

Abstract
Drug–drug interactions can cause unanticipated patient morbidity and mortality. The consequences of drug–drug interactions can be especially severe when anticancer drugs are involved because of their narrow therapeutic index. Veterinary clinicians have traditionally been taught that drug–drug interactions result from alterations in drug metabolism, renal excretion or protein binding. More recently, drug–drug interactions resulting from inhibition of P‐glycoprotein‐mediated drug transport have been identified in both human and veterinary patients. Many drugs commonly used in veterinary patients are capable of inhibiting P‐glycoprotein function and thereby causing an interaction that results in severe chemotherapeutic drug toxicity. The intent of this review is to describe the mechanism and clinical implications of drug–drug interactions involving P‐glycoprotein and anticancer drugs. Equipped with this information, veterinarians can prevent serious drug–drug interactions by selecting alternate drugs or adjusting the dose of interacting drugs.
Keywords: ABCB1, Chemotherapy, Doxorubicin vincristine, Drug Interaction, Ketoconazole, MDR1, Oncology, Spinosad
Abbreviations
- ABC
ATP‐binding cassette superfamily
- ATP
adenosine triphosphate
- MDR
multidrug resistance
- P‐gp
p‐glycoprotein
In the United States, more than 2 million serious adverse drug reactions occur annually in hospitalized human patients with over 100,000 of these resulting in death.1, 2, 3 These statistics do not include adverse drug events in nonhospitalized patient populations. Drug–drug interactions are estimated to represent 3–5% of these events.1 Statistics regarding adverse drug events and drug–drug interactions in veterinary patients are not available. However, it is reasonable to assume that drug–drug interactions are a preventable cause of morbidity and death in veterinary patients. The margin of error for anticancer drugs is extremely low because dosage rates tend to approach the maximum tolerated dose. Thus, any drug–drug interaction that impacts the clearance of anticancer drugs escalates the likelihood of life‐threatening toxicosis.
Understanding the mechanisms involved in drug–drug interactions is an important step in preventing their occurrence. The four mechanisms that are typically cited as causing drug–drug interactions include pharmaceutical interactions, inhibition of drug metabolism, inhibition of renal excretion, and displacement of highly protein bound drugs. More recently, interference with ATP binding cassette (ABC) transporters has been identified as a mechanism responsible for clinically important drug–drug interactions.4 ABC drug transporters play key roles in limiting drug distribution to sensitive tissues (ie, blood–brain barrier)5, 6, 7 and in biliary drug excretion, a key elimination pathway for many anticancer drugs.5, 8, 9, 10 The ABC drug efflux transporter P‐glycoprotein (P‐gp) is especially prone to being involved in serious drug–drug interactions involving anticancer drugs because (i) several classes of anticancer drugs used in veterinary medicine are substrates for P‐gp and (ii) a wide variety of drugs used in veterinary patients can inhibit P‐gp‐mediated drug efflux.4, 10, 11
P‐Glycoprotein Tissue Distribution and Function
P‐gp, the most well characterized drug transporter in the ABC protein superfamily is encoded by the ABCB1, previously named MDR1, gene.12 Among oncologists, P‐gp may be most well‐known for its role in mediating chemotherapeutic multidrug resistance. Justifiably, when P‐gp was first discovered in a multidrug resistant cell line, the gene encoding it was designated the multidrug resistance (mdr) gene. P‐gp causes multidrug resistance by using energy derived from ATP hydrolysis to transport substrates across the plasma membrane often against a steep concentration gradient.13 Because the transport is unidirectional, from within the cell to the extracellular space, tumor cells expressing P‐gp have relatively low intracellular concentrations of anticancer drugs that are transported by (substrates for) P‐gp compared to tumor cells that do not express P‐gp. Thus, tumor cells expressing P‐gp are resistant to a variety of structurally and functionally diverse anticancer drugs that are P‐gp substrates (Table 1).12, 13
Table 1.
| Drug or Drug Class | Based on data in humans or rodents | Based on data/experience in dogs |
|---|---|---|
| Actinomycin D | Yes | |
| Alkylating Agents | No | No |
| Antimetabolites | No | |
| Camptothecins | Yes | Yes |
| Daunorubicin | Yes | No |
| Doxorubicin | Yes | No |
| Epipodophyllotoxins | Yes | Yesa |
| L‐asparaginase | No | |
| Mitoxantrone | No | |
| Platinum compounds | No | |
| Taxanes | Yes | |
| Tyrosine kinase inhibitors | Yes | |
| Vinca Alkaloids | Yes | Yes |
Case study.
Alkylating agents = chlorambucil, cyclophosphamide, lomustine, others.
