

Abstract
While most early (stage I‐II) melanomas are cured by surgery, recurrence is not uncommon. Prognostication by current clinicopathological parameters does not provide sufficient means for identifying patients who are at risk of developing metastases and in need of adjuvant therapy. Actin‐regulating formins may account for invasive properties of cancer cells, including melanoma. Here, we studied formin‐like protein 2 and 3 (FMNL2 and FMNL3) in melanoma by analysing their role in the invasive properties of melanoma cells and by evaluating whether FMNL2 expression is associated with melanoma outcome. Immunohistochemical characterization of FMNL2 in a cohort of 175 primary cutaneous stage I‐II melanomas indicated that high FMNL2 reactivity correlates with poor outcome as evaluated by recurrence free survival (p < 0.0001) or disease specific survival (p < 0.0001). In multivariate analysis, Breslow's thickness (p < 0.05) and FMNL2 expression (p < 0.001) remained as independent prognostic factors. Cellular studies revealed that FMNL2 is a component of filopodia in many melanoma cell lines. Inhibition of either FMNL2 or the closely related FMNL3 affected the maintenance of melanoma cell morphology and reduced migration. Finally, inhibition of the BRAF, PI3K and MAPK oncogenic pathways markedly reduced expression of both FMNL2 and FMNL3 in melanoma cells. The results suggest a major role for FMNL2/FMNL3 formins in melanoma biology and raise the possibility that the novel targeted melanoma drugs may interfere with the cellular properties regulated by these formins.
Keywords: melanoma, formin, actin, FMNL2, FMNL3
Introduction
The incidence of cutaneous melanoma is rapidly rising with 230 000 new cases and 55 000 cancer deaths annually (WHO International Agency of Cancer Research, globocan.iarc.fr). Melanomas are staged clinically based on the level of dissemination: stages I and II being localized, stage III having lymph node metastases and stage IV having distant metastases. The treatment of localized melanoma is surgical, which in most cases is curative. However, melanoma recurrence is not uncommon, sometimes after a decade‐long remission. Recurrence may present locally but more commonly as lymph node metastasis or distant metastasis. With distant metastases, the prognosis is dismal, the 5‐year survival rate being 15–20% (www.cancer.org).
The methods for identifying individuals with adverse prognosis are currently inaccurate and lack any molecular parameters. The evaluation relies on clinical features such as age, gender and location of primary tumour, as well as histological findings. In localized melanoma, these are tumour thickness, presence of ulceration, mitotic count and the presence of tumour‐infiltrating lymphocytes (TILs) 1, 2. There is great need for biomarkers that could help to identify the patients at risk of metastatic progression, as such patients might benefit from adjuvant therapy.
Identification of over‐active signalling pathways in melanoma has guided development of targeted therapies for metastatic disease. The pivotal signalling pathways active in melanoma are the Ras/Raf/MEK/ERK [also known as the MAPK (mitogen activated protein kinase)] and the phosphoinositide‐3‐OH kinase (PI3K) signalling pathways. Abnormal activity of these pathways, induced by oncogenic mutations, leads to increased proliferation and cancer cell survival 3. The most common mutation, present in more than 50% of melanomas, is BRAF V600, which leads to constitutive activation of ERK 4, 5. When present, BRAF inhibitors such as vemurafenib improve survival of patients with metastatic disease. The effect of BRAF inhibition is unfortunately lost by acquired resistance, which typically develops within 6 months 6.
Formins are an actin nucleating protein family with diverse actin‐regulating and potentially pro‐invasive functions 7. Among the sub‐family of diaphanous‐related formins are two members, formin‐like protein 2 (FMNL2 also known as FRL3) and formin‐like protein 3 (FMNL3 also known as FRL2), which share extensive sequence homology. FMNL2 and FMNL3 have both actin polymerizing and bundling capacity 8, 9. The activity of these formins appears to be interconnected as FMNL2 and FMNL3 have been shown to form heterodimers 9. When transfected with cDNA constructs coding for constitutively active FMNL2 or FMNL3 forms, the proteins are targeted to filopodia in mouse melanoma and human T‐cell lymphoma cell lines. Previous in vitro studies have suggested that FMNL2 partakes in melanoma cell invasion 10, 11, whereas no information on FMNL3 exists in this respect. Our earlier studies have shown that FMNL2 is widely expressed in human tissues, and found both in skin keratinocytes and cultured melanoma cells 12. Due to the lack of suitable FMNL3 antibodies, its expression in human tissues has not been characterized.
In the present study, we have characterized the role and interplay of FMNL2/FMNL3 formins in melanoma, both by studying a melanoma cohort with extensive follow‐up and by in vitro methods. By immunohistochemical evaluation, we show that the level of FMNL2 expression is a strong independent indicator of recurrence‐free and melanoma‐specific survival in primarily localized disease. At the cellular level, we show that endogenous FMNL2 and FMNL3 are filopodial components in melanoma cell lines, and show that depletion of FMNL2 and/or FMNL3 leads to altered cell morphology and decreased migration in vitro. Finally, we demonstrate that inhibitors targeting the pathways actively studied in melanoma therapy effectively regulate the expression of both FMNL2 and FMNL3.
