

Abstract
Uterine smooth muscle tumours of uncertain malignant potential (STUMP) are diagnostically and clinically challenging. The alternative lengthening of telomeres (ALT) telomere maintenance mechanism is associated with poor survival in soft tissue leiomyosarcoma. Time to first recurrence and survival were known for 18 STUMP and 43 leiomyosarcomata (LMS). These were screened for ALT telomere maintenance by the presence of ALT‐associated PML bodies (APBs) and for changes associated with the ALT phenotype, namely aberrant p53 expression, isocitrate dehydrogenase 1 mutation (R132H substitution) expression, mutant ATRX (αthalassemia/mental retardation syndrome X‐linked) expression and mutant DAXX (death‐domain‐associated protein) expression by immunohistochemistry (IHC). Overexpression of p16INK4A was examined immunohistologically in a subset of cases. Many of the tumours associated with death or recurrence demonstrated APBs commensurate with ALT telomere maintenance. However, all uterine STUMP (4/4), and vaginal STUMP (2/2) patients, and almost all LMS patients (88.4%, 23/26, including 90% (9/10) of stage 1 LMS cases), who had died of disease or who had recurrent disease, displayed loss of ATRX or DAXX expression. Loss of ATRX or DAXX expression identified poor prognosis (95% CI 2.1 to 40.8, p < 0.003), in the LMS group. Thus, loss of ATRX or DAXX expression in uterine smooth muscle tumours identifies a clinically aggressive molecular subtype of early stage LMS and when histopathological features are problematic such as in STUMP. As ATRX and DAXX IHC is readily performed in diagnostic laboratories these are potentially useful for routine histopathological classification and management.
Keywords: telomere maintenance mechanism, prognosis, leiomyoma, STUMP, leiomyosarcoma, DAXX, ATRX
Introduction
For nearly 50 years uterine smooth muscle tumours fulfilling some, but not all, of the pathological criteria for uterine leiomyosarcoma (LMS) have been diagnostically and clinically challenging 1. Smooth muscle tumours of uncertain malignant potential (STUMP) have histopathological features of malignancy falling short of LMS, variably clinically aggressive course and recurrence rates of 4.6% to 26.7% depending on histopathological criteria employed 2. These characteristics have generated considerable management and treatment uncertainty and necessitated prolonged periods of follow‐up 1, 3 At the molecular level, aberrant immunohistochemical expression of p53 and p16INK4A have been investigated as prognostic and diagnostic tools and are correlated with poorer prognosis in some studies 4
Telomere maintenance is an important aspect of the replicative capacity inherent in neoplasia, and telomerase inhibitors have been developed as potential targeted therapy 5. In the normal cell, progressive telomere shortening to a critical length occurs until cell division ceases. This shortening is circumvented in neoplasia by the activation of telomere maintenance 6. Recent progress in the molecular subtyping of mesenchymal tumours has identified prognostic implications in the type of telomere maintenance mechanism employed. The presence of alternative lengthening of telomeres (ALT) maintenance portends a better prognosis in glioblastoma multiforme compared with telomerase activation in these tumours 7, but a worse prognosis in many soft tissue sarcomata 8. ALT maintenance has been demonstrated in uterine LMS as a poor prognostic indicator 9 10. ALT telomere maintenance tumours may also respond differently to treatment, especially radiotherapy 11.
ALT maintenance in pancreatic neuroendocrine tumours and pediatric glioblastoma multiforme has been shown to closely correlate with the presence of a mutation in ATRX and DAXX genes 12, 13. In glioblastoma multiforme ALT tumours were associated with simultaneous mutations in ATRX‐DAXX, p53 and H3F3A 14. The ATRX and DAXX proteins are demethylases involved in remodeling of chromatin structure. The alteration of chromatin architecture through the inhibition of demethylation by ATRX and DAXX dysfunction has been reported to underlie ALT activation in gliomagenesis 14. Isocitrate dehydrogenase (IDH) mutations have also been identified in association with ALT activation in tumours 15, 16.
ALT TMM produces extremely long and heterogeneous telomere lengths and multiprotein structures called ALT‐associated PML bodies (APBs). Evaluation of ALT by APB detection as well as ATRX and DAXX immunohistochemistry (IHC) was conducted to explore their development as reproducible histopathological tools, thereby enhancing diagnosis and predicting clinical progress of smooth muscle tumours. The profile of the ALT associated R132H IDH1 mutation was studied to provide a biological basis for its application as a radiological diagnostic and therapeutic tool.
We hypothesized that ALT telomere maintenance identifies clinically aggressive tumours prone to recurrence and death and that ALT maintenance was an early tumourigenic change. This would prove useful for diagnostically difficult tumours (STUMP) and clinically useful prior to emergent metastatic disease (stage 1 LMS) for therapeutic management and follow‐up. Tumour protein p53 and p16INK4A IHC was also studied to further define their diagnostic utility in a larger cohort than had previously been examined.
