

Abstract
Background
Mutation analysis of proto‐oncogene c‐kit (c‐kit) is advisable before starting treatment with tyrosine kinase inhibitors in dogs with mast cell tumor (MCT), including those with metastatic disease. Testing is usually performed on primary tumors, assuming that c‐kit mutation status does not change in metastasis.
Hypothesis/Objectives
To give an insight into the mutational processes and to make a recommendation on the use of c‐kit mutational analysis in the clinical setting.
Animals
Twenty‐one client‐owned dogs with metastatic MCT.
Methods
Dogs undergoing resection or biopsy for both primary and matched metastatic MCT were prospectively enrolled. Total RNA or DNA was extracted from primary MCT and corresponding metastases. Exons 8, 9, and 11 were amplified by PCR and sequenced. Genetic features between primary MCT and metastases were compared. Their correlation with clinicopathologic features was investigated.
Results
Concordance (mutated or wild‐type) of mutational status, evaluable in 21 primary and matched metastatic (20 nodal and 1 splenic) MCTs, was 100%. Three new c‐kit mutations were identified. No significant correlation was detected between c‐kit mutation and clinicopathologic features.
Conclusions and Clinical Importance
Proto‐oncogene c‐kit mutational status is conserved between any primary and its matched secondary tumor, suggesting that both can be used for c‐kit mutational testing. Targeted therapies might be also used to treat metastatic disease.
Keywords: Dog, Mastocytoma, Metastasis, Mutation
Abbreviations
- AA
amino acid
- c‐kit
proto‐oncogene c‐kit
- FFPE
formalin‐fixed and paraffin‐embedded
- FNA
fine‐needle aspiration
- GIST
gastrointestinal stromal tumor
- ITD
internal tandem duplication
- MCT
mast cell tumor
- SNP
single nucleotide polymorphism
- TKI
tyrosine kinase inhibitor
- WT
wild‐type
The proto‐oncogene c‐kit (c‐kit), which encodes for the transmembrane receptor KIT, is known to play a critical role in mast cell development and tumors.1 In dogs, approximately 9–30% of mast cell tumors (MCTs) show c‐kit mutations, including internal tandem duplications (ITDs) in the juxtamembrane domain, resulting in constitutive activation of KIT in the absence of ligand binding,1, 2 and activating point mutations in c‐kit extracellular domains (eg, exons 8 and 9).3 In general, ITDs are associated with an increased risk of metastasis and local recurrence, higher tumor proliferation index, and aberrant KIT localization.2, 4, 5, 6
The importance of the mutational status has been elucidated by 2 clinical trials, which showed a lower objective response rate and a shorter survival time when tyrosine kinase inhibitors (TKIs) including toceranib and masitinib, respectively, were administered to dogs with wild‐type (WT) tumors.7, 8 Although TKI‐based therapy is used in dogs with MCT to also treat metastatic disease in the lymph nodes,7 c‐kit status is generally evaluated in the primary lesions because metastatic sites are rarely removed or biopsied before treatment. However, it is still unknown whether c‐kit status differs in metastases compared with primary tumors. The rationale for using small molecule inhibitors of oncogenic proteins as cancer therapies depends, at least in part, on the assumption that metastatic tumors are primarily clonal with respect to the mutant oncogene. If this is not the case, targeted therapies might only be partially efficacious. Therefore, it is of primary importance to verify the correlation between primaries and related metastases with regard to c‐kit status.
In people, controversy exists regarding the stability of mutational status in various tumors throughout the course of the disease, leading to metastases with different mutational status from that of the primary tumor.9, 10, 11 In veterinary medicine, there are only 2 studies comparing immunohistochemical phenotypes between primary mammary carcinomas and their related lymph node metastasis.12, 13 In cats, concordance between primary mammary carcinoma and matched metastasis was detected in 57.1% of cases,12 whereas in dogs in 65% of cases.13
To the authors' knowledge, very few studies have been conducted in dogs on the rate of concordance in terms of c‐kit mutations. One study showed c‐kit ITD heterogeneity in different sites of multiple MCTs in 2 dogs14; in another study, c‐kit ITDs were used to provide evidence of tumor clonality in multiple MCTs developing over 1–2 years in 2 dogs.15
In this study, we prospectively analyzed matched primary and metastatic MCT specimens for c‐kit intra‐ and intertumor heterogeneity (1) to give an insight into the mutational processes; and (2) to make a recommendation on the use of c‐kit mutational analysis in the clinical setting. Moreover, the treatment with TKIs is associated with potential toxicity and high costs; additionally, resistance to certain TKIs is often caused by secondary mutations of c‐kit 7, 16; therefore, it is important to critically review all aspects of the mutational testing to enhance upfront patient selection.