Antimetabolites = Cytarabine, 5‐fluorouracil, gemcitabine, methotrexate, others.
Camptothecins = irinotecan, topotecan.
Epipodophyllotoxins = etoposide, teniposide.
Taxanes = paclitaxel, docetaxel.
Tyrosine kinase inhibitors = imatanib, masitinib, nilotinib, toceranib.
Vinca alkaloids = vinblastine, vincristine, vinorelbine.
Despite its important role in mediating chemotherapeutic drug resistance, it is doubtful that P‐gp actually evolved to protect tumor cells from anticancer drugs. It was not until many years later that researchers began investigating a possible physiologic function for the transporter. Expression of P‐gp was identified in nonneoplastic tissues initially in humans and rodents and much later in companion animal species.5, 14 The highest levels of P‐gp expression by “normal” cells occurs in tissues that either serve as barriers to drug absorption (apical border of intestinal epithelial cells), enhance drug elimination from the body (biliary canalicular or renal tubular epithelial cells), or on capillary endothelial cells at so‐called sanctuary sites (blood–brain barrier; testes; and placenta).12 Because of its strategic location and its highly efficient drug efflux function, P‐gp limits oral absorption, enhances excretion, and prevents entry of substrate drugs into specialized tissues.5, 12, 13 From an evolutionary perspective, it is presumed that P‐gp functions in a protective capacity for mammalian organisms by decreasing their exposure to potentially toxic xenobiotics.12 Thus, it should not be surprising that animals with defective P‐gp function are highly susceptible to toxicosis when treated with drugs that are P‐gp substrates.15, 16, 17 Substrates for P‐gp include not only anticancer drugs (Table 1), but a wide variety of other drugs as well (Table 2).
Table 2.
| Drug class | Based on data in humans, rodents or dogs* |
|---|---|
| Antimicrobial agents | Erythromycin |
| Ketoconazole | |
| Itraconazole | |
| Tetracycline | |
| Doxycycline | |
| Levofloxacin | |
| Sparfloxacin | |
| Antiparasitic agents | Doramectin |
| Ivermectin* | |
| Milbemycin* | |
| Moxidectin* | |
| Selamectin* | |
| Cardiac drugs | Digoxin* |
| Diltiazem | |
| Verapamil | |
| Immunosuppressants | Cyclosporine |
| Tacrolimus | |
| Opioids | Butorphanol* |
| Loperamide* | |
| Miscellaneous | Acepromazine* |
| Ondansetron | |
| Domperidon |
The asterisk indicates drugs for which evidence in the dog (specifically) exists; otherwise data was from human or rodent studies.
Much of what is known about P‐gp's role in drug disposition in veterinary patients has been generated from studies and clinical observations of dogs affected by a well‐characterized mutation in the ABCB1 (MDR1) gene. This particular polymorphism [ABCB1‐1Δ; nt230(del4)MDR1; “the MDR1 mutation”] consists of a 4 base‐pair deletion mutation at the 5′ end of the ABCB1 gene.16 The deletion results in a shift of the reading frame that generates several premature stop codons terminating protein synthesis before 10% of the protein product is synthesized. Dogs with two mutant alleles (MDR1 mutant/mutant) exhibit a P‐gp null phenotype, similar to abcb1 (mdr1) (‐/‐) knockout mice.18 Heterozygotes, dogs with one mutant allele and one wild‐type allele (MDR1 mutant/normal) have an intermediate phenotype with decreased P‐gp function compared to wild‐type (MDR1 normal/normal) dogs.9 Affected dogs include many herding and some sight‐hound breeds (Table 3).19, 20, 21, 22, 23, 24 Intrinsic P‐gp dysfunction in these dogs dramatically illustrates the important role P‐gp plays in the disposition of substrate drugs, particularly drug distribution and biliary drug excretion.6, 9, 17, 25, 26
Table 3.