Methods
Melanoma specimens
The melanoma cohort consists of 175 consecutive archive tissue samples and follow‐up data from patients with primary invasive cutaneous melanoma of the American Joint Committee on Cancer (AJCC) clinical stages I and II, operated at the Turku University Hospital in 1990–2005. Clinical stages I and II together represent localized melanomas of any thickness without clinical nodal involvement or distant metastases 1. At Turku University Hospital, sentinel node biopsy was initiated in 2001. The cases with detected micrometastases in sentinel nodes were included (postoperative AJCC stage III with nodal involvement, n = 11). If a metastasis was histologically sampled during follow‐up, it was also included. This amounted to 34 metastasis samples. The clinicopathological parameters relevant for prognostication of melanoma (ie age, gender, anatomical location, Breslow thickness, Clark level, ulceration, mitosis count, TILs) were re‐evaluated for this study by at least two pathologists. All patients underwent wide local excision with histologically confirmed tumour‐free margins. Suspected metastases were further investigated and mostly treated by surgery, chemotherapy, interferon or radiotherapy. The patients were followed up at the Department of Oncology and Radiotherapy at Turku University Hospital. A recurrence was recorded when a new local tumour, in‐transit metastasis or nodal/distal metastasis appeared. The most recent follow‐up information on the patients was updated from electronic medical records in June 2012. The final follow‐up date of each patient was defined as the date of the most recent hospital call or the date of death. The cause and time of death ware obtained from patient records, autopsy reports or from the Statistics Finland's Archive of Death Certificates.
The Joint Committee on Ethics of the University of Turku and Turku University Hospital approved the use of the tissue collection for this study. According to Finnish legislation, archived tissue samples collected for diagnostic purposes may be used with the permission of the local ethical authority.
Immunohistochemistry
The paraffin‐embedded specimens were stained with a rabbit anti‐human FMNL2 polyclonal antibody (Sigma‐Aldrich Corporation, St Louis, MO) (1:500). The antibody validation procedures and staining protocol have been described previously 12. This antibody detects a single band of approximately 150 kD in western blotting of melanoma cell lines (Figure 1A). Staining intensity was evaluated with the basal layer of skin keratinocytes as internal reference. The following categories were used: negative, weak, moderate (similar to the basal keratinocytes) and strong (Figure 1C). While evaluating staining intensity, the pathologist was blinded for follow‐up data. In eight metastases, BRAF V600E mutation status was studied immunohistochemically, using an antibody from Spring Bioscience (Pleasanton, CA; clone VE1, dilution 1:25). The BRAF staining result was verified by isolating tumour DNA from paraffin sections and genotyping BRAF with allele‐specific PCR.
Figure 1.
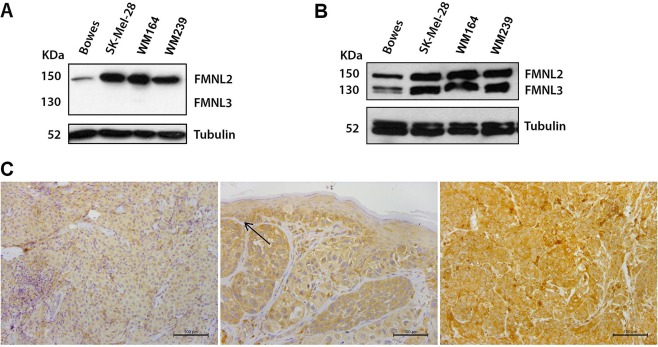
Expression of FMNL2 and FMNL3 in melanoma cell lines and tissues. (A) Western blot analysis of melanoma cell lines with the FMNL2‐specific antibody used for immunohistochemistry. A single 150 kD band corresponding to FMNL2 is detected. (B) A second antibody, used for immunocytochemistry (depicted in Figure 3), reacts with both the 150 kD FMNL2 and the 130 kD FMNL3 formins. Western blot of melanoma cell lines Bowes, SK‐Mel‐28, WM164 and WM239 shows that all cell lines express FMNL2 and FMNL3. (C) Examples of different FMNL2 immunohistochemical staining intensities in primary cutaneous melanomas. Left: Weak cytoplasmic staining of FMNL2. Middle: Moderate FMNL2 staining. Basal keratinocytes express FMNL2 moderately and serve as an internal reference (arrow). Right: Strong FMNL2 staining.
Cell culture
Melanoma cell lines WM239 and WM164 were cultured in RPMI 1640 medium (Gibco‐BRL, UK). SK‐Mel‐28 and Bowes were cultured in minimum essential medium (MEM) and Eagle's minimum essential medium (EMEM) (Invitrogen, Carlsbad, CA). In experiments where signalling pathways were inhibited, the cells were cultured for 3 days in a medium containing 10 μm PI3 kinase inhibitor LY294002 (Tocris Bioscience, Bristol, UK), MEK 1/2 inhibitor U0126 (Cell Signaling Technology, Danvers, MA) or BRAF inhibitor vemurafenib (Santa Cruz Biotechnology, Santa Cruz, CA). PI3K pathway inhibition was verified by immunoblotting with p‐Akt and Akt antibodies (Cell Signaling Technology) and MAPK pathway inhibition by immunoblotting with a p‐ERK 1/2 antibody (Cell Signaling Technology) and an ERK 2 antibody (Santa Cruz Biotechnology), all produced in rabbit.
Transfection with small interfering RNAs
FMNL2 and FMNL3 expression was silenced in SK‐Mel‐28 and Bowes cells using SMARTpool small interfering RNA (siRNA) (Dharmacon Research, Boulder, CA). Non‐targeting Pool siRNA was used as a control. Cells were transfected using Dharmafect 1 or Dharmafect 4 transfection reagent (Dharmacon). FMNL2 or FMNL3 expression was silenced at 50 nm siRNA concentration. For double knockdown of FMNL2 and FMNL3 25 nm of each siRNA was used. The knockdown efficacy was examined 72 h after transfection by immunoblotting.
Western blotting of cell lysates
Western blotting was carried out as described 12. The rabbit‐anti‐FMNL2 antibody (Sigma‐Aldrich) was used at a 1:1000 dilution. Another anti‐FMNL2 antibody, primary mouse monoclonal anti‐FMNL2 (ab56963, Abcam, Cambridge, UK) reacts also with FMNL3 10, but does not cross‐react with Formin‐like protein 1 (FMNL1) 13. This antibody was used at a 1:1000 dilution to detect both FMNL2 and FMNL3 (Figure 1B).
