

Abstract
Background
Evaluation of myocardial function is clinically challenging in dogs with degenerative mitral valve disease (DMVD). Although myocardial dysfunction is caused by pathologic degeneration, histopathologic progression is poorly understood.
Objectives
To characterize myocardial and pulmonary pathologic changes according to severity in dogs with naturally occurring DMVD, and to investigate whether or not pathologic degeneration is reflected by traditional clinical indices.
Animals
One hundred and seventeen dogs with naturally occurring DMVD.
Methods
Prospective observational study. Biopsied left atrium (LA), left ventricle (LV), and lung were evaluated histologically, and an attempt was made to correlate pathologic findings with clinical indices.
Results
Severe myocardial changes were observed in all International Small Animal Cardiac Health Council classes. In the lung, heart failure cell levels were significantly increased in class III patients (P < .0001). In a paired comparison, the LA showed significantly more severe degeneration than the LV, including myocardial fatty replacement, immune cell infiltration, and interstitial fibrosis (P < .0001). In contrast, myocardial cells were more hypertrophied in the LV than in the LA (P < .0001). Left ventricular end‐diastolic dimension (LVEDd) was associated with fatty replacement (P = .033, R 2 = 0.584) and myocardial vacuolization (P = .003, R 2 = 0.588) in the LA.
Conclusions and Clinical Importance
In DMVD, although severe pathologic changes may be evident even in early stages, there may be pathologic discrepancy between the LA and the LV. Myocardial degeneration may be reflected by clinical indices such as LVEDd and EF.
Keywords: Heart failure, Mitral regurgitation, Myocardial degeneration, Myocardial fibrosis
Abbreviations
- DMVD
degenerative mitral valve disease
- EF
ejection fraction
- FS
fractional shortening
- IQR
interquartile ranges
- ISACHC
The International Small Animal Cardiac Heart Council
- LA/Ao
ratio of left atrial‐to‐aortic root diameter
- LA
left atrium
- LVEDd
left ventricular end‐diastolic dimension
- LV
left ventricle
The most common cause of congestive heart failure in small breed dogs is degenerative mitral valve disease (DMVD) characterized by poor coaptation of mitral valve cusps, leading to mitral regurgitation.1, 2 In chronic heart diseases, accurate evaluation of myocardial function is important, because it aids clinicians in planning medical treatment and formulating a prognosis.3, 4 However, assessment of myocardial function is challenging in DMVD because mitral insufficiency induces hemodynamic changes of high volume and low pressure.3, 4 These alterations result in hyperdynamic ventricular wall motion, which can result in misleading echocardiographic measurements.3, 4 Thus, in the presence of DMVD, cardiac function is likely to seem normal, even in the setting of intrinsic myocardial dysfunction that has resulted from pathologic degeneration.
In DMVD, mitral valve leakage leads to a neurohormonal drive to compensate for the decreased forward stroke volume, resulting in cytotoxic environmental conditions such as myocardial ischemia, increased concentration of reactive oxygen species, and disturbances of intracellular calcium cycling.5, 6, 7 These microenvironmental changes worsen myocardial function by inducing pathologic degeneration characterized by loss of myofibrils, myocardial necrosis, and severe deposition of interstitial connective tissue.6, 8, 9, 10, 11 Also, cardiogenic pulmonary congestion, a result of backward failure, causes pulmonary pathologic changes, such as thickened alveolar septa, the presence of heart failure cells, and hyperplasia of type II pneumocytes.12, 13 However, there is little information on myocardial and pulmonary pathologic degeneration in dogs with naturally occurring DMVD. This is because most previous studies on DMVD either have been based on postmortem findings or have focused more on the mitral valvular apparatus than on myocardial integrity.1, 14
Therefore, our study was designed to determine the clinical relevance of microscopic structural degeneration observed on surgical biopsy in dogs with naturally occurring DMVD. The aims of this study were 2‐fold: firstly, to characterize the myocardial and pulmonary pathologic alterations according to clinical severity, and secondly, to investigate whether these pathologic characteristics are reflected in traditionally used clinical indices.
Materials and Methods
Study Population
A total of 117 dogs referred to the Nihon University for mitral valve repair surgery were enrolled in this study. Patients were excluded if they had any evidence of arrhythmias, concurrent congenital cardiac defects, or other systemic diseases. However, relevant cardiovascular diseases that had developed secondary to chronic mitral regurgitation (e.g., pulmonary hypertension) were included. Based on history, physical examination findings, and preoperative cardiac screening, dogs were classified into 3 groups in accordance with the International Small Animal Cardiac Health Council (ISACHC) classification system.15 Written informed consent was obtained from the owners of all dogs, and animal care adhered to the guidelines of the animal ethics committee of Nihon University. The clinical characteristics of the patient population are summarized in Table 1.
Table 1.
