

Abstract
Driver mutations in the two histone 3.3 (H3.3) genes, H3F3A and H3F3B, were recently identified by whole genome sequencing in 95% of chondroblastoma (CB) and by targeted gene sequencing in 92% of giant cell tumour of bone (GCT). Given the high prevalence of these driver mutations, it may be possible to utilise these alterations as diagnostic adjuncts in clinical practice. Here, we explored the spectrum of H3.3 mutations in a wide range and large number of bone tumours (n = 412) to determine if these alterations could be used to distinguish GCT from other osteoclast‐rich tumours such as aneurysmal bone cyst, nonossifying fibroma, giant cell granuloma, and osteoclast‐rich malignant bone tumours and others. In addition, we explored the driver landscape of GCT through whole genome, exome and targeted sequencing (14 gene panel). We found that H3.3 mutations, namely mutations of glycine 34 in H3F3A, occur in 96% of GCT. We did not find additional driver mutations in GCT, including mutations in IDH1, IDH2, USP6, TP53. The genomes of GCT exhibited few somatic mutations, akin to the picture seen in CB. Overall our observations suggest that the presence of H3F3A p.Gly34 mutations does not entirely exclude malignancy in osteoclast‐rich tumours. However, H3F3A p.Gly34 mutations appear to be an almost essential feature of GCT that will aid pathological evaluation of bone tumours, especially when confronted with small needle core biopsies. In the absence of H3F3A p.Gly34 mutations, a diagnosis of GCT should be made with caution.
Keywords: H3F3A, H3F3B, giant cell tumour of bone, malignant giant cell tumour of bone, giant cell granuloma, solid variant of aneurysmal bone cyst, USP6
Introduction
Giant cell tumours of bone (GCT) account for approximately 5% of all primary bone tumours and generally affect the epiphysis of long bones in skeletally mature individuals. They are classified as locally aggressive and rarely metastasising neoplasms 1. When GCT metastasise, they spread to the lungs but notably the morphology remains similar to the primary lesion and the disease is generally controlled by resection alone. Such an event is referred to as ‘benign metastasing’ disease. Although the majority of GCTs presents with a characteristic combination of clinical, radiological and histological findings, some remain diagnostically challenging, particularly on small needle core biopsies 1. Due to varying histological appearances within and between individual GCT and due to morphologic overlap with other bone tumours, the differential diagnoses include osteoclast‐rich osteosarcomas which often represent telangiectatic osteosarcoma, and high grade sarcoma arising within a conventional GCT (malignant GCT), along with several benign mimics comprising aneurysmal bone cyst (ABC), chondroblastoma (CB), nonossifying fibroma (NOF) and giant cell granuloma (GCG) occurring exclusively in the jaws and in the small tubular bones of the hand and feet 2, 3. Less likely, GCT can also be mistaken for osteoid osteoma/osteoblastoma and tenosynovial giant cell tumour.
Recently, we reported the whole genomes of 6 CB which harboured p.Lys36Met (p.K36M) mutations in the paralogous histone 3.3 genes (H3.3), H3F3A (1/6) and H3F3B (5/6). The finding was confirmed in an extended targeted sequencing analysis detecting these mutations in 73/77 CB. Due to similarities in morphology and site of presentation, we also screened a series of GCT for H3.3 mutations. We found H3.3 mutations in 92% of cases (49/53), exclusively in the H3F3A gene which all occurred at glycine 34. H3F3A p.Gly34Trp (p.G34W) alterations were found in all but one case of GCT which harboured a p.G34L mutation. We additionally identified distinct H3.3 mutations in 2/103 osteosarcomas (one p.G34R in H3F3A, and one p.G34R mutation in H3F3B) and in 1/75 chondosarcomas (H3F3A p.K36M) 2. Since then, others have reported a H3F3A mutation in another osteosarcoma (1/10, p.G34W) 3.
In addition to H3F3A and H3F3B mutations, other somatic mutations may potentially be of diagnostic value in the clinic for classifying osteoclast‐rich lesions of bone. Such mutations include rearrangements involving USP6 which forms a fusion gene with a number of partners including CDH11, ZNF9, COL1A1, TRAP150 and OMD. These are found in approximately 75% of primary ABC 4, 5, 6 but have not been found in a screen of 9 GCT 4, 5. In addition, fusions involving macrophage colony‐stimulating factor 1 have been described in tenosynovial giant cell tumours. Unfortunately this is of limited diagnostic value as it is only detectable in a minority of tumour cells 7. Furthermore, IDH2 mutations, which also affect central chondrosarcoma 8, have recently been reported to occur in 80% of GCT (16/20) 9.
In this study, we sequenced the whole genomes of five and whole exome of one GCT to search for driver mutations other than H3.3 mutations. In addition, we sought to define the specificity of p.G34W alterations for GCT by screening 91 GCT as well as a series of 321 osteoclast‐rich lesions including ABC, CB, NOF, GCG, osteoblastoma/osteoid osteoma, TGCT, in addition to malignant osteoclast‐rich tumours of bone.
Materials and methods
Patient samples
Formalin‐fixed paraffin‐embedded cases were retrieved from the diagnostic pathology archives at the Royal National Orthopaedic Hospital (RNOH) and from the Basel Bone Tumour Reference Centre (BBTRC). All samples were reviewed by specialist bone tumour pathologists at RNOH (AMF, MFA, RT) and BBTRC (DB) and diagnosed according to the current WHO classifications 10, 11 (Supporting Information Tables 1 and 2). Individual cases were correlated with clinical and radiological information where available. Tissue sample types are provided in Supporting Information Table 3. In cases in which the histology and mutational analysis did not match, the morphology was re‐evaluated independently by all members of the RNOH pathology team and a consensus diagnosis was subsequently reached by all pathologists.
Table 1.