We hypothesized a discordance of c‐kit mutational status between matched primary and metastatic MCT, thereby recommending the use of c‐kit mutational testing on all involved sites.
Materials and Methods
Case Selection and Tumor Specimens
Inclusion Criteria
Dogs with histologically confirmed MCT undergoing complete clinical staging and total or partial surgical excision of the primary tumor and corresponding metastasis were prospectively recruited. Treatment with neoadjuvant medical treatment (including steroids, chemotherapy, targeted therapy) was not permitted.
Background information recorded for each dog included signalment, body weight, and primary tumor description (location, dimension, presence of ulceration, grade according to Patnaik and Kiupel's sytems).17 Initial staging included history and physical examination, complete blood cell count with differential, serum biochemistry, coagulation profile, cytological evaluation of the cutaneous nodule and regional lymph node, thoracic radiographs (3 views), abdominal ultrasound, fine‐needle aspirates of liver and spleen regardless of their sonographic appearance, and cytologic examination of bone marrow obtained from the iliac crest. Lymph nodes or viscera were considered metastatic, if mast cells appeared in clusters or sheets, in very large numbers or atypical on morphology, as previously documented.18 Histologically, nodal metastatic spread was supported by the localization of mast cells in the subcapsular sinuses; special histochemical stains (Giemsa) were used to detect poorly granulated mast cells. Written informed consent was obtained from all owners.
Tumor Specimens
Tumor samples were obtained by partial or total surgical resection from each primary MCT and matched metastasis before starting any medical treatment. To formulate a histologic diagnosis, samples were fixed in 10% buffered formalin, processed, and embedded in paraffin using a standardized protocol. Slides were reviewed by a single board‐certified pathologist (SDP), and histopathologic criteria for diagnosis were based on those previously published for canine MCT.19, 20
With regard to c‐kit sequencing analysis, either 1 tissue core (2‐mm diameter) or fine‐needle aspirates (FNA) were obtained from each primary MCT sample and matched metastases. Specimens were submersed in a stabilization and storage solution (RNAlater Solution1 ) and refrigerated at −20°C until use. Whenever the primary tumor was surgically excised by other veterinarians, 10‐μm sections of the corresponding formalin‐fixed and paraffin‐embedded (FFPE) block were used for nucleic acid extraction.
Molecular Analysis
Nucleic Acid Extraction
Total RNA was extracted from biopsies and FNA21 with a nucleic acid isolation reagent2 and a commercial kit (High Pure RNA Isolation Kit3), respectively, according to the manufacturer's instructions. Whenever nucleic acids were extracted from FFPE primary tumor sections, another commercial kit4 was used. In this case, the genomic DNA was preferred to RNA because of the poor quality of the extracted RNA.
Nucleic acids yield and purity (260/280 and 260/230 nm absorbance ratios) were measured with a spectrophotometer,5 while their quality was checked by 1% agarose gel electrophoresis. Two micrograms of total RNA was reverse transcribed with a commercial kit (High Capacity cDNA Reverse Transcription Kit1). Both cDNA and DNA were finally stored at −20°C until use.
c‐kit Genotyping
Exons 8, 9, and 11, considered the hot spot regions for activating protein mutations, were screened by PCR and direct sequencing.3, 22 To amplify either c‐kit exons 8, 9, and 11 (starting from cDNA) or exon 11 (from DNA), previously published primers pairs and PCR conditions were used.23 Conversely, exons 8 and 9 primers for genomic DNA amplification were designed ex novo,6 and forward and reverse primers as well as the expected amplicon sizes are reported in Table 1. Amplifications were carried out in a thermocycler,7 with a commercially available PCR kit.8 Two microliters of 5‐fold diluted DNA was used as template, while primer9 concentrations were 16.5 pmol each. Amplicons were visualized in 1.5% agarose gel.
Table 1.
Primers for genomic DNA amplification and sequencing of c‐kit exons 8 and 9
| Exon | Primer Sequence (5′–3′) | Expected Amplicon Size (bp) |
|---|---|---|
| 8 | F: ACTCACTGGTTCCGATGCTC | 408 |
| R: CCCTTAAAAAGCCACATGGA | ||
| 9 | F: CACCCTTGGTTGAAAAAGGA | 458 |
| R: ATATGGCAGGCAGAGCCTAA |
bp, base pairs; F, forward; R, reverse.