Immunofluorescence staining and microscopy
For immunofluorescence, cells were grown on gelatin‐coated coverslips for 24 h and fixed with 4% paraformaldehyde. For detection of FMNL2, coverslips were blocked with 3% bovine serum albumin, 5% dry milk, 0.5% Triton X‐100 in phosphate buffered saline (PBS) and subsequently incubated with rabbit anti‐FMNL2 (1:200; Sigma‐Aldrich Corporation). For detection of both FMNL2 and FMNL3, the monoclonal anti‐FMNL2 antibody from Abcam was used. Cortactin was detected with mouse monoclonal anti‐Cortactin antibody (Millipore, Bedford, MA; p80/85, clone 4F11) (1:300) and proliferating cells with mouse anti‐Ki‐67 antibody (Dako, Hamburg, Germany) (1:100) followed by Alexa Fluor 568 goat anti‐rabbit IgG (Invitrogen) or Alexa Fluor 546 goat anti‐mouse IgG (1:500). Filamentous actin was visualized with Alexa Fluor 488‐conjugated phalloidin (1:50; Invitrogen). Epifluorescence images were analysed with ImageJ software. The proportion of cells with club‐shaped protrusions was evaluated from a minimum of 180 cells in each group.
Wound healing assay
SK‐Mel‐28 and Bowes cells (50 × 103) were grown for 24 h on poly‐l‐lysine (Sigma‐Aldrich) coated 96‐well ImageLock microplates (Essen BioScience, Ann Arbor, MI). Wounds were made with the 96‐pin Wound‐Maker provided with the IncuCyte FLR (Essen Bioscience). Cells were incubated in the IncuCyte FLR with complete medium and wound images were automatically acquired at 1 h intervals for 48 h. The kinetics of the relative wound density was analysed by IncuCyte™ software.
Transwell cell migration analysis
Control, FMNL2 siRNA, FMNL3 siRNA and FMNL2+FMNL3 siRNA treated SK‐Mel‐28 and Bowes cells were used 72 h after transfection. Boyden chambers (Millipore) were placed in 24 well plates containing culture medium, loaded with 150 μl of a suspension of cells (50 × 103) and kept for 48 h. Non‐migrating cells were removed from the upper chamber with a cotton swab and cells adherent to the underside of the filter were fixed with 4% paraformaldehyde, and stained with 0.5% crystal violet (Thermo Fisher Scientific, UK). The inserts were washed with PBS and allowed to dry overnight. The migrated cells were photographed in four random sites by light microscopy at a magnification of 100X. To evaluate the relative amount of migrated cells by an additional method, 1% SDS (400 μl/well) was added to solubilize the cells and the stain. The plate was agitated on an orbital shaker for 30 min until the colour of the membranes was uniform. The absorbance of each sample was measured at 570 nm on a Multiskan FC Machine (Thermo Scientific, Waltham, MA). The experiment was repeated three times.
Statistical analysis
Categorical variables were characterized using frequencies and percent and in case of continuous variables means, range of values were used. Cox's regression analysis was used to determine the significant prognostic factors of disease free survival and recurrence free survival. Disease free survival was calculated from the date of operation to the date of recurrence or to the end of follow up. If an explanatory variable was statistically significant in univariate analysis and unrelated to other explanatory variables, it was included in multivariate analysis. The results of Cox's regression analyses were quantified by calculating hazard ratios with 95% confidence intervals (95% CI). The cumulative percentages for survival were estimated using Kaplan–Meier technique and differences between FMNL2 expression groups were tested using log‐rank test. The p‐values less than 0.05 were considered as statistically significant. Statistical analyses were carried out using SAS system for Windows (SAS Institute Inc., Cary, NC). Unless otherwise stated, analysis of variance (ANOVA) was used to test intergroup differences for significance in cellular studies.
Results
FMNL2 expression and its prognostic association in stage I‐II melanoma
To study the prognostic significance of FMNL2 expression in primary melanomas, 169 stage I‐II melanomas were stained with a previously characterized antibody (Figure 1A) 12. FMNL2 staining could not be performed in 6 of the initially selected 175 cases due to lack of sufficient tissue material. The remaining 169 cases were processed and analysed further. The clinicopathological characteristics of the cohort are presented in Table 1. The mean follow‐up of the cohort was 7.6 years (range: 0–21.9). During follow‐up, 46 patients had a recurrence (27.2%) and 40 patients died of melanoma (23.6%); 54 patients died of other disease (32.0%). Seventy five patients (44.4%) were alive at the end of follow‐up.
Table 1.
Clinical and histological parameters of the studied cases
| Age (years) | ||
| Mean | 63 | |
| Median | 65 | |
| Range | 15–92 | |
| Follow‐up (years) | ||
| Mean | 7.6 | |
| Median | 6.6 | |
| Range | 0–21.9 | |
| Number (%) | ||
| Gender | ||
| Female | 82 (48.5) | |
| Male | 87 (51.5) | |
| Location | ||
| Trunk | 65 (38.4) | |
| Extremities | 64 (37.9) | |
| Head and neck | 40 (23.7) | |
| Breslow thickness | ||
| ≤1 mm | 55 (32.5) | |
| 1.01–2.0 mm | 45 (26.7) | |
| 2.01–4.0 mm | 35 (20.7) | |
| >4.0 mm | 34 (20.1) | |
| Clark level | ||
| II | 35 (20.7) | |
| III | 67 (39.6) | |
| IV | 51 (30.2) | |
| V | 16 (9.5) | |
| Ulceration | ||
| Absent | 96 (56.8) | |
| Present | 73 (43.2) | |
| Dermal mitoses | ||
| <1/mm2 | 115 (68.0) | |
| ≥1/mm2 | 54 (32.0) | |
| Lymphocytic infiltration | ||
| None | 44 (26.0) | |
| Slight | 44 (26.0) | |
| Strong | 81 (48.0) | |
| Regression | ||
| Yes | 46 (27.2) | |
| No | 123 (72.8) | |
| Outcome | ||
| Alive | 75 (44.4) | |
| Died of melanoma | 40 (23.6) | |
| Died of other disease | 54 (32.0) |
All melanoma cases expressed FMNL2; none of the cases were scored as negative. Within the samples, keratinocytes, lymphocytes and endothelial cells served as positive staining controls. The staining intensity of the keratinocytes was used as an internal reference and scored as moderate. The FMNL2 staining intensity varied between the tumours studied, with a cytoplasmic staining pattern in a wide majority of cases. A particularly strong cytoplasmic dot could be seen in some cases, possibly representing the Golgi apparatus. The staining intensity was weak in 54 (32%) cases, moderate in 69 (41%) cases and strong in 46 (27%) cases. Examples of these staining categories are presented in Figure 1C.