Percent of dog breeds that are heterozygous for ABCB1‐1Δ and are presumed to be at highest risk for drug–drug interactions involving P‐glycoprotein
| Breed | Approximate % heterozygotes |
|---|---|
| Australian Shepherd | 40 |
| Border Collie | <5 |
| Collie | 45 |
| English Shepherd | <5 |
| German Shepherd | 10 |
| Herding breed mix | 10 |
| Longhaired Whippet | 50 |
| Miniature Australian Shepherd | 35 |
| Mixed Breed | <5 |
| Old English Sheepdog | <5 |
| Shetland Sheepdog | 10 |
| Silken Windhound | 30 |
Because P‐gp is a component of the blood–brain barrier, distribution of P‐gp substrate drugs to the brain is greatly increased in dogs homozygous for the MDR1 mutation (MDR1 mutant/mutant) dogs and moderately increased in heterozygous dogs (MDR1 mutant/normal).6, 16, 25, 26 Macrocyclic lactones such as ivermectin can cause neurological toxicosis in any animal at high doses (greater than 2 mg/kg).27 MDR1 mutant/mutant dogs experience severe neurological toxicosis at much lower doses (~120 μg/kg) because the defective blood–brain barrier allows ivermectin to accumulate in brain tissue of these animals.16, 28 Other P‐gp substrates (eg, loperamide, ondansetron, acepromazine, butorphanol, milbemycin, selamectin, and moxidectin) are more likely to cause neurological toxicosis in dogs with the MDR1 mutation compared to wild‐type dogs.6, 11, 26 At typical doses, loperamide causes central nervous system depression in MDR1 mutant/mutant dogs, but not in wild‐type dogs.6 Acepromazine and butorphanol cause more profound and prolonged CNS depression in dogs with the MDR1 mutation compared to wild‐type dogs.1 Collectively, these examples underscore P‐gp's critical role in drug disposition at the blood–brain barrier.
Drug excretion, like drug distribution is also an important P‐gp function. P‐gp is expressed on both renal tubular cells and biliary canalicular cells but whether or not it plays a clinically relevant role in renal drug excretion in dogs is not known.5 Conversely, ample evidence exists demonstrating the important role P‐gp plays in biliary drug excretion in dogs. Studies comparing biliary excretion of the radiolabeled P‐gp substrate 99mTc‐sestamibi in MDR1 normal/normal and MDR1 mutant/mutant dogs demonstrate that MDR1 mutant/mutant dogs are unable to excrete this compound into bile.9 99mTc‐sestamibi is essentially undetectable in gallbladders of MDR1 mutant/mutant dogs but is highly concentrated in gallbladders of MDR1 normal/normal dogs (Fig 1). Similarly, in studies involving mdr1 knockout mice, biliary excretion of the P‐gp substrate irinotecan was significantly decreased, roughly 40%, in mdr1 knockout mice compared to wild‐type mice.29 Extrinsic P‐gp deficiency, because of drug‐induced inhibition of P‐gp, can also decrease biliary excretion of P‐gp substrates. In rats for example, concurrent administration of a P‐gp‐inhibitor decreases the biliary clearance of doxorubicin, a P‐gp substrate, resulting in increased plasma concentrations of doxorubicin.8 Similarly, administration of verapamil, a P‐gp inhibitor, to rats decreased biliary excretion of the P‐gp substrate irinotecan by half.30 Decreased biliary excretion of chemotherapy drugs would increase the patient's overall exposure to the drug, with a corresponding increase in the likelihood of drug‐induced toxicity such as myelosuppression and adverse gastrointestinal effects.
Figure 1.
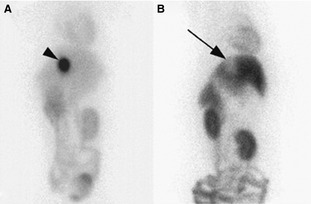
Ventral images of the abdomen acquired at 120 minutes after intravenous injection of 99 mTc‐sestamibi to an MDR1 normal/normal dog (a) and an MDR1 mutant/mutant dog (b). Intense 99mTc‐sestamibi activity (arrow head) is present in the gallbladder in the MDR1 normal/normal whereas a void of activity is observed in the location of the gallbladder in the MDR1 mutant/mutant dog.9 Reproduced from: Coelho JC 1, Tucker R, Mattoon J, Roberts G, Waiting DK, Mealey KL Biliary excretion of technetium‐99m‐sestamibi in wild‐type dogs and in dogs with intrinsic (ABCB1‐1Delta mutation) and extrinsic (ketoconazole treated) P‐glycoprotein deficiency. J Vet Pharmacol Ther. 2009 Oct;32(5):417–421.