Whenever the presence of additional bands of different length (roughly 30 bp) was noticed, these ones were at first individually excised from the agarose gel and, then, purified with a commercial kit (High Pure PCR Cleanup Micro Kit3), according to the manufacturer's instructions. Hence, PCR products were sequenced, using the same PCR primers, with either a capillary electrophoresis machine (ABI Prism 3100 Genetic Analyzer1) or an automatic sequencer (ABI 3730XL DNA Analyzer1).
Sequences were analyzed by a commercially available software.9 Alignments with the WT c‐kit mRNA sequence NM_001003181.1, to discover potential single nucleotide polymorphisms (SNPs), ITDs, or deletions, were performed by an open source software.10
Treatment and Response Criteria
The type of treatment was at the investigator's personal discretion, and included surgery, radiation therapy, chemotherapy, TKI, or a combination of these. Response was determined by RECIST criteria.24 Briefly, disappearance of all lesions was defined as complete response (CR); a decrease of at least 30% in the diameter of a lesion was defined as partial response (PR); the appearance of new MCTs or at least a 20% increase in the diameter of a lesion was defined as progressive disease (PD); <30% reduction or 20% increase in the diameter of a lesion was defined as stable disease (SD).
Statistical Analysis
To evaluate the relationship between c‐kit mutations and clinicopathologic factors, data were analyzed by Fisher's exact test and Pearson χ2 test. For this purpose, the following clinicopathologic features were taken into account: sex (male or female), reproductive status (intact or neutered), breed (purebred or crossbred; predisposition to biologically aggressive MCTs [meaning advanced grade or clinical stage], eg, Shar‐Pei and Labrador Retriever), age (< or ≥10 years), weight (< or ≥10 kg), dimension of the primary lesion (< or ≥3 cm), clinical stage (II or III or IV), substage (a or b), and histologic grading (both Patnaik and Kiupel's systems).17, 25, 26, 27, 28 The anatomic site was categorized as benign or malignant, as some locations have been described as biologically aggressive (eg, inguinal/perineal, head and neck, digit).17 Survival time was defined as the time interval between the initiation of treatment and death. Dogs dead from disease or MCT‐related causes were classified as events; those dead for unrelated causes or lost to follow‐up at the time of the study closure were censored.
Statistical calculations were performed by a commercial software package.11 For all statistical analysis, significance was set at P < .05.
Results
Dogs and MCT Demographics
Between July 2011 and August 2013, 21 dogs met the inclusion criteria and were enrolled. There were 6 Labrador Retrievers, 5 crossbred dogs, 3 Boxers, and one each of the following: Breton, Shih‐Tzu, Shar‐Pei, Beagle, American Staffordshire Terrier, German Hound, and Dogue de Bordeaux. Twelve dogs were spayed females, 3 intact females, 4 intact males, and 2 castrated males. Median age was 8 years (range, 3–14 years), and median body weight was 26.7 kg (range 7.4–50.2 kg).
Eighteen (86%) dogs had single lesions, and 3 (14%) had concurrent multiple tumors. In these latter ones, the biggest MCT was sampled for both histopathologic and mutational analysis. MCTs were in various locations, including 6 (29%) dogs with tumors on distal limbs; 4 (19%) dogs with their tumors on the head; 3 (14%) dogs with digital MCTs; 2 (10%) dogs with tumors on proximal limbs; 2 (10%) dogs with vulvar tumors; and 1 (5%) dog with a MCT on the abdominal wall. All dogs with multiple tumors had them in the same regional areas (axillary region, head, and abdominal wall).
Histopathology was available for all primary MCTs: 14 (66%) dogs had Patnaik's grade 2 MCTs, 6 (29%) dogs had grade 3 MCTs, and 1 (5%) dog had a grade 1 MCT. Regarding the Kiupel's grading system, 11 (52%) tumors were classified as low‐grade MCTs and 10 (48%) as high‐grade MCTs.
All dogs had metastatic disease: 20 (95%) dogs had regional lymph node involvement and, among these; 2 also had hepatic metastasis; 2 had splenic and hepatic metastasis; 1 had hepatic, splenic, and marrow metastasis; 1 had splenic metastasis; and 1 had cutaneous metastasis. One (5%) dog had involvement of liver and spleen without regional lymph node metastasis. Lymph node metastases were confirmed in all 20 dogs by means of histopathology; the remaining dog without lymph node metastasis had only cytologic diagnosis of liver and spleen involvement.