Summary of clinical characteristics of the three groups classified by ISACHC system
| ISACHC I | ISACHC II | ISACHC III | |
|---|---|---|---|
| Mean ± SD | Mean ± SD | Mean ± SD | |
| Signalment | |||
| n (117) | 17 | 30 | 70 |
| Age (year) | 8.60 ± 2.60 | 8.57 ± 2.20 | 9.50 ± 2.20 |
| BW (kg) | 6.49 ± 4.21 | 5.88 ± 3.36 | 6.11 ± 3.37 |
| Sex | M (8), F (9) | M (14), F (16) | M (38), F (32) |
| Breed | Chihuahua (5) | Chihuahua (12) | CKCS (19) |
| Maltese (2) | CKCS (6) | Chihuahua (14) | |
| Mix (2) | Maltese (3) | Maltese (12) | |
| Mastiff bull Terrier (1) | Pomeranian (2) | Mix (5) | |
| CKCS (1) | Shiba (1) | Shih Tzu (4) | |
| Shih Tzu (1) | Papillon (1) | Schnauzer (4) | |
| Shetland Sheepdog (1) | Petit Basset (1) | Pomeranian (3) | |
| Papillon (1) | Shetland Sheepdog (1) | Poodle (2) | |
| Border collie (1) | Beagle (1) | Yorkshire Terrier (2) | |
| Poodle (1) | Shih Tzu (1) | Spitz (1) | |
| Dachshund (1) | Mix (1) | Dachshund (1) | |
| Beagle (1) | |||
| Welsh Corgi (1) | |||
| Papillon (1) | |||
| Clinical indices | |||
| VHS | 11.20 ± 1.30 | 11.90 ± 1.00 | 12.60 ± 1.20 |
| LA/Ao | 1.72 ± 0.49 | 2.10 ± 0.49 | 2.38 ± 0.54 |
| LVEDd (mm) | 32.90 ± 9.12 | 35.23 ± 6.66 | 37.74 ± 8.97 |
| LVEDs (mm) | 17.93 ± 7.41 | 18.09 ± 5.11 | 19.46 ± 7.19 |
| FS (%) | 46.71 ± 7.99 | 49.58 ± 6.97 | 55.42 ± 8.92 |
| EF (%) | 77.98 ± 8.78 | 80.15 ± 5.99 | 77.89 ± 5.13 |
| E (cm/s) | 91.50 ± 27.53 | 109.82 ± 25.90 | 118.62 ± 28.12 |
| A (cm/s) | 64.86 ± 14.73 | 70.83 ± 17.15 | 72.53 ± 31.01 |
Abbreviation: ISACHC, International Small Animal Cardiac Health Council; SD, standard deviation; BW, body weight; VHS, vertebral heart score; LA/Ao, left atrial‐to‐aortic diameter ratio; LVEDd, left ventricular end‐diastolic dimension; LVEDs, left ventricular end‐systolic dimension; FS, Fractional shortening; EF, Ejection fraction; E, velocity of E wave; A, velocity of A wave; CKCS, Cavalier King Charles Spaniel.
Biopsy Tissue Preparation
Left atrium (LA), left ventricle (LV), and lung tissue were obtained during the open‐heart surgery for mitral valve repair. Intraoperative biopsy was performed shortly after left atriotomy after cardiac arrest was induced by cardioplegic drug administration. Myocardial samples were taken transmurally from the LA wall. For the LV, biopsy forceps were used to obtain pinch biopsies from the endocardial tissues of the ventricular free wall between papillary muscles. Also, the dorsal margin of the left caudal lobe of the lung was collected. Each sample used in this study measured 3–4 mm3 and weighed 3–5 mg. The specimens were fixed in 10% formalin and embedded in paraffin wax. The embedded sections were stained in a routine manner with hematoxylin‐eosin and Masson's trichrome.
Histologic Morphometry
Target variables used to quantify myocardial degeneration were fatty replacement, immune cell infiltration, vacuolization, widened perinuclear space, enlarged irregular nuclei, diameter of hypertrophied myocytes, and fraction of interstitial fibrosis (Figs 1, 3). In the lung, thickened alveolar septa, heart failure cells, and hyperplasia of type II pneumocytes were selected as the target lesions (Fig 2).
Figure 1.
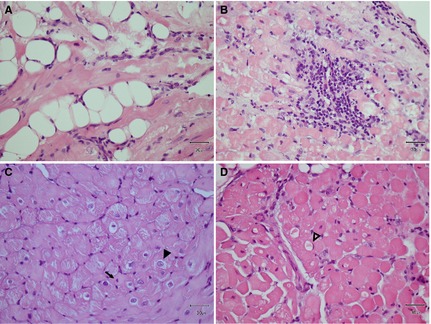
Target variables for myocardial pathologic changes for the quantitative evaluation. (A) Fatty replacement, (B) Inflammatory cell infiltration, (C) Enlarged irregular nuclei and increased perinuclear space (arrow head), distinguished from those of a normal myocardium (arrow), (D) Myocardial vacuolization, often squeezing nucleus to the cell periphery (open arrow head); All hematoxylin‐eosin stain and original magnification (×40).
Figure 3.
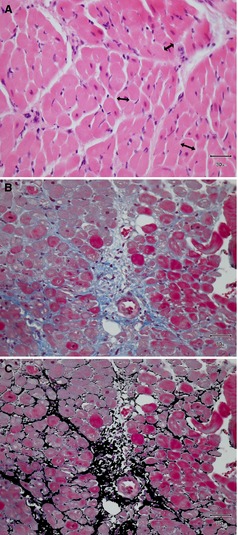
Histologic morphometry of the myocardium by digitization at original magnification (×40). (A) Diameter of myocardium was randomly measured at the level of nucleus (double‐headed arrows), (B) Representative Masson's trichrome stained myocardial section for the quantification of the interstitial fibrous tissue, (C) The same photograph of (B) to illustrate the area of the interstitial connective tissue transformed by computer‐based pixel data.