Overview of giant cell tumours of bone genomes and exomes
| PD21292a | PD21294a | PD21295a | PD21296a | PD7524a | PD9999a | |
|---|---|---|---|---|---|---|
| Genome/exome | Genome | Genome | Genome | Genome | Genome | Exome |
| Tumour coverage | 51.26 | 41.42 | 56.22 | 48.48 | 39.04 | ∼60% covered by at least 30× |
| Normal coverage | 25.93 | 22.56 | 23.28 | 26.1 | 34.26 | ∼60% covered by at least 30× |
| Copy number source | Sequencing reads | Sequencing reads | Sequencing reads | Sequencing reads | SNP6 and sequencing reads | n.a. |
| Structural changes | 0 | 0 | 0 | 0 | LOH 5q and 8p | n.a. |
| Substitutions | 479 | 977 | 739 | 700 | 815 | 7 |
| Precisiona | 0.90 | 0.90 | 0.93 | 0.90 | 0.81 | All reviewed |
| Coding | 5 | 6 | 9 | 11 | 12 | 7 |
| Non‐synonymous | 4 | 4 | 7 | 7 | 8 | 7 |
| Truncating | 0 | 1 | 0 | 1 | 2 | 0 |
| Indels | 54 | 79 | 102 | 46 | 45 | 0 |
| Precisiona | 0.76 | 0.84 | 0.61 | 0.91 | 0.93 | n.a. |
| Coding | 0 | 0 | 0 | 0 | 0 | 0 |
| In frame | 0 | 0 | 0 | 0 | 0 | 0 |
| Out of frame | 0 | 0 | 0 | 0 | 0 | 0 |
| Driver mutation | H3F3A G34W | H3F3A G34Wb | H3F3A G34W | H3F3A G34W | H3F3A G34W | H3F3A G34W |
Based on review of 100 (or less, where fewer mutations were called) randomly selected variants.
Manually called.
n.a.—not applicable.
Table 2.
Results of the targeted genetic anlysis for H3F3A and H3F3B and USP6 rearrangment
| Mutation characteristic of tumour | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tumour type | Original Dx (n) | GCT | CB | ABC | WT | aRevised diagnosis (n) | ||||
| G34W/A (n) | G34L/A (n) | G34R/A (n) | G34M/A (n) | K36/A (n) | K36/B (n) | USP6 (n) | ||||
| GCT | 91 | 83 | 0 | 1 | 1 | 0 | 0 | 2 | 4 | 89 |
| GCT of small bones | 25 | 14 | 2 | 0 | 0 | 0 | 0 | 3 | 6 | 21 |
| CB | 7 | 0 | 0 | 0 | 0 | 5 | 2 | 0 | 0 | 7 |
| ABC | 44 | 2 | 0 | 0 | 0 | 0 | 1 | 32 | 9 | 41 |
| NOF | 66 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 62 | 62 |
| Tenosynovial giant cell Tumour | 51 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 51 | 51 |
| Osteoblastoma | 21 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 21 | 21 |
| Osteoid osteoma | 9 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 9 | 9 |
| Giant cell granuloma of jaw (solitary) | 78 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 78 | 78 |
| Adamantinoma | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 8 | 8 |
| Osteofibrous dysplasia | 6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 6 |
| Total no of mutations | 103 | 2 | 1 | 1 | 5 | 3 | 37 | 252 | ||
Diagnosis revised following review of histology and imaging on discovery of the genetic alterations
Table 3.
Mutation analysis results for the 10 malignant osteoclast cell‐rich tumours
| ID | Year of primary and relapses | Alive/DOD | Age of diagnosis | Gender | Site of primary tumour | Histopathological diagnosis | TP53 | Histone variant | IDH1/2 alterations |
|---|---|---|---|---|---|---|---|---|---|
| S00033676 | 2012 | ALIVE | 33 | M | Talus, left | Malignant osteoclast‐rich tumour | WT | H3F3A/B WT | IDH1/2 WT |
| S00033682 | 2005 | Lost in follow up | 59 | F | Fibula, right, proximal | Osteoclast‐rich dedifferentiated chondrosarcoma | WT | H3F3A/B WT | IDH2 R132S |
| S00033684 | 1998, 2009 | ALIVE | 37 | F | Toe, 1st, left, proximal | Malignant osteoclast‐rich tumour with features of osteosarcoma | WT | H3F3A/B WT | IDH1/2 WT |
| S00033694 | 2006, 2008, 2010, 2011, 2013 | DOD (11/2013) | 52 | F | Tibia, left, proximal | Malignant osteoclast‐rich tumour with features of osteosarcoma | WT | H3F3A/B WT | IDH1/2 WT |
| S00039478 | 2006 | Alive | 11 | M | Metatarsal, 3rd, 4th, 5th, left | Malignant osteoclast‐rich tumour with features of osteosarcoma | WT | H3F3A/B WT | IDH1/2 WT |
| S00039480 | 2002, 2006, 2010 | Lost in follow up | 39 | M | Femur, left, distal | Malignant osteoclast‐rich tumour, multicentric | WT | H3F3A/B WT | IDH1/2 WT |
| S00039484 | 2004 | DOD (10/2007) | 74 | M | Femur, left, distal | Malignant osteoclast‐rich tumour with features of osteosarcoma | WT | H3F3A/B WT | IDH1/2 WT |
| S00039476 | 1998, 2005, 2009 | Alive | 19 | F | Tibia, distal | Malignant osteoclast‐rich tumour with features of osteosarcoma | WT | H3F3A G34W/A | IDH1/2 WT |
| S00038597 | 2010 | DOD | 40 | F | Tibia, right, proximal | Dedifferentiated adamantinoma | WT | H3F3A G34W/A | IDH1/2 WT |
| S00033295 | 2013, 2014 | Alive | 50 | M | Tibia, left, distal | Malignant osteoclast‐rich tumour | WT | H3F3A/B WT | IDH1/2 WT |
Ethical approval was obtained from the Cambridgeshire 2 Research Ethics Service (reference 09/H0308/165), the UCL Biobank for Health and Disease ethics committee (covered by the Human Tissue Authority licence 12055: project EC17.1) and from the Ethikkommission beider Basel (reference 274/12).