Overall, 11 (52%) dogs had stage II disease; 8 (38%) dogs had stage IV disease; and 2 (10%) dogs had stage III disease. Sixteen (76%) dogs were asymptomatic (substage a), and 5 (24%) dogs had signs of systemic effects of MCT (vomiting, diarrhea, pruritus, and regional edema).
c‐kit Mutation Status
All specimens of primary tumors and paired metastases were suitable for c‐kit genotyping. Mutations of c‐kit sequence were detected in 3 (14%) MCTs: 1 in exon 8 (Fig 1); and 2 in exon 11 (Fig 2). Two of them were noticed in dogs with grade 3 MCTs, and 1 in a dog with a grade 2 MCT.
Figure 1.
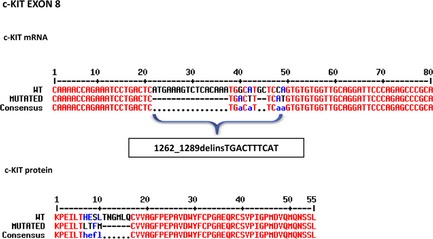
Direct sequencing of c‐kit exon 8 from canine mast cell tumors. Both wild‐type and mutated nucleotide and protein alignments sequences are reported. In cDNA obtained from primary tumor and matched metastasis (the corresponding lymph node), a deletion of 10 amino acids (AAs) (HESLTNGMLQ), associated with an insertion of 4 AAs (Leucine‐Threonine‐Phenylalanine‐Methionine), was detected. This new c‐kit mutation was termed 1262_1289delinsTGACTTTCAT, according to the nomenclature for human sequence variations.25
Figure 2.

Direct sequencing of mutant c‐kit exon 11 from canine mast cell tumors. Alignments of both wild‐type and mutated protein sequences are reported. Two internal tandem duplications (ITDs), namely (A) ITD571‐582 and (B) ITD574‐587, were detected in cDNA obtained from primary tumor and matched metastasis (the corresponding lymph node).
A new 28 amino acids (AAs) deletion affecting 10 AA codons, namely from Histidine‐421 (H421) to Glutamine‐430 (Q430), coupled with a contemporary insertion of 10 base pairs (bp) coding for four AAs (Leucine‐Threonine‐Phenylalanine‐Methionine, LTFM), was detected in exon 8 (Fig 1). This mutation was named 1262_1289delinsTGACTTTCAT, in agreement with the nomenclature for human sequence variations.29 Moreover, 2 new ITDs were found in exon 11 (Fig 2): a first one, consisting in the insertion of 12 AAs at the residue 571 (ITD571–582); and a second one based on an addition of 14 AAs at the residue 574 (ITD574–587).
Furthermore, 2 already known silent SNPs were detected in exon 8 (1275G > A)3 and in exon 11 (1759C > T).30 The relative frequencies were 33% (7/21) and 5% (1/21), respectively.
Noteworthy, the comparison of c‐kit mutations and SNPs in primary tumors and corresponding metastases showed a concordance rate of 100%. Likewise, all dogs with a primary WT c‐kit genotype showed a WT c‐kit in their matched metastases.
Treatment and Clinical Follow‐up
Eleven (52%) dogs (including the 3 dogs with c‐kit mutation) underwent surgical excision of their MCT. Four of these animals also received systemic chemotherapy (vinblastine and prednisone) as front‐line treatment; 3 dogs received vinblastine and TKIs; 1 dog was treated with curative radiation therapy and TKIs; and one with curative radiation therapy and vinblastine. Two (10%) dogs received systemic chemotherapy (vinblastine and prednisone) as their only treatment. Six (28%) dogs were treated with palliative radiation therapy; 4 of these 6 also received vinblastine and prednisone, and 4 other ones TKI. Finally, 2 (10%) dogs were only treated with TKIs.
Overall, 11 (52%) dogs achieved CR, 7 (33%) dogs PR, 2 (10%) dogs SD, whereas 1 (5%) dog did not respond to the treatment and experienced PD. At the end of the study, 10 (48%) dogs were still alive with a median follow‐up of 205 days (range 41–473 days), and 11 (52%) dogs died or were euthanized because of progression of their MCT (n = 10) or for tumor‐unrelated causes (n = 1). The overall median survival was 51 and 149 days for dogs harboring c‐kit mutations and with WT c‐kit gene, respectively.
Relationship between c‐kit Mutational Status and Clinicopathologic Features
No significant correlation was found between primary c‐kit mutation and the considered clinicopathologic characteristics (Table 2).