Figure 2.
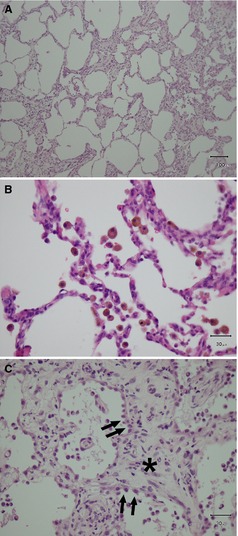
Pathologic lesions for the evaluation of lung tissue. (A) Alveolar septal thickening (×10), (B) Heart failure cells (hemosiderin‐containing macrophages, ×40), (C) Hyperplasia of type II pneumocytes (arrows) with severe interstitial fibrosis (asterisk); All hematoxylin‐eosin stain.
Quantitative evaluation was performed in 2 ways: (i) semiquantitative scoring, and (ii) computer‐based digitizing. The dimensions of the cardiac myocytes and the extent of the interstitial connective tissue were quantified by using a computer‐based image processing program.1 For evaluating myocardial cell hypertrophy, the diameter of cardiac myocytes was determined by measuring the width (axis) of cells on the plane across the nucleus (Fig 3A). One hundred randomly selected myocardial cells were measured in at least 10 different microscopic fields, and the diameters of the myocytes were averaged to determine the value for each sample. Regarding the measurement of interstitial connective tissue, bluish‐stained fibrotic tissue (Fig 3B) in Masson's trichrome specimens was altered into the adopted color that can be digitally identified and quantified (Fig 3C), thus determining the percentage of the total tissue section affected by fibrosis. The area fraction occupied by interstitial fibrous tissue was measured in >10 microscopic views, covering the entire specimen in each sample. All computer‐based digitization was performed at original magnification (40×). The other pathologic findings were assessed using a semiquantitative scoring system. First, each specimen was divided into 4 sections, and 5 nonoverlapping microscopic fields were assessed in each section. When the target pathologic lesions were found within the 5 views, 1 point was counted. As a result, ordinal scales ranging from 0 to 4 were used. The histopathologic characteristics of the biopsy specimens were evaluated without clinical information.
Echocardiography
Standard 2‐dimensional echocardiography2 was performed. The LV dimensions, including fractional shortening (FS) and ejection fraction (EF), were obtained from the right parasternal short‐axis view at the chordae tendineae level, using M‐mode echocardiography. The Teichholz formula was applied to M‐mode images to calculate EF.16 The short‐axis view at the level of the aortic valve was used to measure the ratio of left atrial‐to‐ aortic root diameter (LA/Ao), as described in a previous study.17 Ventricular inflow E and A waves were measured with pulsed wave Doppler in 4‐chamber view of left apical parasternal position. None of the patients was sedated during the examination.
Statistical Analysis
Histograms and Shapiro–Wilk test were used to assess Gaussian distribution of all variables. Clinical data were described as mean and standard deviation; all pathologic data were expressed as median with interquartile ranges (IQR). For the comparisons of the pathologic results among the 3 ISACHC groups, 1‐way ANOVA test was used to analyze continuous variables, followed by Tukey–Kramer posthoc test. For ordinal data, a Kruskal–Wallis ANOVA test was performed, followed by Dunn's multiple comparison. Paired Student's t‐test and Wilcoxon signed ranks test were used to compare the pathologic values between the LA and the LV for continuous and ordinal data, respectively. The influences of pathologic variables upon the clinical indices were investigated by ANCOVA for the ordinal variables and by multiple regression analysis for continuous variables. In multivariate regression analyses, age and body weight were used as covariates to correct imbalances in prognostic factors. The test of homogeneity of regression assumption was satisfied with no interaction between the covariates and the explanatory variables in the prediction of the response variables. Levene's test was performed to test homogeneity of variances. Statistical significance was determined at a value of P < .05. All statistical analyses were performed using a commercially available statistical package.3
Results
Pathologic changes in myocardial and pulmonary tissues were quantitatively investigated in 117 dogs with DMVD (ISACHC I, n = 17; II, n = 30; III, n = 70). Statistically, no difference in all target variables of myocardial degeneration among ISACHC groups was found in both the LA and the LV (Table 2). For pulmonary changes, however, heart failure cells were significantly increased in the ISACHC class III (median, 2; IQR, 1–3; P < .0001) compared with classes I and II (median, 0; IQR, 0–1; Fig 4), whereas other pathologic changes such as thickened alveolar septa and hyperplasia of type II pneumocytes were not significant among ISACHC groups. The proportions of the dogs showing the findings in each group are described as percentages in Table 2.
Table 2.