P‐gp Deficiency and Sensitivity to Anticancer Drugs
Diminished biliary drug excretion is considered to be the mechanism responsible for the increased sensitivity of MDR1 mutant/mutant and MDR1 mutant/normal dogs to the myelosuppressive and gastrointestinal effects of chemotherapeutic drugs that are P‐gp substrates. A prospective study demonstrated that these dogs are significantly more likely to develop hematologic toxicity, both neutropenia and thrombocytopenia, after treatment with the P‐gp substrate vincristine (0.5–0.7 mg/m2) than MDR1 normal/normal dogs.17 The authors considered a similar study assessing doxorubicin toxicosis in dogs with the MDR1 mutation, but abandoned the study because the first two MDR1 mutant/mutant dogs that received doxorubicin (30 mg/m2) died from overwhelming neutropenic sepsis. In dogs, vincristine is eliminated primarily via biliary excretion of parent drug with some urinary excretion of parent drug and metabolites.31 In other species, biliary excretion of doxorubicin is a critical component of its overall clearance.8 Therefore, MDR1 mutant/mutant and MDR1 mutant/normal dogs should receive reduced doses of P‐gp substrate chemotherapeutic agents (eg, doxorubicin, vincristine, vinblastine) in order to avoid severe toxicosis. The fact that MDR1 mutant/mutant and MDR1 mutant/normal dogs tolerate full doses of non‐P‐gp substrate chemotherapeutic drugs such as alkylating agents, platinum compounds and antimetabolites lends credence to the contention that deficient P‐gp‐mediated biliary excretion is the mechanism responsible for the increased sensitivity of MDR1 mutant/mutant and MDR1 mutant/normal dogs to chemotherapeutic drugs that are P‐gp substrates.
Increased brain penetration of P‐gp substrates such as macrocyclic lactones, loperamide, acepromazine, and butorphanol causes greater CNS depression in MDR1 mutant/mutant and MDR1 mutant/normal than in MDR1 normal/normal dogs.6, 11 For these drugs, CNS depression is an adverse effect observed (and expected) when these drugs are administered at high or extremely high doses to any patient. For chemotherapeutic drugs, CNS depression is not a common adverse reaction. However, intrathecal administration of vincristine in human patients is associated with a central neuropathy consisting of ascending progressive radiculomyeloencephalopathy.32 Vincristine‐induced central neurotoxicity occurred after intravenous, not intrathecal, administration in a collie (MDR1 mutant/mutant) described in a recent case report.33 Because extensive work‐up including advanced imaging, cerebrospinal fluid analysis and infectious disease (Neospora, Toxoplasma) titers ruled out other potential causes for the dog's neurological signs, it was concluded that vincristine was able to penetrate this dog's blood–brain barrier because of P‐gp deficiency. Thus, it is important to keep in mind enhanced neurological toxicity as a sequelae of P‐gp deficiency for those chemotherapeutic agents that are both neurotoxic and substrates for P‐gp.
P‐gp Inhibition Mimics Genetically Mediated P‐gp Deficiency
Because P‐gp contributes to multidrug resistant tumor phenotypes in cancer patients, numerous drug candidates have been developed to inhibit P‐gp in an effort to improve outcome in patients treated with chemotherapy.10, 34 In human clinical trials involving P‐gp inhibitors unexpected and undesired pharmacokinetic interactions ensued between the inhibiting agents and the chemotherapeutic drugs. As a result, patients experienced severe adverse effects necessitating discontinuation or dose reductions of the chemotherapeutic drug(s).4, 35 For example, the potent P‐gp inhibitor valspodar was concurrently administered with the P‐gp substrate vinblastine to human patients with advanced malignancies. Because of severe toxicoses (myelosuppression and gastrointestinal) related to relative increases in vinblastine exposure, many patients required significant dose reductions (~50%) for subsequent vinblastine treatments.10 A different clinical trial in human patients with advanced cancer, this one using paclitaxel as the cytotoxic P‐gp substrate and valspodar as the P‐gp inhibitor, yielded similar results. Dose‐limiting neutropenia necessitated paclitaxel dose reduction to compensate for the drug–drug interaction.36 Numerous other studies have documented these types of drug–drug interactions whereby a P‐gp inhibiting drug increases a patient's overall exposure to a P‐gp substrate cytotoxic drug including vinca alkaloids, doxorubicin and paclitaxel (reviewed by Varma).37 The current consensus regarding overcoming the phenomenon of P‐gp‐mediated chemotherapeutic resistance is that pharmacological P‐gp inhibitors cannot discriminate between P‐gp expressed by normal tissues, which protects the patient, and P‐gp expressed in cancerous tissue, which harms the patient.4, 35, 36, 37 Therefore, despite in vitro success in using P‐gp inhibitors to enhance a chemotherapeutic drug's ability to kill tumor cells, this strategy has been abandoned in clinical practice because of enhanced toxicity in the patient.