Methods
Patients with uterine smooth muscle tumours at Dunedin and Tauranga Hospitals, New Zealand, and Queen Mary Hospital, Hong Kong, were studied. Ethical approval was obtained from the New Zealand Health and Disabilities Lower South Regional Ethics Committee. Clinical and pathological data regarding patient's age, tumour size, gross and histological features, tumour stage, treatment and follow‐up were obtained from hospital notes and pathological reports. Eleven cases of cellular or variant leiomyoma (LM), 16 cases of uterine STUMP, two cases of vaginal smooth muscle tumour (fulfilling criteria for uterine STUMP) and 43 cases of LMS were included. The pathological sections were reviewed according to established criteria 4, based on the presence of necrosis, nuclear atypia, margin and mitotic activity. Five of the variant LM were cellular and one was mitotically active but without cytological atypia or tumour cell necrosis. The presence of diffuse or multifocal moderate to severe cytologic atypia and <10 mitoses per 10HPFs, tumour cell necrosis but no other worrisome features or >15 mitoses per 10HPFs but no other worrisome features, defined each STUMP case, and are illustrated in Figure 1. STUMP case 6 was a mixed spindle and myxoid tumour with mitotically active spindle cell component but with low (<0/10HPF) mitotic count in the myxoid component, and thus fell short of criteria for a myxoid LMS. Similarly, case 8 was a mixed spindle and epithelioid tumour, but with less than 50% epithelioid component, thus falling short of criteria for an epithelioid LMS 4. As no criteria exist for the diagnosis of a vaginal STUMP, we have applied the uterine criteria to two vaginal cases in an effort to better characterize these rare tumours. Seven of the 18 cases of STUMP had previously been reported by two of us 2. LMS were classified according to WHO criteria 17.
Figure 1.
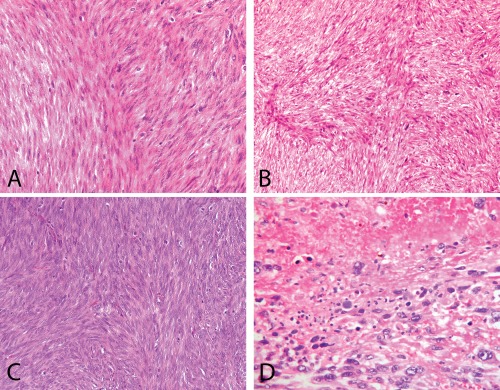
A to C: Three cases of STUMP that were followed by a recurrence. These have moderate to high cellularity, multifocal moderate atypia and <or=<10 mitoses/10 HPFs. D: Leiomyosarcoma with marked atypia and tumour cell necrosis.
IHC analyses
IHC reactions were performed using standard techniques: paraffin embedded tissue sections, heat‐mediated antigen retrieval and antibody detection by EDL (Dako, Glostrup, Denmark) and DAB according to the manufacturers' instructions. p53 expression was determined using DO‐7 (Cell Marque, Rockin, CA) at a 1 in 50 dilution. p16INK4A was detected using 1 in 1000 dilution (2D9A12, Abcam, Cambridge, UK). Mutant IDH1 was detected using the anti‐Human IDH1 R132H specific mouse monoclonal antibody HO‐9 (Dianova, Hamburg, Germany) diluted 1 in 50. ATRX was detected using HPA001906 (Sigma‐Aldrich, St Louis, MO) diluted 1 in 700. DAXX was performed using a 1 in 100 dilution HPA008736 (Sigma‐Aldrich, St Louis, MO). Cells were imaged using light microscopy (DM 2000 microscope, DFC 295 camera and Application Suite software, version 3.5.0, Leica, Solms, Germany).
The slides were assessed by four surgical pathologists (PI, AC, NH and TS) independently. A tumour was considered positive for p53 when either no tumour cell staining was evident, or greater than 80% of the tumour cells were positively stained. IDH1 was considered positive when 10% or more of the tumour nuclei were moderately stained by IHC. IHC for p16INK4A expression was performed in a limited cohort of 36 cases (15/18 STUMP and 24/43 LMS). Greater than 70% nuclear staining at intensity greater than adjacent normal vascular smooth muscle was interpreted as p16INK4A, overexpression (Figure 2). Ten percent or less nuclear staining in more than 50% of the tumour for ATRX or DAXX protein determined loss of expression. Cell nuclei of endothelial and vascular smooth muscle from normal tissue in the sections served as an internal control for ATRX or DAXX protein expression (thus normal cell nuclear staining of these nuclei was expected within the tumour, Figure 2).
Figure 2.