Histopathologic morphometry according to the clinical severity (ISACHC)
| Variable | ISACHC I (n = 17) Median (IQR)/% | ISACHC II (n = 30) Median (IQR)/% | ISACHC III (n = 70) Median (IQR)/% | P Value |
|---|---|---|---|---|
| Fatty replacement | ||||
| LA | 3 (2–4)/82.4% | 3 (2–4)/96.7% | 4 (3–4)/98.6% | 0.07 |
| LV | 0a/11.8% | 0 (0–1)/20.0% | 0a/17.1% | 0.68 |
| Immune cell infiltration | ||||
| LA | 1 (1–3)/58.8% | 1 (0–3)/63.3% | 1 (0–2)/65.6% | 0.64 |
| LV | 0a/5.9% | 0a/6.7% | 0a/12.8% | 0.52 |
| Vacuolization | ||||
| LA | 4a/100% | 4a/100% | 4a/100% | 0.73 |
| LV | 4a/100% | 4a/100% | 4a/100% | 0.53 |
| Increased perinuclear space | ||||
| LA | 4a/100% | 4 (3–4)/100% | 4a/100% | 0.74 |
| LV | 3 (3–4)/94.1% | 3 (3–4)/100% | 4 (3–4)/100% | 0.11 |
| Enlarged irregular nuclei | ||||
| LA | 4a/100% | 4a/100% | 4a/100% | 0.45 |
| LV | 4a/100% | 4a/100% | 4a/100% | 0.19 |
| Hypertrophy of myocytes (μm) | ||||
| LA | 14.86 (11.70–16.76) | 13.84 (12.10–16.06) | 14.26 (13.06–16.05) | 0.10 |
| LV | 17.54 (15.48–20.84) | 18.55 (17.37–20.03) | 18.93 (17.85–20.28) | 0.44 |
| Interstitial fibrosis (%) | ||||
| LA | 13.59 (9.72–20.47) | 12.85 (9.36–17. 17) | 18.03 (11.33–21.98) | 0.51 |
| LV | 2.46 (1.06–4.99) | 3.02 (1.80–5.44) | 5.73 (3.24–6.36) | 0.75 |
| Thickened alveolar septa | 4 (2–4)/100% | 4 (2–4)/100% | 4 (3–4)/100% | 0.68 |
| Heart failure cell | 0 (0–1)/29.4% | 0 (0–1)/30.0% | 2 (1–3)/71.4% | <0.0001 |
| Hyperplasia of Type II pneumocytes | 3 (3–4)/100% | 4 (3–4)/100% | 4 (3–4)/100% | 0.54 |
Abbreviation: ISACHC, International Small Animal Cardiac Health Council; IQR, Interquartile range; LA, left atrium; LV, left ventricle.
IQR could not be described because 1st (Q1) and 3th (Q3) were the same as median.
%. The percentage of patients that have the findings in each group.
Figure 4.
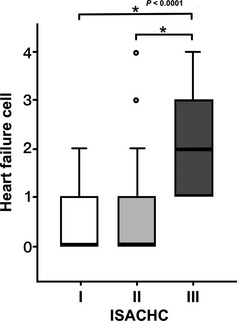
The heart failure cell level in each clinical severity (ISACHC classification). The box plot illustrates the significantly increased heart failure cell in the ISACHC III group than the class I and II with statistical significance marks (asterisk) and P value.
Paired comparisons of myocardial changes between the LA and the LV identified remarkably different pathologic characteristics with respect to fatty replacement, immune cell infiltration, myocardial hypertrophy, and interstitial fibrosis (Fig 5). Compared with the LV, the LA showed significantly increased fatty replacement (median, 3; IQR, 2–4) and immune cell infiltration (median, 1; IQR 0–2), whereas the LV had a value of nearly 0. (Figs 5A,B). The LA also had significantly higher volume fraction of myocardial fibrosis than did the LV (LA: median, 13.38%; IQR, 9.58–20.16% versus LV: median, 2.93%; IQR, 1.31–5.52%; P < .0001; Fig 5D). However, the size of cardiac myocytes was significantly increased in the LV compared with the LA (LA: median, 14.28 μm; IQR, 12.76–16.31 μm versus LV: median, 18.83 μm; IQR, 17.29–20.25 μm; P < .0001; Fig 5C).
Figure 5.
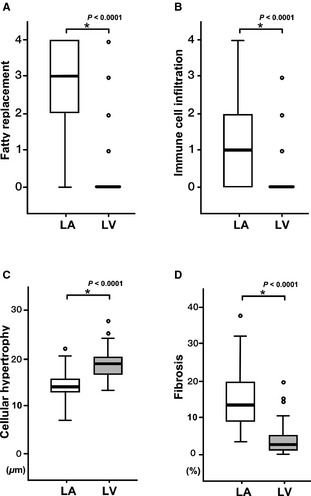
Histopathologic disparity between the LA and the LV. The fatty replacement (A), the immune cell infiltration (B), and the extent of the fibrosis (D) were significantly higher in the LA than in the LV. In particular, the values of the fatty replacement and the inflammatory cell infiltration were nearly zero in the LV. The size of myocytes was significantly larger in the LV than in the LA, indicating cellular hypertrophy (C).