Table 2.
Relationship between c‐kit mutational status and clinicopathologic features in 21 primary mast cell tumors (MCTs)
| Variables | c‐kit Mutation | ||
|---|---|---|---|
| Positive | Negative | P‐Value | |
| Age (years) | |||
| <10 | 2 | 9 | 1.000a |
| >10 | 1 | 9 | |
| Sex | |||
| Male | 2 | 4 | .184a |
| Female | 1 | 14 | |
| Breed | |||
| Pure breed | 3 | 13 | .549a |
| Crossbred | 0 | 5 | |
| Breed predisposition to aggressive MCTs | |||
| Yes | 2 | 5 | .247a |
| No | 1 | 13 | |
| Weight (kg) | |||
| <10 | 1 | 1 | .271a |
| >10 | 2 | 17 | |
| Primary lesion, anatomic site | |||
| Benign | 0 | 11 | .090a |
| Malignant | 3 | 7 | |
| Primary lesion, dimension (cm) | |||
| <3 | 1 | 9 | 1.000a |
| >3 | 2 | 9 | |
| Metastatic lymph node | |||
| Yes | 3 | 17 | 1.000a |
| No | 0 | 1 | |
| Stage | |||
| I–II | 0 | 11 | .097b |
| III | 1 | 1 | |
| IV | 2 | 6 | |
| Substage | |||
| a | 1 | 15 | .128a |
| b | 2 | 3 | |
| Histologic grade (Patnaik) | |||
| I | 0 | 1 | .283b |
| II | 1 | 13 | |
| III | 2 | 4 | |
| Histologic grade (Kiupel) | |||
| Low | 1 | 10 | .586a |
| High | 2 | 8 | |
Fisher exact test.
Pearson χ2 test.
Discussion
In this study, we compared c‐kit mutational status of exons 8, 9, and 11 between primary MCT and matched metastasis, and found a perfect (100%) concordance.
Metastatic MCT represents a major health problem in the canine population, but the introduction of a novel class of targeted antineoplastic agents directed against KIT, TKI, has significantly changed the therapeutic options available for these dogs.7, 8 Indeed, the important role of targeted therapy against molecules contributing to tumor development, progression, and metastasis has attracted considerable attention.31
Because the identification of the mutational status of c‐kit could help to select dogs that have a high probability of benefiting from TKI,7 it is of primary importance to verify the degree of correlation between primaries and related metastases with regard to c‐kit status. Indeed, mutations are mainly evaluated at the primary site and there are little data available regarding the possible concordance in mutational status between the primary tumor and the corresponding metastases.7, 8 However, the death of metastatic cells is the main goal of treatment in a metastatic setting. These cells might be biologically different from the primary tumor, which has implications for the clinical management of MCT.
It is well known that the progression of cancer develops from a single mutated cell, followed by malignant clonal expansion secondary to additional genetic and genomic alterations. As a consequence, the ongoing acquisition of these alterations can result in the emergence of neoplastic subclones with varying genotypes and, consequently, phenotypes,32 leading to discordance between the primary tumor and its metastases. In people, several tumors including melanoma,33 gastrointestinal stromal tumor,34 and lung cancer35 show intratumor and intertumor heterogeneity, indicating the presence of more than one clone of cancer cells within a given neoplastic mass, and the presence of different genetic alterations in different metastatic sites from a single patient, respectively. Therefore, determining if there is homogeneous mutational status between primary tumor and its metastatic sites has important clinical implications, overall to select the appropriate treatment. To our knowledge, the question of mutational status in metastases versus primary MCT has not been addressed so far.
Compared to previously published studies, the mutational status of our case series, including both primary and secondary metastatic tumors, showed a similar proportion of c‐kit mutations.23 Two already known SNPs were found in exon 8 and 113, 30; furthermore, 3 novel mutations (1 in exon 8 and 2 in exon 11), with unknown clinical relevance, were found.
The data presented in this study provide evidence that the WT or mutated c‐kit genotype is conserved in primary MCTs and their matched, concurrent metastases. Although a similar behavior has been reported in human melanomas,22 this result is somewhat surprising, in the light of genomic instability and heterogeneity that characterize most malignant tumors. In fact, it is generally accepted as true that loss of primary mutation, gain of secondary mutation, or both might occur in patients regardless of the use chemotherapy or targeted therapy; such a phenomenon can be explained by the fact that cells with different mutations coexist within the primary tumor, and clonal selection for mutations during tumor progression might lead to different c‐kit mutations status in metastatic sites from that of the primary tumors.14, 36
In the present analysis, discordant cases were not observed, pointing out that in canine MCTs, c‐kit status is maintained in all cases unchanged during the metastatic process.