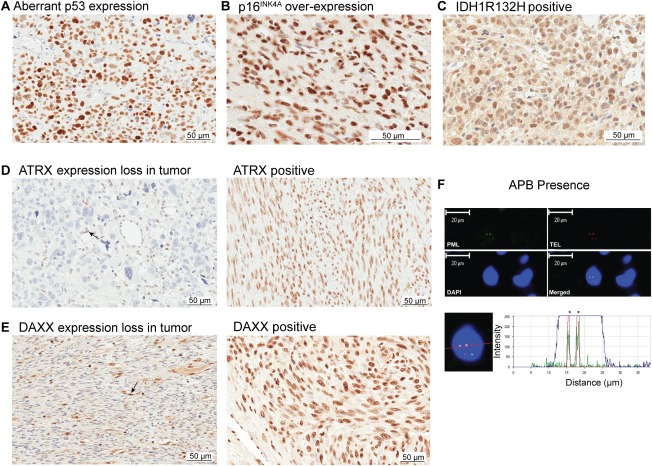
FISH and immunohistochemical staining of uterine smooth muscle tumours. (A) Aberrant p53 expression in a tumour. (B) p16INK4A overexpression in a tumour. (C) IDH1 (isocitrate dehydrogenase 1) mutation (R132H substitution) immunopositive tumour. (D) Left, ATRX (α thalassemia/mental retardation syndrome X‐linked) immunonegative tumour (indicative of loss of expression and surrogate for ATRX mutation). Positive staining is present in associated normal cells (endothelial cells are highlighted with a black arrow). Right, immunopositive ATRX staining (no loss of expression and surrogate for no ATRX mutation). (E) Left, DAXX (DAXX death‐domain‐associated protein) immunonegative tumour (indicative of loss of expression and surrogate for DAXX mutation). Positive staining is present in associated normal cells (positive staining in endothelial cells is highlighted with a black arrow). Right, immunopositive DAXX staining (no loss of expression and surrogate for no DAXX mutation). (F) Co‐localisation of telomere FISH and IHC for promyelocytic protein to detect ALT‐associated PML bodies (APBs). Top left, PML immunofluorescence (green), Top right telomere fluorescence in situ hybridisation (FISH) (red), bottom left DAPI nuclear stain (blue), bottom right combined images. Bottom images, identification of colocalized PML – immunofluorescence image and telomere FISH intensity graph.
APB detection
The APB detection method used in this study is a modified version of the method used by Yeager et al. (1999) and Henson et al. (2005) and our specific methodology has been reported in earlier studies 8, 18, 19. Briefly, slides were incubated an anti‐PML rabbit polyclonal primary antibody (H‐238, Santa Cruz Biotechnology, Santa Cruz SD) using a 1 in 500 dilution and detected with an Alexa Fluor 488 antibody (Life Technologies, Carlsbad, CA). Telomere DNA was detected using a Cy3‐labelled PNA probe 5′‐CCCTAACCCTAACCCTAA‐3′ (Life Technologies, Carlsbad, CA). The cellular nuclei were stained using DAPI. Cells were imaged using confocal microscopy (Zeiss LSM510; Carl Zeiss, Thornwood, NY). For each colocalized signal, the fluorescent signal from each channel was analysed using the software Zeiss LSM Image Examiner (Version 30115; Carl Zeiss Thornwood, NY). A tumour was designated APB positive when a colocalized focus of telomeric DNA and PML protein within the nucleus was identified in ≥ 5% of the cells. One APB in a cell nucleus qualified the cell as positive. APBs were interpreted as bright yellow colocalized areas with a clear peak signal of at least 100 relative fluorescent intensity (Figure 2).
Statistical analyses
Correlations between categorical variables were analysed with the χ2 test with p < 0.05 taken as statistically significant. Descriptive statistics of patients and their tumours, and the frequencies of observing each variant (i.e., prospective marker) among patients in each tumour cohort were prepared. The risk of outcome event among patients with each marker was estimated using univariate Cox regression. Cases with unknown outcome status were considered as censored in these time‐to‐event analyses.
Results
The clinical characteristics of the cases are summarized in Table 1. Patients with LM were treated by hysterectomy or surgical excision alone. None of these patients had a recurrence. Patients with a STUMP diagnosis underwent hysterectomy or surgical excision alone (one case underwent myomectomy only). None of the patients with a uterine STUMP diagnosis received radiation or chemotherapy at the time of diagnosis. In this cohort four women (25%) had first recurrence at mean 49 months, and three women (18.7%) had died of disease at 56, 60 and 140 months post diagnosis. In the LMS cohort, nearly half of the women (49%) had a recurrence or had died, with mean recurrence or death at 32 months. However, the longest recurrence‐free survival was 11 years (case 13).
Table 1.
Clinical details for the LM, STUMP and LMS cohorts
| Leiomyoma | Cellular/Mitotic Leiomyoma | STUMP | LMS | |
|---|---|---|---|---|
| Number of patients | 5 | 6 | 18 | 43 |
| Mean age/years | 50 | 49 | 44 | 47 |
| Postmenopausal at presentation | 2 (40%) | 1 (16%) | 3 (16%) | 10 (23%) |
| Median tumour size cm | 3.5 | 3.9 | 9 | 11 |
| Extrauterine disease at presentation | N/a | N/a | N/a | 45%* |
| LMS Characteristics | Stage | n (%) | Mean survival months | Mean follow‐up months |
| 1 | 23 | 70.2 | 69.3 | |
| 2 | 5 | 55.8 | 55.8 | |
| 3 | 10 | 45.6 | 45.6 | |
| 4 | 3 | 15.3 | 15.3 | |
| Missing | 2 | 19.0 | 19 |
One case unknown.
STUMP, Smooth muscle tumours of uncertain malignant potential;
LMS, Leiomyosarcoma,
N/a not applicable, n: number.