Multivariate linear regression analyses were performed in which the conventional clinical indices (response variables) were analyzed with pathologic findings (explanatory variables) by adjusting with age and body weight (Table. 3). This analysis indicated that left ventricular end‐diastolic dimension (LVEDd) was associated with degeneration of the LA findings such as fatty replacement (R 2 = 0.584, P = .033) and vacuolization (R 2 = 0.588, P = .003). In addition, EF was negatively correlated with the extent of interstitial fibrosis in both LA (R 2 = 0.231, P = .012, β = −0.251) and LV (R 2 = 0.205, P = .036, β = −0.207). Although other indicators (e.g., velocity of E and A wave and LA/Ao) also had correlations with various pathologic findings, their R 2 values were relatively low (Table. 3).
Table 3.
Multivariate analysis adjusted with age and body weight of pathologic characteristics
| LA | LV | Lung | |||||
|---|---|---|---|---|---|---|---|
| Fatty Replacement | Vacuolization | Myocardial Hypertrophy | Interstitial Fibrosis | Increased Perinuclear Space | Interstitial Fibrosis | Heart Failure Cells | |
| LVEDd |
R
2 = 0.584 P = .033 |
R
2 = 0.588 P = .003 |
NS | NS | NS | NS | NS |
| EF | NS | NS | NS |
R
2 = 0.231 P = .012 β = −0.251 |
NS |
R
2 = 0.205 P = .036 β = −0.207 |
NS |
| E Wave | R 2 = 0.105 P = .044 | NS |
R
2 = 0.061 P = .023 β = −0.159 |
NS |
R
2 = 0.148 P = .010 |
R
2 = 0.075 P = .018 β = −0.259 |
R
2 = 0.161 P = .023 |
| A Wave | NS | NS | NS | NS | NS | NS |
R
2 = 0.187 P = .015 |
| LA/Ao |
R
2 = 0.097 P = .040 |
R
2 = 0.104 P = .004 |
R
2 = 0.080 P = .004 |
NS | NS | NS |
R
2 = 0.174 P = .005 |
Abbreviation: LA, left atrium; LV, left ventricle; LVEDd, left ventricular end‐diastolic dimension; EF, Ejection fraction; LA/Ao, left atrial‐to‐aortic diameter ratio; NS, Statistically Insignificant.
β: Standardized coefficients for multiple regressions.
Discussion
Degenerative mitral valve disease (DMVD) characterized by myocardial dysfunction with eccentric cardiac remodeling is the leading cause of heart failure in small breed dogs.1, 2 Myocardial systolic or diastolic dysfunction accrues from histopathologic changes that cause myocardial decompensation, which in turn induces clinical signs such as cough, dyspnea, and exercise intolerance.3, 4, 8, 9, 10 However, knowledge is incomplete regarding the pathologic progression of myocardial degeneration and attendant difficulties in evaluating myocardial dysfunction in DMVD. This study provides useful information on the pathologic characteristics of DMVD observed on surgical biopsy according to severity, and the relationships of these characteristics with the commonly used clinical indicators in DMVD.
The assignment of proper clinical stage plays a fundamental role in planning therapeutic strategy and formulating a prognosis in patients with DMVD.4, 18 However, clinical staging may not adequately reflect intrinsic myocardial condition and structural changes, because guidelines for the classification of heart failure are based on clinical signs.18 Because the clinical signs of DMVD develop slowly over a period of years, they are often recognized only after cardiac remodeling has already occurred.19 Several studies in dogs with experimentally induced mitral valve insufficiency showed severe myocardial degeneration (e.g., myofibrillar loss, increased perinuclear space, and hypertrophy) in the LV as early as 4 months after induction of mild mitral regurgitation.10, 20, 21 Similarly, apoptosis of cardiac myocytes was detected before ventricular hemodynamic impairment or systolic dysfunction in experimental dogs with ventricular pacing.22 Another study using a canine model showed an increase in interstitial fibrosis and infiltration of inflammatory cells in the LA 1 month after partial mitral valve avulsion.23 These previous studies suggest that myocardial pathologic alterations can begin early in both the LA and the LV in the case of mitral valve degeneration, even though the clinical condition of the patient may be stable. Our study showed no significant differences in myocardial degeneration among the different ISACHC classes. This finding suggests that myocardial pathologic changes have already progressed even in the earliest clinical stage of naturally occurring DMVD.
In addition, chronic pulmonary edema leads to damage of the alveolar‐epithelial barrier because of the increased hydrostatic pressure in the pulmonary capillary bed.12, 24 The healing process of the damaged alveolar and interstitial structures involves alveolar septal thickening and mitosis of type II pneumocytes.12, 13, 24 Also, red blood cells that had extravasated into the alveolar space because of the increased pulmonary venous pressure are phagocytosed by alveolar macrophages, which are known as heart failure cells.12, 24 Although there are few studies on pulmonary degeneration resulting from cardiogenic pulmonary congestion in dogs with DMVD, a case report on traumatic mitral valve avulsion described the pathologic findings of lung tissue affected by acute mitral regurgitation.25 In the report, histologic examination identified diffusely thickened alveolar walls, type II pneumocyte hyperplasia, and occasional heart failure cells. These observations are in agreement with the results of our study, which showed highly thickened alveolar septa and increased type II pneumocytes in all ISACHC classes. Considering the chronicity of most cases of DMVD, long‐lasting pulmonary edema in DMVD may cause pathologically more severe degeneration in the lung in comparison with the degeneration associated with acute edema described in the case report. Moreover, this study identified significantly increased heart failure cell level (ordinal score) in the ISACHC III group. This result might be associated with refractory pulmonary edema that often occurs with long‐term diuretic treatment in advanced heart failure.