Another question, still matter of debate, is whether activating c‐kit mutations might be related to a poor prognosis in canine MCTs.23, 37 Based on our results, dogs with c‐kit mutations had a shorter survival time when compared with dogs with WT MCTs. However, because of the different treatments and the limited number of mutated cases, conclusions on the prognostic relevance of c‐kit mutations cannot be drawn. Also, a number of variables, including sex, reproductive status, breed, age, weight, dimension of the primary lesion, clinical stage, substage, and histologic grading, were evaluated to determine whether they were correlated with c‐kit status. None of these variables was found to be significantly associated with the presence of c‐kit mutations, although the small population might have led to an insufficient power to detect differences between subgroups.
Although the current report is limited by the small sample size, our observations indicate that c‐kit mutation in the primary tumors might predict c‐kit mutated metastases with a reasonably high probability, suggesting that c‐kit mutation represents a very early mutational step in MCT pathogenesis and plays a central role in tumor progression. The implication of these results for general oncology practice is that both tissues of primary tumor or metastasis can be used for c‐kit mutation testing. However, the low number of mutated cases analyzed at the present time does not allow drawing any definitive conclusions about the c‐kit asset in synchronous and metachronous metastases, as well as their association with response to treatment.
Clearly, further molecular studies, carried out on dogs with metastatic MCT and receiving chemotherapy, TKI, or both, are needed to clarify whether c‐kit genotype might be somewhat affected by anticancer drugs.
Finally, it must be stressed that the results of our study are valid for lymph node metastases and cannot be extrapolated to other metastatic locations, as only 1 dog with splenic involvement was evaluated here. The lymph node is the predominant site of metastases in the majority of dogs with metastatic MCT; therefore, the results of our study of 20 lymph node metastases provide a reference for clinical decision‐making as to TKI therapy. Nevertheless, as the molecular patterns might differ between metastatic sites,14, 38 and because c‐kit secondary mutations are likely to occur after TKIs administration,16, 39, 40 more results need to be obtained by testing additional metastatic sites, including spleen and liver, before and after targeted therapies. Also, the identification of new c‐kit ITDs underscores the need of further molecular investigations on their prognostic significance.
In conclusion, the mutational status seems to be stable during MCT metastasis, which is encouraging for TKI use in the clinical setting.
Acknowledgments
Part of the financial support for the study (60A08‐1591/13) as well as Dr Zorzan's PhD fellowship was provided by the Università degli Studi di Padova, Italy.
Conflict of Interest Declaration: Authors disclose no conflict of interest.
Findings of this study were presented in part at the European Society of Veterinary Oncology Meeting, Lisbon, 2013.
Footnotes
Life Technologies, Foster City, CA
TRIzol Reagent; Applied Biosystems, Foster City, CA
Roche Applied Science, Indianapolis, IN
AllPrep DNA/RNA FFPE kit, Qiagen, Milan, Italy
Nanodrop ND‐1000 Spectrophotometer, Nanodrop Technologies, Wilmington, UK
Primer3 software, http://primer3.sourceforge.net
TPersonal, Biometra GmbH, Goettingen, Germany
GoTaq Flexi DNA polymerase; Promega Corp., Madison, WI
Eurofins MWG Operon, Ebersberg, Germany
FinchTV Software, Geospiza Inc., Seattle, WA
GraphPad Prism 5, San Diego, CA
References
- 1. Ma Y, Longley BJ, Wang X, et al. Clustering of activating mutations in c‐kit's juxtamembrane coding region in canine mast cell neoplasms. J Invest Dermatol 1999;112:165–170. [DOI] [PubMed] [Google Scholar]
- 2. London CA, Galli SJ, Yuuki T, et al. Spontaneous canine mast cell tumors express tandem duplications in the proto‐oncogene c‐kit. Exp Hematol 1999;27:689–697. [DOI] [PubMed] [Google Scholar]