Whole genome and exome sequencing
DNA from GCT and matching non‐neoplastic tissue, derived from 6 individuals, was subjected to whole genome (n = 5) or exome (n = 1) sequencing as described previously 12, using Illumina Hiseq 2000 analysers (Illumina Inc, CA). Reads were aligned to the reference human genome (NCBI37) by burrows wheeler aligner (BWA) on default settings 13. Unmapped reads and polymerase chain reaction‐derived duplicates were excluded. Relevant coverage metrics are summarised in Table 1.
Targeted next generation sequencing with Ion Personal Genome Machine® (PGM™)
A pooled barcoded amplicon‐tagged library generated using Fluidigm® Access Array™ (Fluidigm Europe B.V, Amsterdam, Netherlands) was diluted for direct input into the emulsion polymerase chain reaction (PCR) with Ion Sphere™ particles with Ion PGM™ Template OT2 200 Kit (Ion OneTouch™ system) following the manufacturer's instructions (Life Technologies Ltd, Paisley, Scotland). The pooled samples were sequenced using 316 chip with Ion PGM™ 200 Sequencing kit on the Ion Torrent PGM™ sequencer following manufacturer instructions (Life Technologies). Targeted hotspot mutations as reported in COSMIC in 14 genes, including EGFR, KRAS, NRAS, BRAF, IDH1, IDH2, KIT, GNAS1, CTNNB1, PTEN, PDGFRA, H3F3A, H3F3B, TP53 (covering in total 50 amplicons) were analysed (Supporting Information Table 4 for primer details and description of hot‐spots targeted).
Reads were aligned to the reference genome hg19 and binary alignment map (BAM) files were generated using Ion torrent suite version 4.0.1. Reads were visualised using integrative genomics viewer (IGV) 14 with the appropriate browser extensible data (BED) files for all 14 genes.
Results
Whole genome/exome sequencing of GCT
To perform a genome‐wide search for driver mutations in GCT, DNA from 5 tumours and blood from the same patients, were studied by whole genome sequencing, and one tumour was subjected to whole exome sequencing. Overall, few somatic changes were seen. In the five genomes, a median of 739 substitutions (range 479–977) and 54 indels (range 45–102) were present (Supporting Information Table 5). Including the additional tumour studied at whole exome level, overall 50 coding substitutions, 37 of which were nonsynonymous and no coding indels were found. Other than H3F3A p.G34W mutations, no driver event was discernible amongst nonsynonymous mutations. No rearrangements or copy number changes were identified, bar two regions of loss of heterozygosity in one of the five genomes (PD7524a) (Supporting Information Figure 1).
Figure 1.
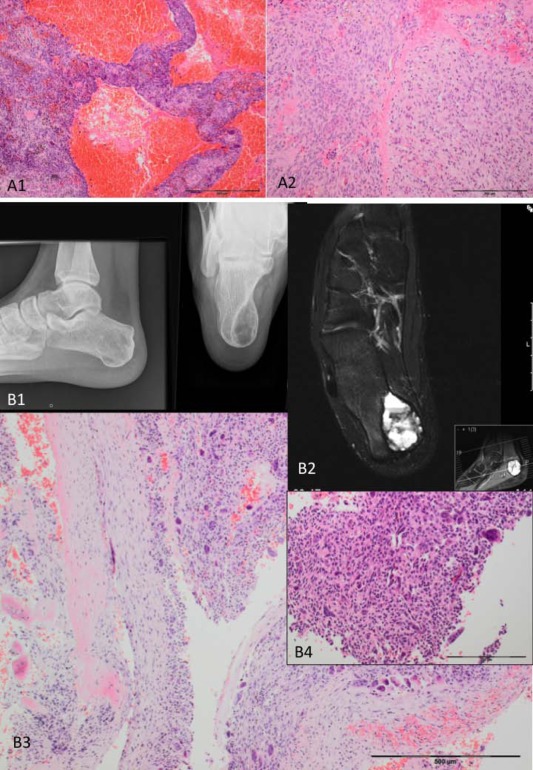
A sacral tumour (S00036624) (A) and a calcaneal tumour (S00036606) (B) originally diagnosed as ABCs. A sacral tumour in an 18‐year‐old female originally diagnosed with an ABC. The tumour shows blood‐filled vascular spaces contained by septa (A1), and scattered solid areas (A2), both of which contain numerous osteoclasts: a USP6 gene rearrangement is not detected. The tumour harboured a H3F3A p.G34W mutation after which the lesion was then reclassified as a giant cell tumour (GCT) with ABC change. A calcaneal tumour in a 22‐year‐old male originally diagnosed with an ABC. The X‐ray reveals a well‐defined lucency (B1), and the MRI shows fluid–fluid levels (B2). The histology shows an osteoclast‐rich predominantly cystic lesion (B3, B4). A H3F3B p.K36M mutation was detected. The histology was reviewed and small areas with features of CB (B4) were detected. The tumour was reclassified as CB with extensive ABC change.
Targeted genetic analysis
Giant cell tumour of bone
A histological diagnosis of GCT was favoured in 91 osteoclast‐rich tumours: 83 were found to harbour a H3F3A p.G34W alteration and alterations represented by p.G34R and p.G34M were detected in two tumours (Table 2). No mutations were detected in the H3F3B gene. The two GCTs (femur S00033722, ulna S00033686) which metastasised to the lung harboured a H3F3A p.G34W mutation. Morphologically, both primary tumours and the metastases showed the typical features of a conventional GCT. Both patients are alive and well and the tumours were classified as ‘benign metastasising’ GCT 15.