In Kaplan–Meyer analysis of both recurrence‐free and melanoma‐specific survival, the outcome was significantly different between FMNL2 expression groups. Recurrence free survival in the strong FMNL2 expression group was significantly shorter than in the weak FMNL2 expression group (p < 0.0001) (Figure 2A). In a similar analysis of melanoma‐specific survival, both moderate and strong FMNL2 expression groups had a significantly worse outcome than the low FMNL2 expressing group (p < 0.0001) (Figure 2B).
Figure 2.
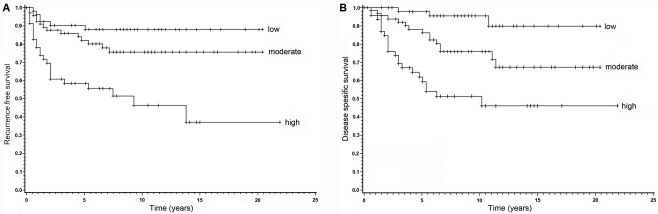
Kaplan–Meyer survival analyses of high, moderate and low FMNL2 expression melanomas. (A) The recurrence free survival is significantly shorter in the group with high FMNL2 expression, when compared to the group with low expression (p < 0.0001). The difference between groups with moderate and low FMNL2 expression is not statistically significant. (B) Melanoma‐specific survival is significantly different between FMNL2 expression groups. Here, both moderate and high FMNL2 expressing groups have a significantly worse outcome than the low FMNL2 expression group (p < 0.0001).
To explore the possible prognostic role of FMNL2 expression, univariate analysis of recurrence‐free and disease‐specific survival was conducted. In the univariate analysis of recurrence‐free survival, the outcome was highly significantly influenced by FMNL2 expression level (p < 0.0001). Other significant parameters were Breslow thickness, AJCC stage, Clark level, presence of ulceration, presence of TILs and dermal mitosis count ≥ 1/mm2. Outcome was also highly significantly influenced by FMNL2 expression in the univariate analysis of melanoma‐specific survival (p < 0.0001). Other significant parameters were Breslow thickness, AJCC stage, Clark level and dermal mitosis count ≥ 1/mm2.
Of these parameters, FMNL2 expression level and clinically used histopathological prognostic markers of melanoma (Breslow thickness, presence of ulceration, dermal mitosis level) were further subjected to multivariate analysis of recurrence‐free and melanoma‐specific survival. Of these, FMNL2 expression level and Breslow thickness came out as significant independent prognostic factors in both recurrence‐free and melanoma‐specific survival (Tables 2 and 3).
Table 2.
Univariate and multivariate Cox proportional hazard model of clinical and histopathological factors for recurrence‐free survival
| Factor | Univariate | Multivariate | ||
|---|---|---|---|---|
| Hazard ratio (95% CI) | p Value | Hazard ratio (95% CI) | p Value | |
| Breslow thickness | <0.0001 | <0.01 | ||
| ≤1 | 1.00 | 1.00 | ||
| 1.01–2.0 | 4.75 (1.33–17.02) | 6.10 (1.65–22.40) | ||
| 2.01–4.0 | 8.05 (2.24–28.91) | 8.75 (2.26–33.86) | ||
| >4.0 | 17.87 (5.23–61.06) | 16.29 (3.87–68.57) | ||
| FMNL2 IHC | <0.0001 | <0.001 | ||
| Weak | 1.00 | 1.00 | ||
| Moderate | 1.97 (0.76–5.13) | 1.92 (0.72–5.08) | ||
| Strong | 5.58 (2.27–13.72) | 5.70 (2.27–14.30) | ||
| Ulceration present | 2.09 (1.14–3.8) | <0.05 | 0.95 (0.49–1.86) | 0.89 |
| Dermal mitoses ≥1/mm2. | 3.5 (1.93–6.50) | <0.001 | 1.17 (0.53–2.56) | 0.70 |
95% CI, 95% confidence interval; IHC, immunohistochemistry.
Table 3.
Univariate and multivariate Cox proportional hazard model of clinical and histopathological factors for melanoma‐specific survival.
| Factor | Univariate | Multivariate | ||
|---|---|---|---|---|
| Hazard ratio (95% CI) | p Value | Hazard ratio (95% CI) | p Value | |
| Breslow thickness | <0.0001 | <0.05 | ||
| ≤1 | 1.00 | 1.00 | ||
| 1.01–2.0 | 3.59 (1.14–11.28) | 4.25 (1.32–13.66) | ||
| 2.01–4.0 | 5.22 (1.64–16.66) | 4.82 (1.42–16.28) | ||
| >4.0 | 11.59 (3.83–35.11) | 10.26 (2.83–37.24) | ||
| FMNL2 IHC | <0.0001 | <0.001 | ||
| Weak | 1.00 | 1.00 | ||
| Moderate | 4.40 (1.27–15.21) | 4.31 (1.23–15.09) | ||
| Strong | 11.22 (3.35–37.53) | 10.77 (3.18–36.52) | ||
| Ulceration present | 1.80 (0.96–3.40) | 0.07 | ||
| Dermal mitoses ≥1/mm2. | 3.3 (1.75–6.13) | <0.001 | 1.11 (0.49–2.55) | 0.80 |
95% CI, 95% confidence interval; IHC, immunohistochemistry.