P‐gp inhibitors used in the human clinical trials just described were specifically synthesized to be used in combination with P‐gp substrate anticancer drugs. However, many drugs commonly used in veterinary patients are also capable of inhibiting P‐gp function (Table 3). Examples include a variety of structurally and functionally unrelated compounds including ketoconazole, spinosad, cyclosporine, omeprazole and others.4, 9, 38 It is essential for veterinarians working with animals receiving cytotoxic P‐gp substrates to understand that pharmacological P‐gp inhibition, so‐called “acquired” or “extrinsic” P‐gp deficiency, can mimic what occurs in dogs with the MDR1 mutation, so‐called “endogenous” or “intrinsic” P‐gp deficiency. For example ketoconazole increases brain penetration of ivermectin in MDR1 normal/normal dogs causing the same neurological toxicity one would expect to see in a Collie with the MDR1 mutation (unpublished data). Ketoconazole's ability to inhibit P‐gp‐mediated biliary excretion of P‐gp substrates has been demonstrated using the radiolabelled P‐gp substrate 99mTc‐sestamibi (Fig 2).9 MDR1 normal/normal dogs had 1.8‐fold greater biliary clearance of 99mTc‐sestamibi before treatment with the P‐gp inhibitor ketonazole than after treatment with ketoconazole. Similar effects would be expected for chemotherapeutic drugs whose clearance involves P‐gp mediated biliary excretion (ie, vinca alkaloids, doxorubicin, taxol). Because these drugs have a narrow therapeutic index, the delayed clearance resulting from the drug–drug interaction would produce unexpectedly severe adverse effects in the patient relative to the dose administered.
Figure 2.
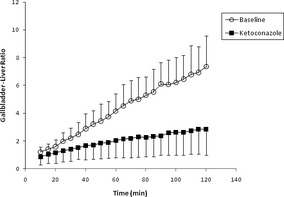
Time–activity curves of gallbladder to liver activity ratios [using mean counts per pixel per regions of interest (ROI)] for ABCB1 wild/wild dogs before (○: mean G/L ratio +SD, n = 6) and after (°: mean G/L ratio −SD, n = 6) administration of ketoconazole (5 mg/kg PO q12 h × 9 doses).9 Reproduced from: Coelho JC 1, Tucker R, Mattoon J, Roberts G, Waiting DK, Mealey KL Biliary excretion of technetium‐99 m‐sestamibi in wild‐type dogs and in dogs with intrinsic (ABCB1‐1Delta mutation) and extrinsic (ketoconazole treated) P‐glycoprotein deficiency. J Vet Pharmacol Ther. 2009 Oct;32(5):417–421.
This is presumed to be what happened to a dog (MDR1 normal/normal) being treated with vinblastine for a grade II mast cell tumor. The dog experienced unexpectedly severe adverse effects (Grade 4 neutropenia and Grade 3 vomiting) after receiving a standard dose of vinblastine (2 mg/m2) concurrently with ketoconazole. Ketoconazole had been prescribed to treat a presumed fungal infection. The severe vomiting and neutropenia led to sepsis, multiple metabolic disturbances and cardiac arrest. In humans, nearly 30% of an intravenous dose of vinblastine is actively excreted into the bile by P‐gp.39 Ketoconazole also inhibits cytochrome P450 (CYP)‐mediated metabolism of vinblastine.2 Thus, this patient experienced an overdose of vinblastine, despite administration of the recommended dose, because of ketoconazole's inhibitory effects on CYP and P‐gp‐mediated vinblastine elimination. Concurrent use of ketoconazole with other P‐gp substrate chemotherapeutic agents (Table 1) would be expected to cause similar adverse effects.
There is currently both anecdotal and experimental evidence that drug–drug interactions involving P‐glycoprotein occur in MDR1 normal/normal dogs. These drug interactions can be severe. However, it is likely that MDR1 mutant/normal dogs (Table 3) would have an even higher risk of clinically significant P‐glycoprotein‐mediated drug–drug interactions because of intrinsically diminished P‐glycoprotein function.
P‐gp Inhibitors
P‐gp inhibitors can act (i) by blocking the drug binding site either competitively, noncompetitive or allosterically; (ii) by interfering with ATP hydrolysis which is required for P‐gp function; or (iii) by altering integrity of cell membrane lipids.4 From a clinical perspective, the most important mechanism involves competitively and noncompetitively blocking drug binding sites. It is essential to note that P‐gp has more than one drug (substrate) binding site, complicating our understanding of drug–drug interactions involving P‐gp. Site‐directed mutagenesis studies have demonstrated that certain mutations alter P‐gp's affinity for some substrates, but not others.40 Thus, it is possible that some P‐gp inhibitors might affect transport of vinblastine, for example, but not doxorubicin. Unfortunately the factors that determine P‐gp binding site selectivity have not yet been determined. Consequently it is not possible to predict which P‐gp substrate drugs in Table 1 would be safe to use concurrently with P‐gp inhibiting drugs in Table 4.