Fluorescence in situ hybridisation (FISH) revealed USP6 break‐apart signals in 2/6 remaining wild type (WT) GCT, both of which were located in the spine (Table 2). In these 2 cases, the histology was re‐evaluated and considered compatible with ABC and the diagnosis revised accordingly. Ten GCT with a H3F3A p.G34W were also tested for USP6 rearrangements and all were negative. Consequently, 85/89 (96%) GCT showed mutations in H3F3A genes (Table 2).
Sixty GCT were screened for hotspot mutations in 12 genes in addition to H3F3A and H3F3B using the Ion Torrent PGM. No mutations, specifically no TP53, IDH1 and IDH2 alterations, were identified (Supporting Information Table 2).
None of the 89 GCT presented in patients with immature skeleton, although 2 female patients presented at the age of 15 and one at 17.
Chondroblastoma
All seven CB analysed by targeted sequencing harboured a p.K36M mutation, five of which were in the H3F3A and two in the H3F3B gene (Table 2). Seven CB were analysed for USP6 rearrangements, including 4/77 cases that were WT for H3F3A and H3F3B in our previous study (Table 1), and the 3/7 cases from this study in which a p.K36M alteration was identified (Table 2). None were detected. No mutations in the hotspots in the Ion Torrent PGM 14 gene panel assay were detected in 19 CB harbouring p.K36M: these included 18 cases from the previous study (Supporting Information Table 1, 2).
Aneurysmal bone cyst
Of 44 tumours where the favoured initial histological diagnosis was ABC, 32 revealed a USP6 rearrangement by FISH: 27 showing break‐apart signals, with 5 showing deletions of the telomeric end (Figure 1, Table 2). One of the remaining cases (1/12) harboured a H3F3B p.K36M mutation which on histological review revealed a tiny fragment with features of a CB (Figure 1B). The diagnosis was, therefore, revised. Two additional cases (2/12) revealed H3F3A p.G34W mutation and were considered to be compatible with GCT on histologic re‐evaluation (Figure 1A). The remaining 9/12 cases were WT for both the USP6 and H3.3 alterations. The USP6 alterations were mutually exclusive with H3.3 mutations. Reclassification of ABC on the basis of the genetic finding in 3/44 lesions resulted in a diagnosis of 41 ABC, 32 (78%) of which demonstrated USP6 rearrangements (Table 2).
Osteoclast‐rich tumours of the small bones of hand and feet and patella
Twenty‐eight osteoclast‐rich tumours of small bones of the hand (n = 13), feet (n = 14) and patella (n = 1) were studied, including three cases from our previous study (Supporting Information Table 1, 2). Sixteen tumours harboured a H3F3A mutation, 14 of which involved a p.G34W alteration. The other two alterations were represented by p.G34L mutations (Supporting Information Table 1, 2). One tumour in the talus harboured a H3F3B p.K36M substitution. On histological review a small focus with features of a CB was seen and the tumour was reclassified accordingly. USP6 break‐apart signals were detected in 3/11 remaining cases that were WT for H3F3A and H3F3B, and these were classified as ABC, leaving 8/28 (29%) cases (patella, bone of hand ×2, bone of foot ×5) without an alteration with which to classify the disease. The USP6 rearrangement was mutually exclusive with H3.3 mutations in these cases. Screening of the mutation hotspots on Ion Torrent PGM failed to reveal any additional mutations in the 24/28 cases.
Nonossifying fibroma, tenosynovial giant cell tumour, osteoblastoma, osteoid osteoma, and giant cell granuloma of the jaw
Details of these tumours are included in Supporting Information Table 2 and genetic alterations in Table 2: 4/66 previously diagnosed NOF harboured H3F3A p.G34W alterations. These were reclassified as GCT following review of histology and imaging. No H3F3A and H3F3B mutations or hotspot mutations were identified in the Ion Torrent screen. Fifteen GCG of the jaws analysed by FISH for USP6 rearrangements did not reveal a break‐apart signal or deletion of one the probes.
We screened 51 tenosynovial giant cell tumours, 21 osteoblastomas, 9 osteoid osteomas, and 78 GCG of the jaw, three cases of genetically proven cherubism and two cases of genetically proven Noonan syndrome. No p.G34 alterations in H3F3A or p.K36 alterations in the H3F3B were identified (Table 2).
H3F3A and H3F3B mutations in malignant osteoclast‐rich tumours
Ten cases classified as malignant osteoclast‐rich tumours were retrieved from the RNOH archives, eight of which were wild type (WT) for H3F3A and H3F3B mutations (Table 3). On histological review, four cases revealed features typical of osteosarcoma in addition to osteoclast‐rich areas. These cases were reclassified as osteosarcoma with osteoclast‐rich areas. Of the four other WT cases, three were composed predominantly of areas of malignant GCT (Figure 2B). The remaining tumour was a predominantly osteoclast‐rich tumour which had been reported as being ‘suspicious for malignancy’ and revealed an IDH2 p.R172S mutation. On review of the biopsy, a small fragment of mature neoplastic cartilaginous tissue in addition to the osteoclast‐rich neoplasm was noted. The tumour was mitotically active (20 mitoses/high power fields). No cartilage was detected in the resection specimen although an area showing a monotonous spindle cell overgrowth was present (Supporting Information Figure 2). This histological observation associated with the IDH2 p.R172S mutation supported a diagnosis of a dedifferentiated chondrosarcoma in which the dedifferentiated component was an osteoclast‐rich neoplasm. The tumour was reclassified accordingly.
Figure 2.
