504. [DOI] [PubMed] [Google Scholar]
- 44. Jolly RD, Hopwood JJ, Marshall NR, et al. Mucopolysaccharidosis type VI in a Miniature Poodle‐type dog caused by a deletion in the arylsulphatase B gene. N Z Vet J 2012;60:183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lohi H, Young EJ, Fitzmaurice SN, et al. Expanded repeat in canine epilepsy. Science 2005;307:81. [DOI] [PubMed] [Google Scholar]
- 46. Rhodes TH, Vite CH, Giger U, et al. A missense mutation in canine C1C‐1 causes recessive myotonia congenita in the dog. FEBS Lett 1999;456:54–58. [DOI] [PubMed] [Google Scholar]
- 47. Finnigan DF, Hanna WJ, Poma R, Bendall AJ. A novel mutation of the CLCN1 gene associated with myotonia hereditaria in an Australian cattle dog. J Vet Intern Med 2007;21:458–463. [DOI] [PubMed] [Google Scholar]
- 48. Lin L, Faraco J, Li R, et al. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell 1999;98:365–376. [DOI] [PubMed] [Google Scholar]
- 49. Hungs M, Fan J, Lin L, et al. Identification and functional analysis of mutations in the hypocretin (orexin) genes of narcoleptic canines. Genome Res 2001;11:531–539. [DOI] [PubMed] [Google Scholar]
- 50. Farias FH, Zeng R, Johnson GS, et al. A truncating mutation in ATP13A2 is responsible for adult‐onset neuronal ceroid lipofuscinosis in Tibetan Terriers. Neurobiol Dis 2011;42:468–474. [DOI] [PubMed] [Google Scholar]
- 51. Wohlke A, Philipp U, Bock P, et al. A one base pair deletion in the canine ATP13A2 gene causes exon skipping and late‐onset neuronal ceroid lipofuscinosis in the Tibetan Terrier. PLoS Genet 2011;7:e1002304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Abitbol M, Thibaud JL, Olby NJ, et al. A canine Arylsulfatase G (ARSG) mutation leading to a sulfatase deficiency is associated with neuronal ceroid lipofuscinosis. Proc Natl Acad Sci USA 2010;107:14775–14780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Melville SA, Wilson CL, Chiang CS, et al. A mutation in canine CLN5 causes neuronal ceroid lipofuscinosis in Border Collie dogs. Genomics 2005;86:287–294. [DOI] [PubMed] [Google Scholar]
- 54. Katz ML, Farias FH, Sanders DN, et al. A missense mutation in canine CLN6 in an Australian Shepherd with neuronal ceroid lipofuscinosis. J Biomed Biotechnol 2011;2011:198042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Katz ML, Khan S, Awano T, et al. A mutation in the CLN8 gene in English Setter dogs with neuronal ceroid‐lipofuscinosis. Biochem Biophys Res Commun 2005;327:541–547. [DOI] [PubMed] [Google Scholar]
- 56. Chen X, Johnson GS, Schnabel RD, et al. A neonatal encephalopathy with seizures in Standard Poodle dogs with a missense mutation in the canine ortholog of ATF2 . Neurogenetics 2008;9:41–49. [DOI] [PubMed] [Google Scholar]
- 57. Fyfe JC, Al‐Tamimi RA, Liu J, et al. A novel mitofusin 2 mutation causes canine fetal‐onset neuroaxonal dystrophy. Neurogenetics 2011;12:223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bruun CS, Jaderlund KH, Berendt M, et al. A Gly98Val mutation in the N‐Myc downstream regulated gene 1 (NDRG1) in Alaskan Malamutes with polyneuropathy. PLoS ONE 2013;8:e54547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Baranowska I, Jaderlund KH, Nennesmo I, et al. Sensory ataxic neuropathy in Golden Retriever dogs is caused by a deletion in the mitochondrial tRNATyr gene. PLoS Genet 2009;5:e1000499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nadon NL, Duncan ID, Hudson LD. A point mutation in the proteolipid protein gene of the ‘shaking pup’ interrupts oligodendrocyte development. Dev Suppl 1990;110:529–537. [DOI] [PubMed] [Google Scholar]