The follow‐up period totaled 2567 person‐months without recurrence, 1243.5 person‐months with recurrence, 2742 person‐months death not observed and 1199.5 person‐months among patients who died of disease. The STUMP cases were followed for a mean of 70 months (median 58 months, range 1–140 months) until recurrence or death, and 85.4 months (median 85 months, range 6–168 months) for no evidence of disease. The LMS cases were followed a mean of 35 months (median 22 months, range 1–138 months) until recurrence or death, and 78 months (median 78 months, range 1–156 months) for no evidence of disease. The uterine STUMP cases recurred later (mean 49 months, 95% CI 47, 51) compared to recurrent LMS cases (mean 22.3 months, 95% CI 6, 38, p = 0.02), but both succumbed in a similar period (STUMP 76 months, 95% CI 9, 143 versus LMS mean 98 months, 95% CI 78–117, p = 0.07). For patients who presented in stage 1 LMS at diagnosis (n = 23), 35% had died by 10 years follow‐up (114 months) and two patients were alive with disease at 23 and 78 months follow‐up.
In the STUMP cohort the predominant histopathological type was spindle cell, except for two mixed spindle cases (with <50% area of epithelioid component, and myxoid admixed). The LMS cases were also predominantly spindle cell type (72%, with epithelioid, myxoid or mixed types in the remaining). Neither age nor tumour size was significantly different between the STUMP and LMS cases (p = 0.10 and p = 0.18). The mitotic rate, however, was significantly lower in the STUMP group (p < 0.0001) as expected with required diagnostic criterion of less than 10 mitoses per 10HPF. The individual features are given in Tables 2 and 3.
Table 2.
Clinical, pathological and molecular characteristics of the STUMP cohort
| Clinical Features | Histopathological Features | Molecular Features | Clinical Outcome | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case | Age/yrs | Clinical Status | Tumour size/cm | Cytological Atypia | Necrosis | Mitotic Count/10HPF | ATRX or DAXX Expression | APB Presence | Aberrant p53 Expression# | p16INK4A Overexpression | IDH1 R132H Expression | Time to Recurrences or DOD (months) | Follow‐up (months) |
| 1 | 39 | DOD | 5 | mod/severe | nil | 5 | LOSS | Present | Present | Present | neg | 48, 56 | — |
| 2 | 45 | DOD | 8 | mod/severe | nil | 6 | LOSS | Present | Present | n/a | neg | 50, 60 | — |
| 3 | 50 | DOD | 10 | Mild | TCN | 4 | LOSS | Present | neg | n/a | neg | 51, 123, 132, 140 | — |
| 4 | 40 | AWD Treated Recurrence** | 10 | mod/severe | nil | 7 | LOSS | Present | Present | Present | neg | 48 | 72 |
| 5 | 26 | NED | 1.5 | mod/severe | nil | 5 | pos | neg | Present | Present | neg | nil | 56 |
| 6* | 57 | NED | 1·5 | mod/severe | nil | 1 | pos | neg | Present | Present | neg | nil | 34 |
| 7 | 54 | NED | 4 | mod/severe | nil | 9 | pos | neg | Present | neg | neg | nil | 168 |
| 8* | 39 | NED | 12 | mod/severe | nil | 1 | pos | neg | neg | Present | neg | nil | 75 |
| 9 | 41 | NED | 6·5 | mod/severe | nil | 2 | pos | neg | neg | Present | neg | nil | 56 |
| 10 | 48 | NED | 17 | Mild | TCN | 5 | pos | neg | neg | neg | neg | nil | 107 |
| 11 | 46 | NED | 15 | Mild | TCN | 2 | pos | neg | neg | n/a | neg | nil | 126 |
| 12 | 52 | NED | 7·5 | Mild | TCN | 3 | pos | neg | neg | neg | neg | nil | 120 |
| 13 | 40 | NED | 8·5 | Mild | TCN | 4 | pos | neg | neg | neg | neg | Lost to follow‐up | 6 |
| 14 | 42 | NED | 10 | Mild | TCN | 0 | pos | neg | neg | neg | neg | nil | 124 |
| 15 | 35 | NED | 8·5 | Mild | TCN | 5 | pos | neg | neg | neg | neg | nil | 122 |
| 16 | 42 | NED | 4 | Mild | TCN | 4 | pos | neg | neg | neg | neg | nil | 95 |
| 17VAGINAL | 71 | DOD | Unk | Mild | TCN | 9 | LOSS | Present | neg | neg | neg | 56 | — |
| 18VAGINAL | 38 | NED‐Treated Recurrences | 4.2 | Mild | TCN | 1 | LOSS | neg | neg | neg | neg | 1, 13 | 35 |
^No STUMP tumour had more than 10 mitoses per 10HPFs
*Histopathological subtype case 6 mixed spindle (mitotic component) and myxoid (nonmitotic), case 8 mixed spindle and epithelioid,
** Debulking and radiotherapy
# ≥80% or 0% staining
Results: Bold text and box shading to highlight variant cases
NED, No evidence of disease; AWD, Alive with disease; DOD, Dead of disease.
Mod/severe: Moderate to severe cytological atypia
Neg, Negative IHC staining as defined in methods; Pos, Positive IHC staining as defined in methods.
ATRX, α thalassemia/mental retardation syndrome X‐linked; DAXX, Death‐domain‐associated protein.