Table 4.
| Drug class | Based on data in humans or rodents |
|---|---|
| Antidepressants | Fluoxetine |
| Paroxetine | |
| Antimicrobial agents | Erythromycin |
| Ketoconazolea | |
| Itraconazole | |
| Cardiac drugs | Diltiazem |
| Nicardepine | |
| Quinidine | |
| Verapamil | |
| Immunosuppressants | Cyclosporine |
| Tacrolimus | |
| Miscellaneous | Spinosada |
| Tamoxifen |
Evidence of P‐gp‐mediated drug–drug interactions in dogs at clinically used doses.
Conclusion
Many years passed between the initial discovery of P‐gp and the realization that P‐gp contributed substantially to the disposition of chemotherapeutic drugs not only within tumor cells, but in the patient. The importance of P‐gp mediated drug–drug interactions has only recently been identified as a clinically important problem in human patients. Thus, drug package inserts may not contain information about P‐gp‐mediated drug–drug interactions. Improved understanding of P‐gp drug binding sites and P‐gp dependent drug clearance and distribution mechanisms will enhance our ability to more specifically predict P‐gp‐mediated drug–drug interactions. Until then, veterinarians will need to take a proactive role in considering potential drug interactions involving P‐gp.
Acknowledgments
The authors thank Beryl Swanson for summarizing the medical record for one of the cases described in this report.
Conflict of Interest Declaration: Authors disclose no conflict of interest.
Off‐label Antimicrobial Declaration: Authors declare no off‐label use of antimicrobials.
Footnotes
Deshpande E, Gieseg M, Hill K, Bridges, K Chambers P. The pharmacogenetic effects of the MDR1‐1D mutation on sedation of dogs with acetylpromazine. ACVIM Forum abstract 2014
Vinblastine package insert
References
- 1. Leape LL, Bates DW, Cullen DJ, et al. Systems analysis of adverse drug events. ADE Prevention Study Group. J Am Med Assoc 1995;274:35–43. [PubMed] [Google Scholar]
- 2. Raschetti R, Morgutti M, Menniti‐Ippolito F, et al. Suspected adverse drug events requiring emergency department visits or hospital admissions. Eur J Clin Pharmacol 1999;54:959–963. [DOI] [PubMed] [Google Scholar]
- 3. Lazarou J, Pomeranz B, Corey PN. Incidence of adverse drug reactions in hospitalized patients: A meta‐analysis of prospective studies. J Am Med Assoc 1998;279:1200–1205. [DOI] [PubMed] [Google Scholar]
- 4. Marchetti S, Mazzanti R, Beijnen JH, Schellens JHM. Concise review: Clinical relevance of drug‐drug and herb‐drug interactions mediated by the ABC Transporter ABCB1 (MDR1, P‐glycoprotein). Oncologist 2007;12:927–941. [DOI] [PubMed] [Google Scholar]
- 5. Leslie EM, Deeley RG, Cole SP. Multidrug resistance proteins: Role of P‐glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol 2005;204:216–237. [DOI] [PubMed] [Google Scholar]
- 6. Mealey KL, Greene S, Bagley R, et al. P‐glycoprotein contributes to the blood‐brain, but not blood‐cerebrospinal fluid, barrier in a spontaneous canine p‐glycoprotein knockout model. Drug Metab Dispos 2008;36:1073–1079. [DOI] [PubMed] [Google Scholar]
- 7. Choo EF, Leake B, Wandel C, et al. Pharmacological inhibition of P‐glycoprotein transport enhances the distribution of HIV‐1 protease inhibitors into brain and testes. Drug Metab Dispos 2000;28:655–660. [PubMed] [Google Scholar]
- 8. Iyer L, Ramirez J, Shepard DR, et al. Biliary transport of irinotechan and metabolites in normal and P‐glycoprotein deficient mice. Cancer Chemother Pharmacol 2002;49:336–341. [DOI] [PubMed] [Google Scholar]
- 9. Coelho JC, Tucker R, Mattoon J, et al. Biliary excretion of technetium‐99 m‐sestamibi in wild‐type dogs and in dogs with intrinsic (ABCB1‐1Delta mutation) and extrinsic (ketoconazole treated) P‐glycoprotein deficiency. J Vet Pharmacol Ther 2009;32:417–421. [DOI] [PubMed] [Google Scholar]