When the staining intensity of 34 metastases was compared to the primary tumours of the same patient, no significant difference was found (not shown). This indicates that FMNL2 expression is a determinant of the primary tumour, not a property obtained during metastatic dissemination. Eight metastasis samples were further tested for BRAF mutation status. Four BRAF V600E positive and negative samples were compared for FMNL2 staining intensity. No correlation between BRAF mutation status and FMNL2 staining could be established.
Expression of FMNL2 and FMNL3 in melanoma cell lines
To analyse whether FMNL2 or the closely related FMNL3 is expressed in cultured melanoma cells, a Western blot analysis of melanoma cell lines Bowes, SK‐Mel‐28, WM164 and WM239 was carried out. An anti‐FMNL2 antibody (ab56963) detects both FMNL2 (150 kD) and FMNL3 (120 kD) as described by Block et al., and further characterized by us 10, 13. Using this antibody, Western blotting showed that all tested cell lines express FMNL2 and FMNL3 (Figure 1B). Strong expression of both FMNL2 and FMNL3 was seen in SK‐Mel‐28, a cell line derived from a primary cutaneous melanoma 14. We chose this cell line, together with WM164 and Bowes for further morphological and functional studies. The cell lines SK‐Mel‐28 and WM164 harbour a BRAF V600E mutation. All cell lines were analysed in the imaging studies with similar results. The SK‐Mel‐28 and Bowes cells were selected for functional assays.
We have shown previously that FMNL2 is a filopodial component in the WM164 cell line 12. We considered whether this might be a common phenomenon in melanoma cell lines. Immunostaining of melanoma cells revealed that using both the antibody that detects FMNL2 and the antibody that detects both FMNL2 and FMNL3, the staining pattern is identical. The results showed that FMNL2 is localized at filopodia tips, as well as granules in the cytoplasm and diffusely in a majority of nuclei (Figure 3). As the staining patterns with both antibodies were identical, the results indicate indirectly that FMNL2 and FMNL3 are located at the same sub‐cellular loci.
Figure 3.
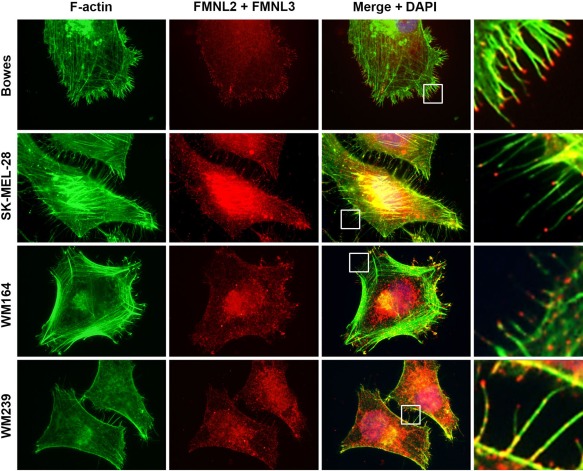
Sub‐cellular localization of FMNL2 and FMNL3 in melanoma cell lines. Immunocytochemical staining of F‐actin (green) and FMNL2 and FMNL3 (red) with an antibody that detects both FMNL forms in four melanoma cell lines. FMNL2 and FMNL3 staining is seen at filopodial tips (insets at right panel), but also along actin filaments, in the cytoplasm and in nuclei.
The effect of FMNL2 and FMNL3 knockdown on cellular features
To analyse the potential role of FMNL2 and FMNL3 in the morphology of melanoma cells and formation of filopodia, their transcripts were transiently silenced in SK‐Mel‐28 cells. Treatment with FMNL2, siRNA altered cell morphology. The number of filopodia was slightly reduced, and some altered cellular protrusions were noted. As a majority of filopodia were morphologically unchanged, we considered whether FMNL3 could be either up‐regulated or functionally compensating for the FMNL2 loss. Therefore, we depleted both FMNL2 and FMNL3, either separately and simultaneously. Knockdown efficacy was verified by Western blotting (Figure 4A). The siRNA‐treated formins were reduced by more than 80% compared to cells treated with non‐targeting siRNA. Notably, silencing FMNL2 and FMNL3 individually did not influence the expression of the related counterpart.
Figure 4.
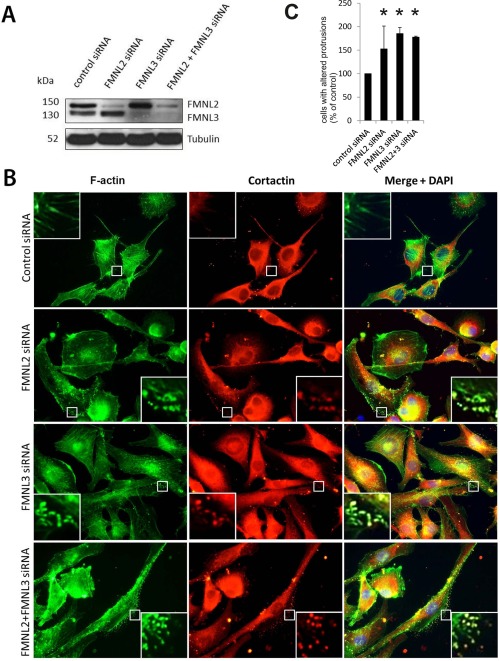
FMNL2 and FMNL3 regulate actin cytoskeleton and cellular protrusions in the SK‐Mel‐28 melanoma cell line. (A) Western blotting of SK‐Mel‐28 cells confirms efficient knockdown of FMNL2, FMNL3 or both. (B) FMNL2, FMNL3 and FMNL2+FMNL3 silenced cells have reduced numbers of filopodia, and instead display altered actin‐rich protrusion at the lateral aspect of cells, mostly at the lamellipodia. These protrusions are enriched with cortactin (insets), not typically present in the filopodia of control cells. (C) The proportion of cells with club‐shaped lateral protrusions in different treatment groups. Bars indicate means, error bars indicate standard error. *p < 0.05.