Mitral regurgitation can injure the LA myocardial cells by mechanical stretching of the atrial chamber and the shear stress created by retrograde jet flow.14, 23 This study demonstrated the disparity of pathologic characteristics between the LA and the LV, where fatty replacement, inflammatory cell infiltration and fibrosis were remarkably severe in the LA. These pathologic findings are very closely associated with inflammatory and reparative processes in the following manner: (i) migration of leukocytes (chemotaxis for phagocytosis), (ii) formation of scar tissue (fibrosis for healing), and (iii) fatty replacement (lipomatous metaplasia).11, 26, 27 A study on mitral regurgitation in humans also has described such pathologic characteristics, with the LA showing more frequent and considerable degenerative changes than the LV.28 In addition, a study on ventricular pacing in a canine model showed more severe pathologic changes in the LA (findings similar to our study) because of the functional mitral regurgitation in congestive heart failure.29 Thus, more severe pathologic changes in the LA probably are a result of the regurgitant flow‐induced chronic and mechanical injury to the LA and the subsequent reparative processes. Conversely, the LV showed not only very few inflammation‐related findings but also significantly higher cellular hypertrophy in this study. The larger size of myocytes in the LV may be explained by compensatory myocardial hypertrophy that is determined by filling volume.30 These findings are consistent with a study that used an animal model of chronic volume overload induced by infrarenal aorto‐caval fistula, in which the compensatory stage of heart failure was characterized by ventricular hypertrophy without necrosis, inflammatory infiltrates, or scar tissue.31 However, some of the histopathologic lesions were not different between the LA and the LV. This finding may be because of the fact that cellular hypertrophy per se is a contributing factor to myocardial degeneration.28, 30, 31, 32 Many studies determined that the alterations in cellular metabolic processes resulting from compensatory myocardial hypertrophy cause swelling or lysis of cellular organelles. This effect is manifested by vacuolization, enlarged irregular nuclei, and increased perinuclear space.6, 28, 30, 31, 32 Therefore, the pathologic discrepancy between the chambers may be attributed to the compensatory myocardial hypertrophy in the LV as well as the mechanical injury and subsequent repair in the LA.
Multiple linear regression analyses adjusted by age and body weight were performed to investigate whether the commonly used clinical indices can reflect myocardial and pulmonary degeneration in DMVD. The results showed that some clinical indices were statistically related to several pathologic variables. Particularly, LVEDd was strongly correlated with the pathologic variables of the LA, such as fatty replacement and vacuolization, suggesting that increase in LVEDd may imply progression to myocardial degeneration in the LA. Increased LVEDd can be useful for identifying volume overload and increased ventricular filling pressure, which also is evidence of the increased pressure in the LA.3, 4 Hence, the association of LVEDd and the degeneration of the LA may be a result of the increased LA filling pressure because of mitral regurgitation. On the other hand, LA/Ao had a significant but weak relationship with several degenerative events in the LA, suggesting that LA/Ao is not sufficient to reflect myocardial degeneration. This finding might be because LA/Ao does not properly reflect the actual situation of degeneration as it can be visualized in 3‐dimensional irregular dilatation of the LA.
Excessive fibrosis has been considered as a cause of myocardial dysfunction.28 In heart diseases, chronic neurohormonal stimulation can result in the deposition of interstitial connective tissue (i.e., reactive fibrosis) as well as myocardial necrosis followed by fibrous tissue formation (i.e., reparative fibrosis).33, 34 A study in humans with dilated cardiomyopathy indicated that deposition of interstitial collagen may cause deterioration in ventricular function. In 1 study, a patient group with increased interstitial fibrosis showed significantly lower EF than a group without increased interstitial fibrosis.35 Another study showed that the volume fraction of myocardial fibrosis was inversely correlated with EF.9 Similarly, in the present study, EF was negatively correlated with myocardial fibrosis of the LV. This result suggests that fibrosis may have a negative effect on the stroke volume ejected from the LV in DMVD. It contradicts the results of other studies using canine models of mitral regurgitation, which found that volume overload triggers degradation of interstitial connective tissue for eccentric cardiac remodeling in the LV.20, 36 However, previous studies were conducted over a short period (i.e., a few weeks) that may have been insufficient to identify the chronic change of myocardial fibrosis in DMVD, which is affected by neurohormonal stress over many years. Further study with advanced echocardiographic techniques such as tissue Doppler or 2‐planimetric method‐based EF is warranted to better understand the alteration of systolic function related to myocardial fibrosis.
Our study also showed that EF was statistically correlated with myocardial fibrosis in the LA. However, it may be a coincidental finding that can be related to the period of time over which myocardial cells of the LA have been affected by regurgitant flow. Moreover, EF and E wave had significant but relatively weak goodness‐of‐fit statistics compared with LVEDd in the regression analysis. This finding might be because the chosen echocardiographic indices do not show steady upward or downward trends over the progression of DMVD.