- 10. Callaghan R, Luk F, Bebawy M. Inhibition of the multidrug resistance P‐glycoprotein: Time for a change of strategy. Drug Metab Disp 2014;42:623–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mealey KL. Therapeutic implications of the MDR‐1 gene. J Vet Pharmacol Ther 2004;27:257–264. [DOI] [PubMed] [Google Scholar]
- 12. Dean M, Annilo T. Evolution of the ATP‐binding cassette (ABC) transporter superfamily in vertebrates. Annu Rev Genomics Hum Genet 2005;6:123–142. [DOI] [PubMed] [Google Scholar]
- 13. DeGorter MK, Xia CQ, Yang JJ, Kim RB. Drug transporters in drug efficacy and toxicity. Annu Rev Pharmacol Toxicol 2012;52:249–273. [DOI] [PubMed] [Google Scholar]
- 14. Ginn PE. Immunohistochemical detection of P‐glycoprotein in formalin‐fixed and paraffin‐embedded normal and neoplastic canine tissues. Vet Pathol 1996;33:533–541. [DOI] [PubMed] [Google Scholar]
- 15. Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Adv Drug Deliv Rev 2003;55:3–29. [DOI] [PubMed] [Google Scholar]
- 16. Kerb R. Implications of genetic polymorphisms in drug transporters for pharmacotherapy. Cancer Lett 2006;234:4–33. [DOI] [PubMed] [Google Scholar]
- 17. Mealey KL, Bentjen SA, Gay JM, Cantor GH. Ivermectin sensitivity in collies is associated with a deletion mutation of the mdr1 gene. Pharmacogenetics 2001;11:727–733. [DOI] [PubMed] [Google Scholar]
- 18. Mealey KL, Fidel J, Gay JM, et al. ABCB1‐1Delta polymorphism can predict hematologic toxicity in dogs treated with vincristine. J Vet Intern Med 2008;22:996–1000. [DOI] [PubMed] [Google Scholar]
- 19. Schinkel AH. Pharmacological insights from P‐glycoprotein knockout mice. Int J Clin Pharmacol Ther 1998;36:9–13. [PubMed] [Google Scholar]
- 20. Mealey KL, Meurs KM. Breed distribution of the ABCB1‐1Delta (multidrug sensitivity) polymorphism among dogs undergoing ABCB1 genotyping. J Am Vet Med Assoc 2008;233:921–924. [DOI] [PubMed] [Google Scholar]
- 21. Gramer I, Leidolf R, Doring B, et al. Breed distribution of the nt230(del4) MDR1 mutation in dogs. Vet J 2011;189:67–71. [DOI] [PubMed] [Google Scholar]
- 22. Kawabata A, Momoi Y, Inoue‐Murayama M, Iwasaki T. Canine mdr1 gene mutation in Japan. J Vet Med Sci 2005;67:1103–1107. [DOI] [PubMed] [Google Scholar]
- 23. Mealey KL, Munyard KA, Bentjen SA. Frequency of the mutant MDR1 allele associated with multidrug sensitivity in a sample of herding breed dogs living in Australia. Vet Parasitol 2005;131:193–196. [DOI] [PubMed] [Google Scholar]
- 24. Tappin SW, Goodfellow MR, Peters IR, et al. Frequency of the mutant MDR1 allele in dogs in the UK. Vet Rec 2012; 171: 72. [DOI] [PubMed] [Google Scholar]
- 25. Neff MW, Robertson KR, Wong AK, et al. Breed distribution and history of canine mdr1‐1Delta, a pharmacogenetic mutation that marks the emergence of breeds from the collie lineage. Proc Natl Acad Sci U S A 2004;101:11725–31170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nelson OL, Carsten E, Bentjen SA, Mealey KL. Ivermectin toxicity in an Australian Shepherd dog with the MDR1 mutation associated with ivermectin sensitivity in Collies. J Vet Intern Med 2003;17:354–356. [PubMed] [Google Scholar]
- 27. Sartor LL, Bentjen SA, Trepanier L, Mealey KL. Loperamide toxicity in a collie with the MDR1 mutation associated with ivermectin sensitivity. J Vet Intern Med 2004;18:117–118. [DOI] [PubMed] [Google Scholar]
- 28. Paul AJ, Tranquilli WJ, Seward RL, et al. Clinical observations in collies given ivermectin orally. Am J Vet Res 1987;48:684–685. [PubMed] [Google Scholar]
- 29. Tranquilli WJ, Paul AJ, Seward RL. Ivermectin plasma concentrations in collies sensitive to ivermectin‐induced toxicosis. Am J Vet Res 1989;50:769–770. [PubMed] [Google Scholar]