The depletion of each formin resulted in the presence of club‐shaped or curved protrusions, located at the lateral periphery of cells, and especially in lamellipodia. Conventional slender shafted, needle‐like filopodia were still abundant. The curved protrusions were enriched in filamentous actin and cortactin (Figure 4B). The proportion of cells with these irregular cell processes was evaluated in SK‐Mel‐28 cells in each treatment group. All siRNA‐treated groups had a significant increase of cells with club‐shaped lateral protrusions when compared to control cells. Interestingly, the depletion of FMNL2 and FMNL3 simultaneously did not further increase the proportion of cells with such projections (Figure 4C). The proliferation was assessed by Ki‐67‐staining. The Ki‐67 staining index was not significantly altered by FMNL2 and/or FMNL3 silencing (not shown).
The effect of FMNL2 or FMNL3 knockdown on cell migration
As filopodia are considered to facilitate directional migration, we next subjected SK‐Mel‐28 and Bowes cells treated with FMNL2 and/or FMNL3 siRNAs and control siRNAs to a wound healing experiment. All the knockdown groups had significantly reduced wound density at 36 h (Figure 5A). This indicates that both FMNL2 and FMNL3 have a modest but undisputable role in melanoma cell migration in two‐dimensional cell culture.
Figure 5.
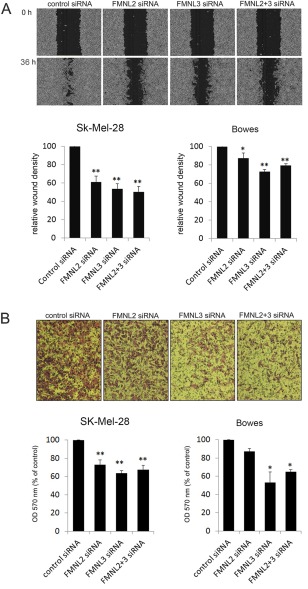
The effect of FMNL2 and FMNL3 silencing on migration of melanoma cells. (A) In wound healing experiments, FMNL2, FMNL3 and FMNL2 + 3 silenced SK‐Mel‐28 and Bowes melanoma cells migrate inferiorly compared to control cells. Depicted images were taken from SK‐Mel‐28 cells at 0 and 36 h. (B) In transwell migration assay, FMNL2 silencing consistently reduced migration, albeit not to a statistically significant degree in Bowes cells. FMNL3 and FMNL2 + 3 silenced cells migrated to the bottom chamber significantly less effectively than control cells. Co‐silencing of both formins did not give an additive effect. The depicted images represent SK‐Mel‐28 cells. Bars in the graphs indicate standard error. *p < 0.05, **p < 0.01.
The wound healing experiments may not measure the requirements needed for invasive cellular movement through a three‐dimensional environment. Movement through the extracellular matrix (ECM) requires complex rearrangement of the actin cytoskeleton. We considered whether FMNL2 and FMNL3 depletion would also impair such single cell migration, as examined by migration through pores in a membrane. To test this possibility, a Boyden chamber assay was performed. In this migration assay, both FMNL2 and FMNL3 silencing in SK‐Mel‐28 and Bowes cells indeed reduced the amount of cells crossing the membrane. In FMNL2 silenced groups, the reduction was statistically highly significant in SK‐Mel‐28 cells, but did not reach statistical significance in Bowes cells. The reduced migration was statistically significant or highly significant in both FMNL3 silenced groups. FMNL2 and FMNL3 co‐silencing did not have an additive effect (Figure 5B).
FMNL2 and FMNL3 expression is dependent on cancer‐promoting signalling pathways
To investigate whether FMNL2 or FMNL3 expression is dependent on MAPK or PI3K signalling pathways, SK‐Mel‐28 and Bowes cells were treated with the MEK1/2 inhibitor UO126, the BRAF inhibitor vemurafenib, the PI3 kinase inhibitor LY294002 or the vehicle DMSO as control. Effective inhibition of the pathways was confirmed by Western blotting of MAPK and Akt phosphorylation states, respectively. Concurrently with the inhibition of either MAPK or PI3K pathway, FMNL3 expression was reduced to a nearly undetectable level and FMNL2 expression was reduced to a lesser extent in both cell lines. However, vemurafenib reduced MAPK phosphorylation and FMNL2/3 expression only in the SK‐Mel‐28 cell line (Figure 6). This is explained by the BRAF status of the cell lines, as SK‐Mel‐28 has an activating BRAF mutation required for vemurafenib efficacy. Bowes cells are BRAF wild type, and therefore rely on other mechanisms to upregulate MAPK signalling. Taken together, the results suggest that the expression of both FMNL2 and FMNL3 in melanoma cells is reliant on the activity of both MAPK and PI3K signalling pathways.
Figure 6.