APB, ALT associated PML bodies; IDH1, Isocitrate dehydrogenase 1 R132H substitution;
p53, Tumour protein 53; Nd: Not determined, Unk: unknown
Table 3.
Clinical, pathological and molecular characteristics of the LMS cohort grouped by TNM Stage
| Clinical Features | Histopathological Features | Clinical Outcome | Treatment at Diagnosis | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case | Stage (TNM) | Age/yrs | Clinical Status | Tumour Size (cm) | Tumour Cytology | Mitotic Count/10HPF | ATRX or DAXX Expression | APB Presence | Aberrant p53 Expression | p16INK4A Overexpression | IDH1 R132H Expression | Time to Recurrence/s or DOD (months) | Follow‐up/months | |
| 1 | Stage 1 | 76 | DOD | 7 | Spindle | 10 | LOSS | Present | Present | Present | neg | 61,74 | — | None |
| 2 | 47 | DOD | Unk | Spindle | 31 | LOSS | Present | Present | neg | neg | 8, 52 | — | Gemcitabine, taxotere and radiotherapy | |
| 3 | 35 | DOD | 8 | Epithelioid | 15 | LOSS | Present | neg | Present | neg | 22,30 | — | Radiotherapy | |
| 4 | 46 | DOD | 5 | Spindle | 10 | LOSS | Present | neg | Present | neg | 24, 27 | — | Radiotherapy | |
| 5 | 39 | DOD | 5 | Spindle | 12 | LOSS | neg | Present | nd | Present | 56 | — | None | |
| 6 | 63 | DOD | 10 | Epithelioid | 8 | LOSS | neg | Present | nd | Present | 15,44 | — | Radiotherapy | |
| 7 | 50 | DOD | 4 | Spindle | 4 | LOSS | neg | neg | nd | neg | 114 | — | None | |
| 8 | 35 | DOD | 13 | Spindle | 40 | pos | neg | neg | neg | neg | 77.5 | — | None | |
| 9 | 53 | Treated Recurrence | 7.8 | Spindle | 30 | LOSS | neg | Present | Present | neg | 23 | 43 | None | |
| 10 | 35 | AWD | Unk | Epithelioid | 5 | pos | neg | neg | Present | neg | nil | 23 | Radiotherapy | |
| 11 | 47 | NED | 7 | Spindle | 7 | LOSS | Present | Present | nd | neg | nil | 78 | None | |
| 12 | 44 | NED | 10 | Spindle | 18 | LOSS | Present | neg | neg | neg | nil | 59 | Radiotherapy | |
| 13 | 50 | NED | 4.5 | Spindle | 11 | LOSS | Present | neg | neg | neg | nil | 156 | None | |
| 14 | 53 | NED | 4 | Spindle | 4 | LOSS | Present | neg | nd | neg | nil | 66 | Radiotherapy | |
| 15 | 39 | NED | 4 | Epithelioid | 0 | pos | neg | Present | Present | Present | nil | 68 | Radiotherapy | |
| 16 | 47 | NED | 8 | Epithelioid | 15 | pos | neg | Present | nd | neg | nil | 4 | Radiotherapy | |
| 17 | 40 | NED | 4 | Epithelioid | 0 | pos | neg | neg | neg | neg | nil | 66 | Radiotherapy | |
| 18 | 47 | NED | 10 | Spindle | 10 | pos | neg | neg | neg | neg | nil | 80 | Radiotherapy | |
| 19 | 47 | NED | 20 | spindle | 17 | pos | neg | neg | neg | neg | nil | 84 | unknown | |
| 20 | 56 | NED | 13 | spindle | 10 | pos | neg | neg | neg | neg | nil | 83 | unknown | |
| 21 | 39 | NED | 11 | Spindle | 10 | pos | neg | neg | nd | neg | nil | 64 | Radiotherapy | |
| 22 | 52 | NED | 14.5 | Spindle | 10 | pos | neg | neg | nd | neg | nil | 120 | Radiotherapy | |
| 23 | 57 | NED | 10 | Spindle | 17 | pos | neg | neg | nd | neg | nil | 146 | None | |
| 24 | Stage 2 | 49 | DOD | 11 | Spindle | 14 | LOSS | Present | neg | neg | neg | 71,84, 98 | — | Radiotherapy |
| 25 | 45 | AWD | 16.5 | Epithelioid | 21 | LOSS | Present | neg | Present | neg | lost to follow‐up | 1 | Radiotherapy | |
| 26 | 45 | AWD | 29 | Epithelioid & myxoid | 3 | LOSS | neg | Present | nd | neg | lost to follow‐up | 1 | Unknown | |
| 27 | 48 | NED | 7 | Spindle | 24 | pos | Present | neg | Present | Present | nil | 103 | Radiotherapy, megace | |
| 27 | 39 | NED | 21 | Spindle | 9 | pos | neg | Present | nd | neg | nil | 76 | Radiotherapy | |
| 29 | Stage 3 | 52 | DOD | 8.5 | Epithelioid | 43 | LOSS | Present | Present | Present | neg | 2 | — | Gemcitabine, taxotere |
| 30 | 45 | DOD | 5 | Spindle | 25 | LOSS | Present | Present | nd | neg | 37, 48 | — | Radiotherapy | |
| 31 | 51 | DOD | 17 | Spindle | 7 | LOSS | Present | neg | nd | Present | 23 | — | VAC+AC, followed by ERT & adriamycin | |
| 32 | 79 | DOD | 13 | Epithelioid | 13 | LOSS | neg | Present | nd | neg | 1 | — | None | |
| 33 | 42 | DOD | 4 | Spindle | 12 | LOSS | neg | neg | Present | neg | 10,14 | — | VAC‐vincristine, actinomycin‐D and cyclophosphamide | |
| 34 | 44 | DOD | 9 | Spindle | 28 | LOSS | neg | neg | nd | neg | 3 | — | None | |