- 61. Li FY, Cuddon PA, Song J, et al. Canine spongiform leukoencephalomyelopathy is associated with a missense mutation in cytochrome b. Neurobiol Dis 2006;21:35–42. [DOI] [PubMed] [Google Scholar]
- 62. Gill JL, Capper D, Vanbellinghen JF, et al. Startle disease in Irish Wolfhounds associated with a microdeletion in the glycine transporter GlyT2 gene. Neurobiol Dis 2011;43:184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Martin DR, Rigat BA, Foureman P, et al. Molecular consequences of the pathogenic mutation in feline GM1 gangliosidosis. Mol Genet Metabol 2008;94:212–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Martin DR, Cox NR, Morrison NE, et al. Mutation of the GM2 activator protein in a feline model of GM2 gangliosidosis. Acta Neuropathol 2005;110:443–450. [DOI] [PubMed] [Google Scholar]
- 65. Muldoon LL, Neuwelt EA, Pagel MA, Weiss DL. Characterization of the molecular defect in a feline model for type II GM2‐gangliosidosis (Sandhoff disease). Am J Pathol 1994;144:1109–1118. [PMC free article] [PubMed] [Google Scholar]
- 66. Martin DR, Krum BK, Varadarajan GS, et al. An inversion of 25 base pairs causes feline GM2 gangliosidosis variant. Exp Neurol 2004;187:30–37. [DOI] [PubMed] [Google Scholar]
- 67. Kanae Y, Endoh D, Yamato O, et al. Nonsense mutation of feline beta‐hexosaminidase beta‐subunit (HEXB) gene causing Sandhoff disease in a family of Japanese domestic cats. Res Vet Sci 2007;82:54–60. [DOI] [PubMed] [Google Scholar]
- 68. Bradbury AM, Morrison NE, Hwang M, et al. Neurodegenerative lysosomal storage disease in European Burmese cats with hexosaminidase beta‐subunit deficiency. Mol Genet Metabol 2009;97:53–59. [DOI] [PubMed] [Google Scholar]
- 69. Berg T, Tollersrud OK, Walkley SU, et al. Purification of feline lysosomal alpha‐mannosidase, determination of its cDNA sequence and identification of a mutation causing alpha‐mannosidosis in Persian cats. Biochem J 1997;328:863–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. He X, Li CM, Simonaro CM, et al. Identification and characterization of the molecular lesion causing mucopolysaccharidosis type I in cats. Mol Genet Metabol 1999;67:106–112. [DOI] [PubMed] [Google Scholar]
- 71. Yogalingam G, Litjens T, Bielicki J, et al. Feline mucopolysaccharidosis type VI. Characterization of recombinant N‐acetylgalactosamine 4‐sulfatase and identification of a mutation causing the disease. J Biol Chem 1996;271:27259–27265. [DOI] [PubMed] [Google Scholar]
- 72. Fyfe JC, Kurzhals RL, Lassaline ME, et al. Molecular basis of feline beta‐glucuronidase deficiency: An animal model of mucopolysaccharidosis VII. Genomics 1999;58:121–128. [DOI] [PubMed] [Google Scholar]
- 73. Somers KL, Royals MA, Carstea ED, et al. Mutation analysis of feline Niemann‐Pick C1 disease. Mol Genet Metabol 2003;79:99–103. [DOI] [PubMed] [Google Scholar]
- 74. Fyfe JC, Menotti‐Raymond M, David VA, et al. An approximately 140‐kb deletion associated with feline spinal muscular atrophy implies an essential LIX1 function for motor neuron survival. Genome Res 2006;16:1084–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. O'Brien DP. Genetic diseases In: Platt S, Olby N, eds. BSAVA Manual of Canine and Feline Neurology, 4th ed Quedgeley: British Small Animal Veterinary Association; 2012:107–116. [Google Scholar]