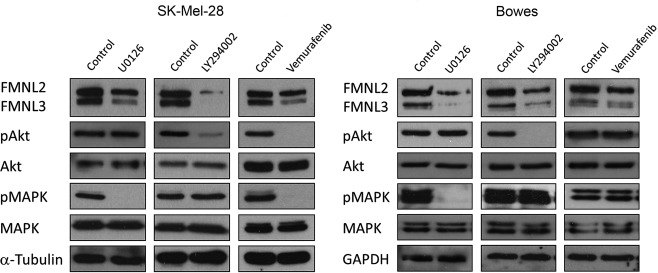
MAPK‐ and PI3K‐signalling regulates FMNL2 and FMNL3 expression in melanoma cells. SK‐Mel‐28 and Bowes cells were treated with MAPK inhibitor UO126, Akt inhibitor LY294002, BRAF inhibitor vemurafenib or DMSO control. The efficacy of inhibitors was verified by detection of pMAPK or pAkt. Inhibition of MAPK and PI3K signalling was accompanied by marked reduction of FMNL2 and FMNL3 expression. Vemurafenib only reduced MAPK signalling and FMNL2/3 expression in the BRAF‐mutated cell line SK‐Mel‐28.
Discussion
Although early stage melanomas are typically cured by surgery, recurrence is not uncommon, and the prognosis of metastatic melanoma is poor. At present, there are no clinically applicable biomarkers to predict melanoma behaviour, and prognostication is done solely in the basis of clinical and histopathological markers. Here, we show that increased expression of the formin family member FMNL2 is a significant and independent predictor of poor outcome as measured by recurrence‐free survival or melanoma‐specific survival. Interestingly, while the conventional prognostic markers Breslow's thickness, ulceration and dermal mitosis count were all predictive in univariate analysis, the only other independent predictor, apart from FMNL2 expression, was Breslow. As FMNL2 expression was already upregulated in the primary tumours, and not further altered in recurrence during follow‐up, our interpretation is that the increase in FMNL2 expression is an early event in melanoma tumourigenesis, and not related to clinical progression.
The role of FMNL2 in cancer is poorly investigated and there are no previous reports on its expression in melanoma. In colorectal cancer, high FMNL2 has been associated with likelihood for lymphatic metastases 15, while in hepatocellular carcinoma reduced FMNL2 expression was suggested as a marker of poor outcome 16. The strong prognostic correlation between FMNL2 and melanoma outcome is likely to boost analysis of FMNL2 in other tumour types. Even less is known about the expression of FMNL3 in cancers. We could unfortunately not address this issue in melanoma due to lack of antibodies suitable for immunohistochemistry.
The activity of FMNL2 and FMNL3 is regulated by well characterized mechanisms 17. Autoinhibition is considered to be relieved by binding to a member of the family Rho GTPases. FMNL2 is activated by the RhoGTPases RhoC and Cdc42 8, 10 and FMNL3 by RhoC 18. Importantly, both Cdc42 and RhoC regulate metastatic dissemination of melanomas as evidenced by in vitro and in vivo studies 19, 20. In line with our results on the involvement of FMNL2 and FMNL3 in melanoma biology, the expression levels of Cdc42 and RhoC associate with metastasis and poor prognosis in primary cutaneous melanoma 21, 22. The negative prognostic effect mediated by high FMNL2 may thus be augmented by high expression of its activators.
We found endogenous FMNL2 at the tips of filopodia of all melanoma cell lines and further showed that FMNL2 and/or FMNL3 silencing was accompanied by alteration of cellular extensions. The filopodia are finger‐like protrusions of the cell membrane that probe the environment during migration and invasion 23, 25. Our findings thus complement previous studies, in which exogenously overexpressed FMNL2 (and FMNL3) were located at cell membrane protrusions or filopodial tips and induced cell protrusions 10, 11, 17.
We found that FMNL2 and particularly FMNL3 contribute to migration of melanoma cells. FMNL2 involvement in the invasive capacity of melanoma cells in vitro has been previously reported 8, 10. Melanoma cells are able to utilize the amoeboid form of migration for invasion, squeezing through the ECM without protein degradation 24. Consistent with this, the melanoma cells in our experiments did not degrade gelatin (not shown). In cellular motility experiments, FMNL2 and/or FMNL3 silencing reduced migration in conventional wound healing assays as well as in Transwell chambers. The latter experiment more closely models amoeboid migration. In this experimental setup, migration requires that the cells identify pores in a membrane and squeeze through as single cells. The exact mechanism by which FMNL2 and FMNL3 silencing reduces this kind of migration remains unexplained. It is possible that a subtle disruption of cell membrane structures affects their ability to sense the serum gradient, identify the pores or form focal complexes for adhesion.
In all in vitro experiments, silencing of either FMNL2 or FMNL3 produced a similar effect. No additive effect was detected by co‐silencing of both proteins. As silencing of one FMNL did not modify expression of the other, the most likely explanation is that FMNL2 and FMNL3 act in concert to exert their functions. Indeed, there is evidence for heterodimerization of FMNL2 and FMNL3 9, which could account for the lack of additive effect in co‐silencing. Another potential scenario is that the formins are involved in different steps of the same process, eg filopodia initialization and elongation.
The BRAF, MAPK and PI3K signalling pathways are constitutively activated in melanoma, through diverse molecular mechanisms. The efficacy of inhibiting these pathways in the treatment of metastatic melanoma is being investigated in several ongoing studies 3. We found that inhibiting either the upstream BRAF tyrosine kinase or the more downstream MEK1/2 or PI3K kinases markedly reduced both FMNL2 and FMNL3 expression in melanoma cells. Taken together, it is reasonable to argue that the FMNL2 expression in melanomas is controlled downstream of these crucial signalling pathways and reflects their activity. Further research will be needed to clearly define whether the prognostic impact of FMNL2 expression in clinical melanoma relates to a mere bystander effect or to a direct mechanism such as actin assembly in specific invasion‐related membrane structures.
Author Contributions
MG, VDH and OC conceived the experiments and analysed data. VDH carried out experiments. IK established and updated the melanoma databank and analysed data. MK analysed data/contributed analysis tools. All authors were involved in writing the paper and had final approval of the submitted version.