Although this study provides insight into the histopathologic characteristics in dogs with naturally occurring DMVD and their clinical relevance, it has some limitations. Firstly, it was impractical to compare patients with DMVD to normal, healthy dogs because of the invasiveness of the biopsy procedure. Further study should be carried out to clarify the effect of myocardial senescence caused by aging by comparing normal dogs similar in age. Secondly, the study population was categorized by the ISACHC classification because using a newer staging system (American College of Veterinary Internal Medicine) would have worsened the imbalance of the population size among groups. Nonetheless, the authors believe that the use of the ISACHC system does not decrease the informative value of this study. Thirdly, the biopsied samples, because of their small size, may not represent the entirety of the cardiac muscles and the lung. However, there is no reported evidence that myocardial degeneration occurs only at specific sites in heart failure. Also, pulmonary changes probably are not localized, because all the patients manifested the findings of thickened alveolar septa and hyperplasia of pneumocytes. Fourthly, the Teichholz method, an ejection fraction estimator that was weakly related to myocardial fibrosis in this study, has been shown to be inadequate in dogs with DMVD because of its likelihood to overestimate the volume of the LV, although the method is still being used in individual follow‐ups to monitor disease progression. Further research with advanced techniques for the measurement of EF may be able to more clearly elucidate the relationship between EF and myocardial fibrosis. Lastly, medication provided to each individual was not considered in this study. However, all patients had been managed according to the standard therapeutic protocol for heart failure until surgery.
In conclusion, myocardial and pulmonary pathologic degeneration may be significantly advanced even in early clinical stages in dogs with DMVD. Myocardial degeneration may be attributed to the wound healing processes in the LA and compensatory responses in the LV. Also, these pathologic conditions might be reflected by some clinical indices such as LVEDd and EF.
Acknowledgments
The authors thank Professor Chaewoong Lim (Chonbuk National University) for advice on the pathologic review.
Conflict of Interest Declaration: Authors disclose no conflict of interest.
Off‐label Antimicrobial Declaration: Authors declare no off‐label use of antimicrobials.
Footnotes
ImageJ 1.47v, Bethesda, MD
Toshiba Aprio, Tokyo, Japan.
IBM SPSS V22.0.0, New York, NY.
References
- 1. Detweiler DK, Patterson DF, Hubben K, et al. The prevalence of spontaneously occuring cardiovascular disease in dogs. Am J Public Health Nations Health 1961;51:228–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Borgarelli M, Savarino P, Crosara S, et al. Survival characteristics and prognostic variables of dogs with mitral regurgitation attributable to myxomatous valve disease. J Vet Intern Med 2008;22:120–128. [DOI] [PubMed] [Google Scholar]
- 3. Bonagura JD, Schober KE. Can ventricular function be assessed by echocardiography in chronic canine mitral valve disease? J Small Anim Pract 2009;50:12–24. [DOI] [PubMed] [Google Scholar]
- 4. Chetboul V, Tissier R. Echocardiographic assessment of canine degenerative mitral valve disease. J Vet Cardiol 2012;14:127–148. [DOI] [PubMed] [Google Scholar]
- 5. Gladden JD, Ahmed MI, Litovsky SH, et al. Oxidative stress and myocardial remodeling in chronic mitral regurgitation. Am J Med Sci 2011;342:114–119. [DOI] [PubMed] [Google Scholar]
- 6. Meehan JT. Myotoxins In: Cheville NF, ed. Ultrastructural Pathology: The Comparative Cellular Basis of Disease, 2nd ed Iowa: Wiley‐Blackwell; 2009:635–658. [Google Scholar]
- 7. Gustafsson AB, Gottlieb RA. Heart mitochondria: Gates of life and death. Cardiovasc Res 2007;77:334–343. [DOI] [PubMed] [Google Scholar]
- 8. Urabe Y, Mann DL, Kent RL, et al. Cellular and ventricular contractile dysfunction in experimental canine mitral regurgitation. Circ Res 1992;70:131–147. [DOI] [PubMed] [Google Scholar]
- 9. Schwarz F, Mall G, Zebe H, et al. Quantitative morphologic findings of the myocardium in idiopathic dilated cardiomyopathy. Am J Cardiol 1983;51:501–506. [DOI] [PubMed] [Google Scholar]
- 10. Tsutsui H, Spinale FG, Nagatsu M, et al. Effects of chronic beta‐adrenergic blockade on the left ventricular and cardiocyte abnormalities of chronic canine mitral regurgitation. J Clin Invest 1994;93:2639–2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Su L, Siegel JE, Fishbein MC. Adipose tissue in myocardial infarction. Cardiovasc Pathol 2004;13:98–102. [DOI] [PubMed] [Google Scholar]
- 12. Majno G, Joris I. Disturbances of fluid exchange In: Majno G, Joris I, eds. Cells, Tissues & Disease: Principles of General Pathology, 2nd ed New York: Oxford University Press; 2004:613–629. [Google Scholar]