| 35 | 52 | DOD | 22 | Spindle | 22 | LOSS | neg | neg | nd | neg | 138 | — | adriamycin | |
| 36 | 74 | DOD | 20 | Spindle | 8 | pos | neg | Present | Present | neg | 2 | — | None | |
| 37 | 38 | NED | 24 | Epithelioid | 8 | pos | neg | POS | nd | neg | nil | 147 | DDP/Adriamycin | |
| 38 | 47 | NED | 30 | Myxoid | 1 | pos | neg | neg | Present | neg | nil | 78 | ERT | |
| 39 | Stage 4 | 54 | DOD | 11 | Epithelioid | 15 | LOSS | Present | Present | Present | Present | 29 | — | Gemcitabine, taxotere |
| 40 | 77 | DOD | 10.5 | Spindle | 20 | LOSS | neg | neg | Present | neg | 2 | — | None | |
| 41 | 41 | DOD | Unk | Spindle | 10 | LOSS | neg | neg | nd | neg | 15 | — | DDP and adriamycin | |
| 42 | Unknown | 52 | DOD | 8 | spindle | 15 | LOSS | Present | Present | neg | neg | 16 | — | unknown |
| 43 | 65 | DOD | 4.5 | Spindle | 20 | LOSS | Present | Present | nd | neg | 22 | — | None | |
Results: Bold text and box shading to highlight variant cases
NED, No evidence of disease; AWD, Alive with disease; DOD, Dead of disease.
Neg, Negative IHC staining as defined in methods;
Pos, Positive IHC staining as defined in methods.
ATRX, α thalassemia/mental retardation syndrome X‐linked; DAXX, Death‐domain‐associated protein.
APB, ALT associated PML bodies; IDH1, Isocitrate dehydrogenase 1 R132H substitution;
p53, Tumour protein 53; nd: Not determined
Unk‐Unknown
All tumours were screened for APB presence, p53 aberrant expression (a surrogate for p53 mutation), IDH1 R132H mutation expression and ATRX or DAXX expression loss to determine the ALT phenotype. A subset of 24 LMS and 15 STUMP were also screened for p16INK4A overexpression by IHC. We refer to each of these as the molecular variants. No molecular variant was identified as specific to the spindled, epithelioid or myxoid subtypes. Examples of p16INK4A overexpression, p53 expression, IDH1 R132H mutation expression, loss of expression of ATRX or DAXX, no loss of expression for ATRX or DAXX (no evidence of mutation), and APB presence determination are given in Figure 2A–F. None of the LM cases demonstrated a molecular variant, except for one case with IDH1 R132H mutation expression.
The ALT phenotype was found in STUMP and LMS but not in LM or variant LM cases
Amongst the 22 STUMP and LMS cases that were ALT positive as determined by APB presence, 20 had ATRX or DAXX loss (p < 0.0001). Thus, ALT was highly associated with ATRX and DAXX expression loss commensurate with the presence of mutations. Only one LMS case was APB positive with no loss of ATRX or DAXX expression. Another 19.6% (12/61) of STUMP and LMS showed ATRX or DAXX expression loss without APB presence. Only three LMS cases were DAXX negative and the remaining cases were ATRX negative. No STUMP and 14% (6/43) of LMS cases showed IDH1 R132H mutation expression.
ATRX or DAXX expression loss was associated with older patient age, higher mitotic index and cytological atypia
Overall, loss of ATRX or DAXX expression correlated with older patient age (mean 51years CI 46.9, 55.0, versus mean 45 years, CI 41.4, 48.8, p = 0.03), and a higher mitotic count (mean 14/10HPF, CI 4.7, 11.3, versus mean 8/10HPF, CI 10.5, 17.2, p = 0.01). Similarly, diffuse or multifocal moderate to severe cytological atypia was identified in 80% of tumours with ATRX or DAXX expression loss, compared to only 56% of tumours that maintained expression of either ATRX or DAXX IHC. There was no correlation between histological type and molecular markers. No correlation was found with tumour size.
In STUMP and LMS, ATRX or DAXX expression loss best predicted poor prognosis
All STUMP (6/6) patients and 23/26 (88.4%) LMS patients, who had died or had recurrent disease, had ATRX or DAXX expression loss in their tumours. One patient of the three with neither ATRX nor DAXX expression loss, also showed no variation in any other molecular feature assessed, and died at 77.5 months post diagnosis, aged 41 years (LMS case 8). One patient with an ALT positive, non‐p53‐aberrant tumour was without disease at 156 months following resection of a polypoid LMS, and this morphology may have contributed to prolonged survival (LMS case 13).