Acknowledgements
The authors wish to thank Sinikka Kollanus and Paula Merilahti for skilful technical assistance and Jaakko Liippo for help with photography.
All authors declare that they have no conflict of interest.
Contract/grant details: This study was supported by funding from the Academy of Finland, Finska Läkaresällskapet, the Sigrid Juselius Foundation, the K Albin Johansson Foundation, the Perklén Foundation, the Finnish Medical Society Duodecim, the Finnish Cancer Organizations, and Turku University Hospital Research Funds. MG has during this study been supported by the National School of Clinical Investigation.
References
- 1. Balch CM, Gershenwald JE, Soong SJ, et al Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol 2009; 27: 6199–6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mandala M, Imberti GL, Piazzalunga D, et al Clinical and histopathological risk factors to predict sentinel lymph node positivity, disease‐free and overall survival in clinical stages I‐II AJCC skin melanoma: outcome analysis from a single‐institution prospectively collected database. Eur J Cancer 2009; 45: 2537–2545. [DOI] [PubMed] [Google Scholar]
- 3. Russo A, Ficili B, Candido S, et al Emerging targeted therapies for melanoma treatment (review). Int J Oncol 2014; 45: 516–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Davies H, Bignell GR, Cox C, et al Mutations of the BRAF gene in human cancer. Nature 2002; 417: 949–954. [DOI] [PubMed] [Google Scholar]
- 5. Libra M, Malaponte G, Navolanic PM, et al Analysis of BRAF mutation in primary and metastatic melanoma. Cell Cycle 2005; 4: 1382–1384. [DOI] [PubMed] [Google Scholar]
- 6. Chapman PB, Hauschild A, Robert C, et al Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364: 2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Faix J, Grosse R. Staying in shape with formins. Dev Cell 2006; 10: 693–706. [DOI] [PubMed] [Google Scholar]
- 8. Kitzing TM, Wang Y, Pertz O, Copeland JW, Grosse R. Formin‐like 2 drives amoeboid invasive cell motility downstream of RhoC. Oncogene 2010; 29: 2441–2448. [DOI] [PubMed] [Google Scholar]
- 9. Vaillant DC, Copeland SJ, Davis C, Thurston SF, Abdennur N, Copeland JW. Interaction of the N‐ and C‐terminal autoregulatory domains of FRL2 does not inhibit FRL2 activity. J Biol Chem 2008; 283: 33750–33762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Block J, Breitsprecher D, Kuhn S, et al FMNL2 drives actin‐based protrusion and migration downstream of Cdc42. Curr Biol 2012; 22: 1005–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harris ES, Gauvin TJ, Heimsath EG, Higgs HN. Assembly of filopodia by the formin FRL2 (FMNL3). Cytoskeleton (Hoboken) 2010; 67: 755–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gardberg M, Talvinen K, Kaipio K, et al Characterization of diaphanous‐related formin FMNL2 in human tissues. BMC Cell Biol 2010; 11: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gardberg M, Heuser VD, Iljin K, Kampf C, Uhlen M, Carpen O. Characterization of leukocyte formin FMNL1 expression in human tissues. J Histochem Cytochem 2014; 62: 460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carey TE, Takahashi T, Resnick LA, et al Cell surface antigens of human malignant melanoma: mixed hemabsorptions assays for humoral immunity to cultured autologous melanoma cells. Proc Nat Acad Sci USA 1976; 73: 3278–3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhu XL, Liang L, Ding YQ. Overexpression of FMNL2 is closely related to metastasis of colorectal cancer. Int J Colorectal Dis 2008; 23: 1041–1047. [DOI] [PubMed] [Google Scholar]
- 16. Liang L, Guan J, Zeng Y, et al Down‐regulation of formin‐like 2 predicts poor prognosis in hepatocellular carcinoma. Human Pathology 2011; 42: 1603–1612. [DOI] [PubMed] [Google Scholar]
- 17. Moriya K, Yamamoto T, Takamitsu E, et al Protein N‐myristoylation is required for cellular morphological changes induced by two formin family proteins, FMNL2 and FMNL3. Biosci Biotechnol Biochem 2012; 76: 1201–1209. [DOI] [PubMed] [Google Scholar]
- 18. Vega FM, Fruhwirth G, Ng T, Ridley AJ. RhoA and RhoC have distinct roles in migration and invasion by acting through different targets. J Cell Biol 2011; 193: 655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature 2000; 406: 532–535. [DOI] [PubMed] [Google Scholar]
- 20. Stengel K, Zheng Y. Cdc42 in oncogenic transformation, invasion, and tumorigenesis. Cell Signal 2011; 23: 1415–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tucci MG, Lucarini G, Brancorsini D, et al Involvement of E‐cadherin, beta‐catenin, Cdc42 and CXCR4 in the progression and prognosis of cutaneous melanoma. Br J Dermatol 2007; 157: 1212–1216. [DOI] [PubMed] [Google Scholar]
- 22. Boone B, Van Gele M, Lambert J, Haspeslagh M, Brochez L. The role of RhoC in growth and metastatic capacity of melanoma. J Cutan Pathol 2009; 36: 629–636. [DOI] [PubMed] [Google Scholar]
- 23. Arjonen A, Kaukonen R, Ivaska J. Filopodia and adhesion in cancer cell motility. Cell Adh Migr 2011; 5: 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sanz‐Moreno V, Gadea G, Ahn J, et al Rac activation and inactivation control plasticity of tumor cell movement. Cell 2008; 135: 510–523. [DOI] [PubMed] [Google Scholar]
- 25. Ridley AJ. Life at the leading edge. Cell 2011; 145: 1012–1022. [DOI] [PubMed] [Google Scholar]