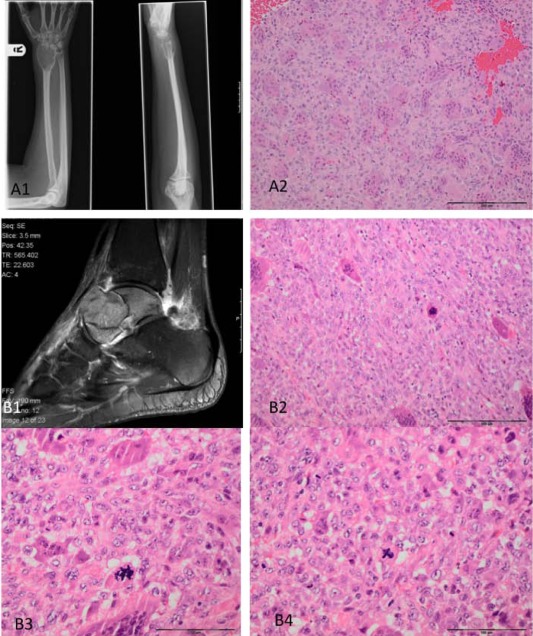
Photomicrographs and X‐rays of a conventional (S00033682) (A), and malignant giant cell tumour (GCT) (S00030176) (B). A central lytic tumour of the distal radius (A1). The histology is typical for GCT in which the stromal cells show no significant cytological atypia (A2). An aggressive lytic lesion of the talus, which has broken through the cortex (B1). Microscopy shows features of a malignant osteoclast‐rich tumour in which the stromal cell are enlarged with nuclear pleomorphism and numerous mitoses, including atypical forms (B3 and B4). Geographic‐type tumour necrosis was also present (not shown).
Of the three patients whose tumours showed predominantly a malignant GCT component, one was sited in the talus and the patient is well having experienced no recurrence following amputation with 24 months of follow‐up (Figure 2B). The second was a multicentric malignant GCT diagnosed in 2002. This patient developed brain and lung metastasis and died 9 years after diagnosis. The third tumour developed in the distal tibia and within 6 months of diagnosis and amputation, the patient developed recurrent disease at the amputation stump and metastatic disease in the pelvis.
One osteoclast‐rich malignant tumour which harboured a H3F3A p.G34W mutation originated in the distal tibia and within a short interval presented with multifocal disease in the vertebrae, which revealed unequivocal malignant features. The patient was treated with resection and radiotherapy and is alive with disease 20 years after diagnosis. Neither tissue nor imaging from the original tumour was available for review.
The second osteoclast‐rich malignant tumour which harboured a H3F3A p.G34W mutation was classified as an unusual, high grade osteoclast‐rich tumour with adamantinoma‐like features located in the right proximal tibia. Histological features typical of a conventional adamantinoma were present and this component merged with a high grade spindle cell sarcomatous area partly infiltrated with large numbers of osteoclasts. Sited in the subarticular space it was considered more in keeping with a GCT because adamantinomas generally occur in the diaphysis (Supporting Information Figure 2). No TP53 mutations were detected. The tumour was resected en bloc: the patient developed multiple spinal metastases and died of disease 7 months later. The different histological areas were microdissected and all revealed the H3F3A p.G34W alteration (Table 3). H3.3 mutations were not detected in six other cases of osteofibrous dysplasia and eight cases of adamantinoma (Table 2).
Overall our observations suggest that the presence of H3F3A p.G34W mutations does not entirely exclude malignancy in osteoclast‐rich tumours. However, H3F3A p.G34W mutations appear to be an almost essential feature of GCT that will aid pathological evaluation of bone tumours, especially when confronted with small needle core biopsies (see algorithm‐based for diagnosis decision Supporting Information Figure 3).
Discussion
Whole genome/exome sequencing of 6 GCT showed remarkably few somatic changes, other than the occurrence of H3F3A mutations. In this way, GCT genomes showed similarities with previously reported CB genomes 2 which reflects overlapping features of the two tumour types, including the osteoclast‐rich morphology, their site of occurrence and their biological behaviour. The notable difference between these tumours is the association of cartilaginous differentiation in CB with the H3.3 K36M alteration, the majority of which occur in H3F3B, whereas 96% of the mutations in GCT occur in H3F3A involving p.G34. The finding of highly recurrent H3.3 substitutions in the absence of other recurrent alterations in both primary and recurrent disease also implies that these mutations in both GCT and CB represent essential oncogenic drivers. It is noteworthy that in the two cases of ‘benign’ metastatic disease no driver mutation other than p.G34W alterations were found in the primary and distant lesions.
In our previous publication 2, we remarked on the exquisite specificity of individual H3.3 mutations for different bone and brain tumour types. Here, we confirm that the vast majority (83/89) of GCT alterations occur in H3F3A and involve p.G34W. However, we also detected an overlap of specific H3.3 mutations and tumour types in a minority of cases. We found that p.G34 substitutions in GCT involve on rare occasions H3F3A p.G34R which is commonly seen in paediatric glioblastoma. In addition, p.G34M was also detected in a single GCT, a substitution previously reported only once in diffuse intrinsic pontine glioma 16. In contrast to the occurrence of p.G34R and p.G34M in aggressive brain tumours, in which TP53 mutations are also seen 17, the 3 GCT with these alterations followed a typical clinical course and did not harbour TP53 mutations, with 2/3 having at least 5 years follow‐up (p.G34R—2009; p.G34M—2004). Finally, we also confirm that p.G34L mutations are rare and 2/3 occurred in small bones of the hands and feet. This mutation, like p.G34W, appears to be confined to bone tumours, having not been reported in primary brain tumours.