For the LMS group, loss of ATRX expression was found in all but three cases, which showed loss of DAXX expression. In four cases, with loss of neither ATRX nor DAXX expression, no disease was yet evident at follow‐up periods of 59, 66, 78 and 156 months. Patient outcomes are detailed in Tables 2 and 3. At least one molecular variant was found in the majority of tumours with recurrence or death.
The STUMP group was too small for the statistical analysis of survival. Univariate Cox regression showed that, for overall survival in the LMS group, loss of ATRX or DAXX expression identified poor prognosis (CI 2.1 to 40, p < 0.003) and, for progression free survival, loss of ATRX or DAXX expression was statistically significant (CI 2.2 to 25, p = 0.001). We were limited in our analysis of p16INK4A IHC due to tissue availability but p16INK4A overexpression was narrowly significant (CI, 0.3 to 0.9, p = 0.047). Aberrant p53 expression did not reach significance.
Discussion
The homologous recombination based ALT mechanism is found predominantly in mesenchymal malignancies 20. This is the largest study to date examining ALT maintenance molecular markers with recurrence and survival time in smooth muscle tumours of the uterus. In particular, we examine early stage LMS and tumours that are diagnostically problematic.
Although APB determination is a well‐tested marker of ALT TMM, the detection method involves combined FISH and IHC techniques, requiring particular expertise and time and some exceptions do occur 19. Furthermore, in our hands, APB presence was not as significant as ATRX or DAXX expression loss. ATRX or DAXX loss appears to occur early in tumourigenesis and this could explain why some tumours with ATRX or DAXX expression loss were not ALT positive or p53 expression aberrant. The association of ATRX or DAXX expression loss with a higher proliferation rate (p = 0.0003) and pronounced cytological atypia provides a molecular basis for the histopathological assessment.
Immunohistochemical staining is widely used as a surrogate for ATRX and DAXX mutation 12, 13, 21, 22. This does not prove mutation and is less sensitive on small‐sized tissue microarrays compared to sequence based methods to identify ATRX mutations directly 22. However, with awareness of tumour heterogeneity, interpretation is straightforward and IHC is a practical test in the diagnostic workup 12, 13.
Tumours that lose ATRX or DAXX but maintain wild‐type p53 may respond better to DNA damaging agents. In a recent study loss of ATRX sensitized cells to 5‐fluorouracil and cisplatin treatment by p53 mediated cell death 11. In our study one uterine STUMP and 14 LMS had loss of ATRX or DAXX expression and wild‐type p53 IHC expression, and thus both recurrent STUMP and LMS could potentially respond to DNA damaging treatment. However, for patients with ATRX or DAXX expression loss, the associated mutant p53 in their tumours may make such treatment ineffective.
The acquisition of the R132H IDH1 mutation leads to an alteration in catalytic ability and the production of an oncometabolite, R(‐)‐2‐hydroxyglutarate (2‐HG) that inhibits histone demethylases and indirectly affects H3F3A methylation 23, 24, 25. Furthermore, 2‐HG has been identified by noninvasive imaging 26, and tumours with the mutant enzyme may be susceptible to chemotherapy 27. The R132H IDH1 mutation precedes mutation of p53, which appears to be the lynch pin facilitating recombination and the emergence of the gross deregulated ALT telomere length phenotype 28. IDH1(R132H) status by IHC was not informative in our cohort, but our analysis did not capture all mutations in IDH. Other mutations in the isoforms of IDH1 or IDH2 may be involved. The R132H IDH1 mutation is the most prominent IDH1 mutation in glioma 29, 30, but in cartilaginous tumours other mutations in IDH1 were more frequent than R132H 31, 32. We identified IDH1 R132H mutation in a LM, which has not been previously reported.
The use of p16INK4A IHC as a diagnostic marker indicative of changes in the p16INK4A‐cyclinD1‐pRB‐E2F‐cyclinE cell cycle control pathway has been investigated in LMS and STUMP with mixed results 2, 33, 34. With no difference in proliferation between the p16INK4A overexpressed and nonover‐expressed tumours, it is unlikely to reflect enhanced p16INK4A function. In the absence of a known viral oncogene, overexpression in these tumours probably reflects the mutational or epigenetic status of the retinoblastoma gene. Indeed, alterations of the retinoblastoma gene locus play an important role in the pathogenesis of LMS 35. Understanding alterations in the p16INK4A cell cycle control pathway will have treatment implications 35. That p16INK4A overexpressing STUMP were also associated with diffuse or multifocal moderate to severe cytological atypia may reflect a greater degree of genetic alteration compared to tumours that were not overexpressing p16INK4A and validates the use of cytological atypia in the histopathological assessment. In practice, however, p16INK4A IHC is of limited value in distinguishing between LM with bizarre nuclei and STUMP containing diffuse, bizarre cells, as this marker has also been expressed in a large number of the former cases 34.