- 13. Ackermann MR. Toxicity of respiratory systems In: Cheville NF, ed. Ultrastructural Pathology: The Comparative Cellular Basis of Disease, 2nd ed Iowa: Wiley‐Blackwell; 2009:717–736. [Google Scholar]
- 14. Fox PR. Pathology of myxomatous mitral valve disease in the dog. J Vet Cardiol 2012;14:103–126. [DOI] [PubMed] [Google Scholar]
- 15. Recommendations for the diagnosis and the treatment of heart failure in small animals In: Fox PR, Sisson D, Moise NS, eds. Textbook of Canine and Feline Cardiology, 2nd ed Philadelphia, PA: WB Saunders; 1999:883–901. [Google Scholar]
- 16. Teichholz LE, Kreulen T, Herman MV, et al. Problems in echocardiographic volume determinations: Echocardiographic‐angiocardiographic correlations in the presence or absence of asynergy. Am J Cardiol 1976;37:7–11. [DOI] [PubMed] [Google Scholar]
- 17. Rishniw M, Erb HN. Evaluation of four 2‐dimensional echocardiographic methods of assessing Left atrial size in dogs. J Vet Intern Med 2000;14:429–435. [DOI] [PubMed] [Google Scholar]
- 18. Atkins C, Bonagura J, Ettinger S, et al. Guidelines for the diagnosis and treatment of canine chronic valvular heart disease. J Vet Intern Med 2009;23:1142–1150. [DOI] [PubMed] [Google Scholar]
- 19. Dillon AR, Dell'Italia LJ, Tillson M, et al. Left ventricular remodeling in preclinical experimental mitral regurgitation of dogs. J Vet Cardiol 2012;14:73–92. [DOI] [PubMed] [Google Scholar]
- 20. Pat B, Killingsworth C, Denney T, et al. Dissociation between cardiomyocyte function and remodeling with beta‐adrenergic receptor blockade in isolated canine mitral regurgitation. Am J Physiol Heart Circ Physiol 2008;295:H2321–H2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pat B, Chen Y, Killingsworth C, et al. Chymase inhibition prevents fibronectin and myofibrillar loss and improves cardiomyocyte function and LV torsion angle in dogs with isolated mitral regurgitation. Circulation 2010;122:1488–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cesselli D, Jakoniuk I, Barlucchi L, et al. Oxidative stress‐mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ Res 2001;89:279–286. [DOI] [PubMed] [Google Scholar]
- 23. Verheule S, Wilson E, Everett T 4th, et al. Alterations in atrial electrophysiology and tissue structure in a canine model of chronic atrial dilatation due to mitral regurgitation. Circulation 2003;107:2615–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lopez A. Respiratory system, mediastinum, and pleurae In: Zachary JF, McGavin MD, eds. Pathologic Basis of Veterinary Disease, 5th ed Missouri: Mosby; 2012:458–465. [Google Scholar]
- 25. Miller LM, Keirstead ND, Snyder PS. Traumatic mitral valve avulsion from the annulus fibrosis producing acute left heart failure in a dog. Can Vet J 2004;45:761–763. [PMC free article] [PubMed] [Google Scholar]
- 26. Miller LM, Van Vleet JF, Gal A. Cardiovascular system and lymphatic vessels In: Zachary JF, McGavin MD, eds. Pathologic Basis of Veterinary Disease, 5th ed Missouri: Mosby; 2012:539–588. [Google Scholar]
- 27. Baroldi G, Silver MD, De Maria R, et al. Lipomatous metaplasia in left ventricular scar. Can J Cardiol 1997;13:65–71. [PubMed] [Google Scholar]
- 28. Thiedemann KU, Ferrans VJ. Left atrial ultrastructure in mitral valvular disease. Am J Pathol 1977;89:575–604. [PMC free article] [PubMed] [Google Scholar]
- 29. Hanna N, Cardin S, Leung T‐K, et al. Differences in atrial versus ventricular remodeling in dogs with ventricular tachypacing‐induced congestive heart failure. Cardiovasc Res 2004;63:236–244. [DOI] [PubMed] [Google Scholar]
- 30. Lorell BH, Carabello BA. Left ventricular hypertrophy: Pathogenesis, Detection, and Prognosis. Circulation 2000;102:470–479. [DOI] [PubMed] [Google Scholar]
- 31. Brower GL, Janicki JS. Contribution of ventricular remodeling to pathogenesis of heart failure in rats. Am J Physiol Heart Circ Physiol 2001;280:H674–H683. [DOI] [PubMed] [Google Scholar]
- 32. Hilfiker‐Kleiner D, Landmesser U, Drexler H. Molecular mechanisms in heart failure focus on cardiac hypertrophy, inflammation, angiogenesis, and apoptosis. J Am Coll Cardiol 2006;48:A56–A66. [Google Scholar]
- 33. de Jong S, van Veen TA, de Bakker JM, et al. Biomarkers of myocardial fibrosis. J Cardiovasc Pharmacol 2011;57:522–535. [DOI] [PubMed] [Google Scholar]
- 34. Tan LB, Jalil JE, Pick R, et al. Cardiac myocyte necrosis induced by angiotensin II. Circ Res 1991;69:1185–1195. [DOI] [PubMed] [Google Scholar]
- 35. Nakayama Y, Shimizu G, Hirota Y, et al. Functional and histopathologic correlation in patients with dilated cardiomyopathy: An integrated evaluation by multivariate analysis. J Am Coll Cardiol 1987;10:186–192. [DOI] [PubMed] [Google Scholar]
- 36. Dell'Italia LJ, Balcells E, Meng QC, et al. Volume‐overload cardiac hypertrophy is unaffected by ACE inhibitor treatment in dogs. Am J Physiol 1997;273:H961–H970. [DOI] [PubMed] [Google Scholar]