In contrast to the high prevalence of H3.3 mutations in GCT and CB, these are rare in malignant primary bone tumours. Specifically, H3.3 mutations have previously been reported to occur in 3/113 osteosarcoma, 1/15 clear cell chondrosarcoma and 1/75 central chondrosarcoma 2, 3. A novel finding in this study is the presence of a H3F3A p.G34W alteration in an unusual high grade osteoclast‐rich tumour with adamantinoma‐like features thereby providing further evidence to link an osteoclast‐rich phenotype with a H3.3 mutation. However, the mutation was present in both osteoclast‐rich and osteoclast‐deficient areas. Further research is required to explain the genotype–phenotype association between H3F3A p.G34W and osteoclast recruitment. This is only the second time that the p.G34W alteration has been associated with a malignant bone tumour. The first was reported in an osteosarcoma but no histological or clinical details of this case were reported 3. We have previously reported 2 osteosarcomas harbouring a H3.3 mutation, namely p.G34R in H3F3A and in H3F3B. Taken together these findings indicate that malignancy in bone tumours cannot be excluded on detection of H3.3 mutations. In contrast, diagnosis of a GCT in the absence of a H3F3A alteration should be made with considerable caution in view of the high prevalence of H3F3A mutations in GCT, 95% (132/139 from current and previous study).
The role of H3.3 genes in human neoplasms has been brought to the fore by large scale sequencing studies undertaken on paediatric brain tumours 18. One of the most intriguing aspects of these mutations is the specificity of mutations for age and anatomical location of these tumours. Similarly, our work presented here confirms our previous observation of tumour type specificity of H3.3 mutations in further bone tumour subtypes. Mutations at p.G34 were confined to tumours harbouring cells of the osteoclastic lineage, whereas mutations in p.K36 occurred solely in bone tumour of chondroblastic lineage. Furthermore, we found that mutations of H3F3B at G34 are exceedingly rare and have to date only been described in a single case of osteosarcoma. Finally, the association of the rare p.G34L mutation with tumours of small bones, caused by a double substitution, is noteworthy. The point mutations of H3F3A in brain tumours localise close to sites of post‐translational modification of p.K27 and p.K36 resulting in methylation of H3K27 (transcriptional repression) and H3K36 (transcriptional elongation and splicing). It is, therefore, likely that these mutations, accompanied by subsequent post‐translational modifications, exert their oncogenic effects through altered gene expression profiles in specific cells and in a particular developmental context. This proposition is supported by in vitro experimental evidence using glioma cell lines showing a clear link between histone mutation, methylation, gene expression and tumourigenesis 18, 19. The specific roles that H3.3 p.G34 mutations may have in tumourigenesis are still not known.
Kato Kaneko et al recently reported a high prevalence of IDH2 alterations in GCT (16/20 cases): 13 were detected by direct Sanger sequencing and 3 by subcloning the PCR product and subsequent sequencing 9. By contrast, we did not find any IDH1 or IDH2 mutations in the majority of 84 GCT and 22 osteoclast‐rich tumours of the small bones of the hands and feet. A canonical IDH2 mutation was detected in one of our cases but, on histological re‐evaluation, we considered this case to represent a dedifferentiated chondrosarcoma. Although a rare finding, this could soon have therapeutic implications in view of the development of IDH1/2 inhibitors, and a vaccine that targets mutant IDH1 20, 21, and therefore, this case highlights the utility of genotyping tumours for diagnostic purposes. The reason for the findings by Kato Kaneko et al is unclear: we consider it is unlikely to be explained by a different cohort of tumours in view of the numbers of cases that we have analyzed. It is unlikely to be a sensitivity issue as the Ion Torrent platform is very sensitive. DNA contamination is a possibility, particularly if subcloning of the IDH1/2 genes has been performed previously in this laboratory.
The presence of H3F3A p.G34 alterations in 96% of GCT along with their absence in other benign bone tumours including CB, osteoid osteoma, osteoblastoma, tenosynovial giant cell tumour and chondromyxoid fibroma provides evidence that H3F3A p.G34 mutations are a highly sensitive and specific marker for GCT. It is particularly valuable for distinguishing between those tumours with mutually exclusive alterations such as GCT and CB, and GCT and ABC, which harbour USP6 fusion genes in 75% of cases 5, 22. In contrast, there is no genetic alteration characteristic of NOF. Although H3F3A p.G34W alterations were detected in 4/66 cases, we propose that these cases represent misdiagnoses, based on a multidisciplinary review of these cases. Indeed, in all cases studied here in which genotype was discordant with the original pathological diagnosis, we reviewed histology and imaging. In all but one case, we altered our original diagnosis in favour of the genetic findings. Nevertheless, using the detection of H3.3 alterations in clinical practice would be more robust, if mutually exclusive markers were available for each of these tumour types, or if a second recurrent alteration was present in these tumours.
GCG of the small bone of the hands and feet and GCG of the jaws are considered to be distinct from conventional GCT (WHO classification 1). It has been argued that these lesions represent a ‘solid variant’ of a primary ABC 23. Agaram et al recently provided molecular evidence for this proposition by demonstrating USP6 rearrangements in 8/9 GCG of the hands and feet 4. They also reported no USP6 alterations in eight conventional GCT, two of which were located in the finger. The combined data from our current and previous study 2 provide further evidence for the occurrence of solid variants of ABC in the bones of the hands and feet, and show for the first time that GCT also represents a significant number of osteoclast‐rich tumours at these sites, highlighting the benefit of using these mutually exclusive genetic alterations for distinguishing these entities.
It is noteworthy that neither H3.3 nor USP6 alterations were detected in any of the 78 GCG of the jaw in our study indicating that GCG of the jaws are distinct from both GCT and solid variants of primary ABC. From a clinical perspective, it is important to note that osteoclast‐rich lesions of the jaw also occur in patients with Noonan syndrome, neurofibromatosis (NF1) and cherubism (SH2BP2), and that these should be excluded before provision of a diagnosis of GCG 24, 25. Not all GCG included, in this study had been analysed for these alterations and it is, therefore, possible that some of the cases diagnosed as GCG represent syndrome‐related lesions.