- 3. Letard S, Yang Y, Hanssens K, et al. Gain‐of‐function mutations in the extracellular domain of KIT are common in canine mast cell tumors. Mol Cancer Res 2008;6:1137–1145. [DOI] [PubMed] [Google Scholar]
- 4. Downing S, Chien MB, Kass PH, et al. Prevalence and importance of internal tandem duplications in exons 11 and 12 of c‐kit in mast cell tumors of dogs. Am J Vet Res 2002;63:1718–1723. [DOI] [PubMed] [Google Scholar]
- 5. Webster JD, Yuzbasiyan‐Gurkan V, Kaneene JB, et al. The role of c‐KIT in tumorigenesis: Evaluation in canine cutaneous mast cell tumors. Neoplasia 2006;8:104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Webster JD, Yuzbasiyan‐Gurkan V, Miller RA, et al. Cellular proliferation in canine cutaneous mast cell tumors: Associations with c‐KIT and its role in prognostication. Vet Pathol 2007;44:298–308. [DOI] [PubMed] [Google Scholar]
- 7. London CA, Malpas PB, Wood‐Follis SL, et al. Multi‐center, placebo‐controlled, double‐blind, randomized study of oral toceranib phosphate (SU11654), a receptor tyrosine kinase inhibitor, for the treatment of dogs with recurrent (either local or distant) mast cell tumor following surgical excision. Clin Cancer Res 2009;15:3856–3865. [DOI] [PubMed] [Google Scholar]
- 8. Hahn KA, Ogilvie G, Rusk T, et al. Masitinib is safe and effective for the treatment of canine mast cell tumors. J Vet Intern Med 2008;22:1301–1309. [DOI] [PubMed] [Google Scholar]
- 9. Gancberg D, Di Leo A, Cardoso F, et al. Comparison of HER‐2 status between primary breast cancer and corresponding distant metastatic sites. Ann Oncol 2002;13:1036–1043. [DOI] [PubMed] [Google Scholar]
- 10. Scartozzi M, Bearzi I, Berardi R, et al. Epidermal growth factor receptor (EGFR) status in primary colorectal tumors does not correlate with EGFR expression in related metastatic sites: implications for treatment with EGFR‐targeted monoclonal antibodies. J Clin Oncol 2004;22:4772–4778. [DOI] [PubMed] [Google Scholar]
- 11. Italiano A, Vandenbos FB, Otto J, et al. Comparison of the epidermal growth factor receptor gene and protein in primary non‐small‐cell‐lung cancer and metastatic sites: Implications for treatment with EGFR‐inhibitors. Ann Oncol 2006;17:981–985. [DOI] [PubMed] [Google Scholar]
- 12. Brunetti B, Asproni P, Beha G, et al. Molecular phenotype in mammary tumours of queens: Correlation between primary tumour and lymph node metastasis. J Comp Pathol 2013;148:206–213. [DOI] [PubMed] [Google Scholar]
- 13. Beha G, Brunetti B, Asproni P, et al. Molecular portrait‐based correlation between primary canine mammary tumor and its lymph node metastasis: Possible prognostic‐predictive models and/or stronghold for specific treatments? BMC Vet Res 2012;8:219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Amagai Y, Tanaka A, Matsuda A, et al. Heterogeneity of internal tandem duplications in the c‐kit of dogs with multiple mast cell tumours. J Small Anim Pract 2013;54:377–380. [DOI] [PubMed] [Google Scholar]
- 15. Zavodovskaja R, Chien MB, London CA. Use of kit internal tandem duplications to establish mast cell tumor clonality in 2 dogs. J Vet Intern Med 2004;18:915–917. [DOI] [PubMed] [Google Scholar]
- 16. Gao J, Tian Y, Li J, et al. Secondary mutations of c‐KIT contribute to acquired resistance to imatinib and decrease efficacy of sunitinib in Chinese patients with gastrointestinal stromal tumors. Med Oncol 2013;30:522. [DOI] [PubMed] [Google Scholar]
- 17. Blackwood L, Murphy S, Buracco P, et al. European consensus document on mast cell tumours in dogs and cats. Vet Comp Oncol 2012;10:e1–e29. [DOI] [PubMed] [Google Scholar]
- 18. Stefanello D, Valenti P, Faverzani S, et al. Ultrasound‐guided cytology of spleen and liver: A prognostic tool in canine cutaneous mast cell tumor. J Vet Intern Med 2009;23:1051–1057. [DOI] [PubMed] [Google Scholar]
- 19. Patnaik AK, Ehler WJ, MacEwen EG. Canine cutaneous mast cell tumor: Morphologic grading and survival time in 83 dogs. Vet Pathol 1984;21:469–474. [DOI] [PubMed] [Google Scholar]