- 30. Kiso S, Cai SH, Kitaichi K, et al. Inhibitory effect of erythromycin on P‐glycoprotein‐mediated biliary excretion of doxorubicin in rats. Anticancer Res 2000;20:2827–2834. [PubMed] [Google Scholar]
- 31. Bansal T, Mishra G, Jaggi M, et al. Effect of P‐glycoprotein inhibitor, verapamil, on oral bioavailability and pharmacokinetics of irinotecan in rats. Eur J Pharm Sci 2009;36:580–590. [DOI] [PubMed] [Google Scholar]
- 32. Castle MC, Margileth DA, Oliverio VT. Distribution and excretion of (3H)vincristine in the rat and the dog. Cancer Res 1976;36:3684–3689. [PubMed] [Google Scholar]
- 33. Reddy GK, Brown B, Nanda A. Fatal consequences of a simple mistake: How can a patient be saved from inadvertent intrathecal vincristine? Clin Neurol Neurosurg 2011;113:68–71. [DOI] [PubMed] [Google Scholar]
- 34. Krugman L, Bryan JN, Mealey KL, Chen A. vincristine‐induced central neurotoxicity in a collie homozygous for the ABCB1Δ mutation. J Sm An Pract 2012;53:185–187. [DOI] [PubMed] [Google Scholar]
- 35. Dantzig AH, deAlwis DP, Burgess M. Considerations in the design and develoment of transport inhibitors as adjuncts to drug therapy. Adv Drug Deliv Rev 2003;55:133–150. [DOI] [PubMed] [Google Scholar]
- 36. Bates S, Kang M, Meadows B, et al. A phase I study of infusional vinblastine in combination with the p‐glycoprotein antagonist PSC 833 (valspodar). Cancer 2001;92:1577–1590. [DOI] [PubMed] [Google Scholar]
- 37. Chico I, Kang MH, Bergan R, et al. Phase I study of infusional paclitaxel in combination with the P‐glycoprotein antagonist PSC 833. J Clin Oncol 2001;19:832–842. [DOI] [PubMed] [Google Scholar]
- 38. Varma MVS, Ashokraj Y, Dey CS, Panchagnula R. P‐glycoprotein inhibitors and their screening: A perspective from bioavailability enhancement. Pharm Res 2003;48:347–359. [DOI] [PubMed] [Google Scholar]
- 39. Dunn ST, Hedges L, Sampson KE, et al. Pharmacokinetic interaction of the antiparasitic agents ivermectin and spinosad in dogs. Drug Metab Dispos 2011; 39: 789–795. [DOI] [PubMed] [Google Scholar]
- 40. Zhou XJ, Rahmani R. Preclinical and clinical pharmacology of vinca alkaloids. Drugs 1992;44S:1–16. [DOI] [PubMed] [Google Scholar]
- 41. Hafkemeyer P, Dey S, Ambudkar SV, et al. Contribution to substrate specificity and transport of nonconserved residues in transmembrane domain 12 of human P‐glycoprotein. Biochemistry 1998;37:16400–16409. [DOI] [PubMed] [Google Scholar]
- 42. Bradley G, Ling V. P‐glycoprotein, multidrug resistance and tumor progression. Cancer Metastasis Rev 1994;13:223–233. [DOI] [PubMed] [Google Scholar]
- 43. Eadie LN, Hughes TP, White DL. Interaction of the efflux transporters ABCB1 and ABCG2 with imatinib, nilotinib and dasatinib. Clin Pharmacol Ther 2014;95:294–306. [DOI] [PubMed] [Google Scholar]
- 44. Fromm MF. The influence of MDR1 polymorphisms on P‐glycorptoein expression and function in humans. Adv Drug Deliv Rev 2002;54:1295–1310. [DOI] [PubMed] [Google Scholar]
- 45. Sakaeda T, Nakamura T, Okumura K. MDR1 genotype‐related pharmacokinetics and pharmacodynamics. Biol Pharm Bull 2002;25:1391–1400. [DOI] [PubMed] [Google Scholar]
- 46. Page RL, Hughes CS, Huyan S, et al. Modulation of P‐glycoprotein‐mediated doxorubicin resistance in canine cell lines. Anticancer Res 2000;20:3533–3538. [PubMed] [Google Scholar]
- 47. Ford JM, Hait WN. Pharmacological circumvention of multidrug resistance. Cytotechnology 1993;12:171–212. [DOI] [PubMed] [Google Scholar]
- 48. Binkhathlan Z, Lavasanifar A. P‐glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: Current status and future perspectives. Curr Cancer Drug Targets 2013;13:326–346. [DOI] [PubMed] [Google Scholar]
- 49. Ford JM. Modulators of multidrug resistance. Preclinical studies. Hematol Oncol Clin North Am 1995;9:337–361. [PubMed] [Google Scholar]