The introduction of screening of bone tumours for H3.3 alterations along with other common bone cancer genes including IDH1/2 alterations in cartilaginous tumours 8, 9, GRM1 rearrangements of chondromyxoid fibromas 26 and USP6 alterations in ABC 5, 6, 22 adds to the armoury with which pathologists can provide accurate diagnoses. Although H3.3 alterations does not stratify patients to therapeutic agents at this time, in the future this may be the case 20, 21. The study also highlights the changes required in laboratory practices if these new findings and those yet to be discovered are to be introduced effectively into clinical medicine.
Author contributions
The concept was conceived by AMF. Review of the pathology was undertaken by histopathologists AMF, MFA, RT, DB and GJ. Genome sequencing was performed at the CR‐UK Oxford Molecular Pathology Institute (ABH) and analysed at Wellcome Trust Sanger by DW, PVL, SB, PT, and PC. Preparation of the samples and DNA was performed by BK. The experimental design was performed by NP, MFA, RH, AMF and DB. Analysis of the data was performed by NP, MFA, and AMF. The MS was written by AMF, DB, SB, NP and MFA.
Sequencing data have been deposited at the European Genome‐Phenome Archive (EGA, http://www.ebi.ac.uk/ega/), which is hosted by the European Bioinformatics Institute (EBI); accession number pending.
Supporting information
SUPPLEMENTARY MATERIAL ON THE INTERNET
The following supplementary material may be found in the online version of this article:
Supplementary methods:
Table S1. Samples of giant cell tumour (GCT) and chondroblastoma (CB) with H3.3 alterations published in the previous study [2] with additional results to the original table published for USP6 analysis and targeted sequencing.
Table S2. Samples with diagnoses, clinical and genetic data from this study.
Table S3. Sample type and distribution.
Table S4. Primer sequences and description of the hot‐spot targeted with PGM sequencer.
Table S5. Somatic substitutions and indels.
Figure S1. Genome‐wide copy number profile of five giant cell tumours of bone. 4/5 tumours show essentially no copy number changes. Bottom: PD7524a harbours telomeric LOH in chromosomes 5q and 8p (arrows) assessed by Affymetrix SNP6. The smaller changes seen are likely to represent ‘noise’ as the sample was shown to be of low cellularity (33%). X‐axis: genomic position (ascending). Y‐axis: copy number. Numbers in plot refer to chromosome. Red and green: major and minor allele, respectively.
Figure S2. (A) Photomicrographs of a tumour presenting in a 59 year old originally diagnosed as a GCT. The biopsy showed a fragment of mature hyaline cartilage and an osteoclast‐rich mitotically active tumour. The tumour was wild type for H3F3 and an IDH2 alteration was detected. Following re‐evaluation and in the context of the genetics a diagnosis of a dedifferentiated chondrosarcoma was made. (B) Radiograph and photomicrographs of subarticular lytic tumour in the proximal tibia. The imaging suggested a GCT but the histology showed a mitotically active spindle cell tumour with epithelioid differentiation, supported by the cytokeratin expression (inset) and an osteoclast‐rich area. The tumour was classified as an unusual high grade osteoclast‐rich tumour with adamantinoma‐like features.
Figure S3. Algorithm‐based schema illustrating the approach to reaching a diagnosis of osteoclast‐rich tumours.
Acknowledgements
We are grateful to the patients for participating in the research and to the clinicians and support staff in the London Sarcoma Service involved in their care. We are grateful to Siobhan Roche, RNOH, for her technical support and to Mrs Ru Grinnell and Miss Dee Ibrahim for consenting the patients to the RNOH Musculoskeletal Research Programme and Biobank.
Contract/grant details: The research was funded by Skeletal Cancer Action Trust (SCAT), UK. Support was provided to by the National Institute for Health Research, UCLH Biomedical Research Centre (AMF), and the National Institute for Health Research, Musculoskeletal BRU Oxford Sarcoma Theme, and the CRUK UCL Experimental Medicine Cancer Centre (AMF), and the EuroBoNeT consortium, a European Commission granted Network of Excellence for studying the pathology and genetics of bone tumours. P.J.C. is personally funded through a Wellcome Trust Senior Clinical Research Fellowship (grant reference WT088340MA). SB is funded through the Wellcome Trust PhD Programme for Clinicians. DB and GJ were supported by the Foundation for the Preservation of the Basel Bone Tumor Reference Center.
The authors have declared no conflicts of interest.