- 20. Kiupel M, Webster JD, Bailey KL, et al. Proposal of a 2‐tier histologic grading system for canine cutaneous mast cell tumors to more accurately predict biological behavior. Vet Pathol 2011;48:147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kobayashi M, Sugisaki O, Ishii N, et al. Canine intestinal mast cell tumor with c‐kit exon 8 mutation responsive to imatinib therapy. Vet J 2012;193:264–267. [DOI] [PubMed] [Google Scholar]
- 22. Torres‐Cabala CA, Wang W‐L, Trent J, et al. Correlation between KIT expression and KIT mutation in melanoma: A study of 173 cases with emphasis on the acral‐lentiginous/mucosal type. Modern Pathol 2009;22:1446–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Giantin M, Vascellari M, Morello EM, et al. c‐KIT messenger RNA and protein expression and mutations in canine cutaneous mast cell tumors: Correlations with post‐surgical prognosis. J Vet Diagn Invest 2012;24:116–126. [DOI] [PubMed] [Google Scholar]
- 24. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–247. [DOI] [PubMed] [Google Scholar]
- 25. White CR, Hohenhaus AE, Kelsey J, Procter‐Gray E. Cutaneous MCTs: Associations with spay/neuter status, breed, body size, and phylogenetic cluster. J Am Anim Hosp Assoc 2011;47:210–216. [DOI] [PubMed] [Google Scholar]
- 26. Dobson JM. Breed‐predispositions to cancer in pedigree dogs. ISRN Vet Sci 2013;2013:941275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kiupel M, Webster JD, Miller RA, Kaneene JB. Impact of tumour depth, tumour location and multiple synchronous masses on the prognosis of canine cutaneous mast cell tumours. J Vet Med A 2005;52:280–286. [DOI] [PubMed] [Google Scholar]
- 28. Murphy S, Sparkes AH, Blunden AS, et al. Effects of stage and number of tumours on prognosis of dogs with cutaneous mast cell tumours. Vet Rec 2006;158:287–291. [DOI] [PubMed] [Google Scholar]
- 29. den Dunnen JT, Antonarakis SE. Nomenclature for the description of human sequence variations. Hum Genet 2001;109:121–124. [DOI] [PubMed] [Google Scholar]
- 30. Zemke D, Yamini B, Yuzbasiyan‐Gurkan V. Mutations in the iuxtamembrane domain of c‐KIT are associated with higher grade mast cell tumors in dogs. Vet Pathol 2002;39:529–535. [DOI] [PubMed] [Google Scholar]
- 31. London CA. Tyrosine kinase inhibitors in veterinary medicine. Top Companion Anim Med 2009;24:106–112. [DOI] [PubMed] [Google Scholar]
- 32. Fidler IJ, Kripke ML. Metastasis results from preexisting variant cells within a malignant tumor. Science 1977;197:893–895. [DOI] [PubMed] [Google Scholar]
- 33. Katona TM, Jones TD, Wang M, et al. Genetically heterogeneous and clonally unrelated metastases may arise in patients with cutaneous melanoma. Am J Surg Pathol 2007;31:1029–1037. [DOI] [PubMed] [Google Scholar]
- 34. Liegl B, Kepten I, Le C, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol 2008;216:64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Taniguchi K, Okami J, Kodama K, et al. Intratumor heterogeneity of epidermal growth factor receptor mutations in lung cancer and its correlation to the response to gefitinib. Cancer Sci 2008;99:929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dai B, Cai X, Kong Y‐Y, et al. Analysis of KIT expression and gene mutation in human acral melanoma: With a comparison between primary tumors and corresponding metastases/recurrences. Hum Pathol 2013;44:1472–1478. [DOI] [PubMed] [Google Scholar]
- 37. Takeuchi Y, Fujino Y, Watanabe M, et al. Validation of the prognostic value of histopathological grading or c‐kit mutation in canine cutaneous mast cell tumours: A retrospective cohort study. Vet J 2013;196:492–498. [DOI] [PubMed] [Google Scholar]
- 38. Klein CA. Parallel progression of primary tumours and metastases. Nat Rev Cancer 2009;9:302–312. [DOI] [PubMed] [Google Scholar]
- 39. Ando K, Oki E, Sugiyama M, et al. Secondary resistance of extra‐gastrointestinal stromal tumors to imatinib mesylate: Report of a case. Surg Today 2011;41:1290–1293. [DOI] [PubMed] [Google Scholar]
- 40. Wang W‐L, Hornick JL, Mallipeddi R, et al. Cutaneous and subcutaneous metastases of gastrointestinal stromal tumors: A series of 5 cases with molecular analysis. Am J Dermatopathol 2009;31:297–300. [DOI] [PubMed] [Google Scholar]