References
- 1. Athanasou NA, Bansal M, Forsyth R, et al Giant cell tumour of bone In WHO Classification of Tumours of Soft Tissue and Bone, (4th edn), Fletcher CDM, Bridge JA, Hogendoorn PCW, et al (eds). IARC Press: Lyon, France, 2013; 321–324. [Google Scholar]
- 2. Behjati S, Tarpey PS, Presneau N, et al Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet 2013; 45: 1479–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Joseph CG, Hwang H, Jiao Y, et al Exomic analysis of myxoid liposarcomas, synovial sarcomas, and osteosarcomas. Genes Chromosomes Cancer 2014; 53: 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Agaram NP, LeLoarer FV, Zhang L, et al USP6 gene rearrangements occur preferentially in giant cell reparative granulomas of the hands and feet but not in gnathic location. Hum Pathol 2014; 45: 1147–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Oliveira AM, Hsi BL, Weremowicz S, et al USP6 (Tre2) fusion oncogenes in aneurysmal bone cyst. Cancer Res 2004; 64: 1920–1923. [DOI] [PubMed] [Google Scholar]
- 6. Oliveira AM, Perez‐Atayde AR, Inwards CY, et al USP6 and CDH11 oncogenes identify the neoplastic cell in primary aneurysmal bone cysts and are absent in so‐called secondary aneurysmal bone cysts. Am J Pathol 2004; 165: 1773–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. West RB, Rubin BP, Miller MA, et al A landscape effect in tenosynovial giant‐cell tumor from activation of CSF1 expression by a translocation in a minority of tumor cells. Proc Natl Acad Sci USA 2006; 103: 690–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Amary MF, Bacsi K, Maggiani F, et al IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol 2011; 224: 334–343. [DOI] [PubMed] [Google Scholar]
- 9. Kato Kaneko M, Liu X, Oki H, et al Isocitrate dehydrogenase mutation is frequently observed in giant cell tumor of bone. Cancer Sci 2014; 105: 744–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barnes L, Eveson JW, Reichart P, et al World Health Organization Classification of Tumours: Pathology and Genetics of Head and Neck Tumours. IARC Press: Lyon, France, 2005. [Google Scholar]
- 11. Fletcher CDM, Bridge JA, Hogendoorn PCW, et al (eds) WHO Classification of Tumours of Soft Tissue and Bone. IARC Press: Lyon, France, 2013. [Google Scholar]
- 12. Behjati S, Tarpey PS, Sheldon H, et al Recurrent PTPRB and PLCG1 mutations in angiosarcoma. Nat Genet 2014; 46: 376–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 2009; 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high‐performance genomics data visualization and exploration. Brief Bioinform 2013; 14: 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Siebenrock KA, Unni KK, Rock MG. Giant‐cell tumour of bone metastasising to the lungs. A long‐term follow‐up. J Bone Joint Surg Br 1998; 80: 43–47. [DOI] [PubMed] [Google Scholar]
- 16. Ballester LY, Wang Z, Shandilya S, et al Morphologic characteristics and immunohistochemical profile of diffuse intrinsic pontine gliomas. Am J Surg Pathol 2013; 37: 1357–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schwartzentruber J, Korshunov A, Liu XY, et al Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012; 482: 226–231. [DOI] [PubMed] [Google Scholar]
- 18. Lewis PW, Muller MM, Koletsky MS, et al Inhibition of PRC2 activity by a gain‐of‐function H3 mutation found in pediatric glioblastoma. Science 2013; 340: 857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chan KM, Fang D, Gan H, et al The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev 2013; 27: 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rohle D, Popovici‐Muller J, Palaskas N, et al An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013; 340: 626–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang F, Travins J, DeLaBarre B, et al Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 2013; 340: 622–626. [DOI] [PubMed] [Google Scholar]
- 22. Amary MF, Ye H, Berisha F, et al Detection of USP6 gene rearrangement in nodular fasciitis: an important diagnostic tool. Virchows Arch 2013; 463: 97–98. [DOI] [PubMed] [Google Scholar]
- 23. Oliveira AM, Chou MM. USP6‐induced neoplasms: the biologic spectrum of aneurysmal bone cyst and nodular fasciitis. Hum Pathol 2014; 45: 1–11. [DOI] [PubMed] [Google Scholar]
- 24. Reichenberg EJ, Flanagan AM. Cherubism In WHO Classification of Tumours of Soft Tissue and Bone, Fletcher CDM, Bridge JA, Hogendoorn PCW, et al (eds). IARC Press: Lyon, France, 2013; 374. [Google Scholar]
- 25. Flanagan AM, Tirabosco R, Panagiotis DG. Osteoclast‐rich lesions of bone: a clinical and molecular view In Bone Cancer Progression and Therapeutic Approaches, (2nd edn) Heymann D. (ed). Academic Press, Elsevier Inc., USA. 2015; 257–272. [Google Scholar]
- 26. Nord KH, Lilljebjorn H, Vezzi F, et al GRM1 is upregulated through gene fusion and promoter swapping in chondromyxoid fibroma. Nat Genet 2014; 46: 474–477. [DOI] [PubMed] [Google Scholar]
- 27. Van Loo P, Nordgard SH, Lingjaerde OC, et al Allele‐specific copy number analysis of tumors. Proc Natl Acad Sci U S A 2010; 107: 16910–16915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY MATERIAL ON THE INTERNET
The following supplementary material may be found in the online version of this article:
Supplementary methods:
Table S1. Samples of giant cell tumour (GCT) and chondroblastoma (CB) with H3.3 alterations published in the previous study [2] with additional results to the original table published for USP6 analysis and targeted sequencing.
Table S2. Samples with diagnoses, clinical and genetic data from this study.
Table S3. Sample type and distribution.
Table S4. Primer sequences and description of the hot‐spot targeted with PGM sequencer.
Table S5. Somatic substitutions and indels.
Figure S1. Genome‐wide copy number profile of five giant cell tumours of bone. 4/5 tumours show essentially no copy number changes. Bottom: PD7524a harbours telomeric LOH in chromosomes 5q and 8p (arrows) assessed by Affymetrix SNP6. The smaller changes seen are likely to represent ‘noise’ as the sample was shown to be of low cellularity (33%). X‐axis: genomic position (ascending). Y‐axis: copy number. Numbers in plot refer to chromosome. Red and green: major and minor allele, respectively.
Figure S2. (A) Photomicrographs of a tumour presenting in a 59 year old originally diagnosed as a GCT. The biopsy showed a fragment of mature hyaline cartilage and an osteoclast‐rich mitotically active tumour. The tumour was wild type for H3F3 and an IDH2 alteration was detected. Following re‐evaluation and in the context of the genetics a diagnosis of a dedifferentiated chondrosarcoma was made. (B) Radiograph and photomicrographs of subarticular lytic tumour in the proximal tibia. The imaging suggested a GCT but the histology showed a mitotically active spindle cell tumour with epithelioid differentiation, supported by the cytokeratin expression (inset) and an osteoclast‐rich area. The tumour was classified as an unusual high grade osteoclast‐rich tumour with adamantinoma‐like features.
Figure S3. Algorithm‐based schema illustrating the approach to reaching a diagnosis of osteoclast‐rich tumours.