The two vaginal cases posed particular difficulties diagnostically. In case 17, mild atypia accompanied 9/10HPF mitoses and tumour cell necrosis. The presence of necrosis alone is viewed with caution in this area 17, and in addition to the mitotic rate (9/10HPF) this tumour could have been considered malignant (unfortunately tumour size was not available). The other vaginal tumour (case 18) also demonstrated tumour cell necrosis, but with low mitotic rate, small size (4.2 cm) and mild atypia. Of note, neither tumour demonstrated another molecular variant, such as p53 aberrant expression or p16INK4A overexpression. These tumours highlight the importance of identifying specific criteria for a specific tumour, in different anatomical sites, but also suggest loss of ATRX or DAXX expression may prove similarly prognostic as in uterine tumours.
In summary, in this uterine STUMP cohort we show a statistically significant longer time to first recurrence than LMS, but that both STUMP and LMS cases with loss of ATRX or DAXX expression succumb at a similar time. All STUMP and 88.5% of LMS associated with death or recurrence displayed loss of ATRX or DAXX expression, including 90% of stage 1 LMS cases. Commensurate with poor prognosis these tumours had a higher rate of mitotic activity and diffuse or multifocal moderate to severe cytological atypia. That loss of ATRX or DAXX expression is a stronger diagnostic marker than APB presence suggests that demethylase loss occurs prior to APB induction in tumourigenesis. Determination of ATRX or DAXX expression by IHC in smooth muscle tumours is readily applicable diagnostically and these markers are potentially useful for histopathological classification and clinical management.
Author contributions
With great sadness we mourn the loss of our co‐author, beloved Obstetrician and dear friend, Bill Clow, who never failed to encourage, inspire and ask the difficult questions. NH, HH, IP, AC, JR and TLS conceived experiments and analysed data. NH, IP, AC, PS, WC, TS and CD recruited cases. TLS and HH prepared and performed the technical work. NH, IP, AC, TLS, TS AS and JR made the diagnoses and interpreted the results. AS performed the statistical analyses. All authors were involved in writing the paper and had final approval of the submitted and published versions with deep regret.
Acknowledgements
With great sadness we mourn the loss of our co‐author, beloved Obstetrician and dear friend, Bill Clow, who never failed to encourage, inspire and ask the difficult questions. We thank Miss Alisha Shaw for her technical assistance at the University Of Otago Department Of Pathology Histology Unit, and Miss Esther Wong of Queen Mary Hospital Histology Department, Hong Kong. Pathlab Tauranga, New Zealand is thanked for their histological preparations. We also wish to thank Andrew McNaughton and facilities at the Otago Centre for Confocal Microscopy.
Contract/grant details: The Cancer Society of New Zealand
References
- 1. Abell MR, Hertig AT, Norris HJ, et al. The Uterus. Williams & Wilkins: Baltimore, 1973. [Google Scholar]
- 2. Ip PP, Cheung AN, Clement PB. Uterine smooth muscle tumors of uncertain malignant potential (STUMP): a clinicopathologic analysis of 16 cases. Am J Surg Pathol 2009; 33: 992–1005. [DOI] [PubMed] [Google Scholar]
- 3. Bell SW, Kempson RL, Hendrickson MR. Problematic uterine smooth muscle neoplasms. A clinicopathologic study of 213 cases. Am J Surg Pathol 1994; 18: 535–558. [PubMed] [Google Scholar]
- 4. Ip PP, Cheung AN. Pathology of uterine leiomyosarcomas and smooth muscle tumours of uncertain malignant potential. Best Pract Res Clin Obstet Gynaecol 2011; 25: 691–704. [DOI] [PubMed] [Google Scholar]
- 5. Ruden M, Puri N. Novel anticancer therapeutics targeting telomerase. Cancer Treat Rev 2013; 39: 444–456. [DOI] [PubMed] [Google Scholar]
- 6. Bryan TM, Englezou A, Dalla‐Pozza L, et al. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor‐derived cell lines. Nat Med 1997; 3: 1271–1274. [DOI] [PubMed] [Google Scholar]
- 7. Hakin‐Smith V, Jellinek DA, Levy D, et al. Alternative lengthening of telomeres and survival in patients with glioblastoma multiforme. Lancet 2003; 361: 836–838. [DOI] [PubMed] [Google Scholar]
- 8. Henson JD, Hannay JA, McCarthy SW, et al. A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin Cancer Res 2005; 11: 217–225. [PubMed] [Google Scholar]
- 9. Lee YK, Park NH, Lee H. Prognostic value of alternative lengthening of telomeres‐associated biomarkers in uterine sarcoma and uterine carcinosarcoma. Int J Gynecol Cancer 2012; 22: 434–441. [DOI] [PubMed] [Google Scholar]
- 10. Liau JY, Tsai JH, Jeng YM, et al. Leiomyosarcoma With Alternative Lengthening of Telomeres Is Associated With Aggressive Histologic Features, Loss of ATRX Expression, and Poor Clinical Outcome. Am J Surg Pathol 2015; 39: 236–244. [DOI] [PubMed] [Google Scholar]
- 11. Conte D, Huh M, Goodall E, et al. Loss of Atrx sensitizes cells to DNA damaging agents through p53‐mediated death pathways. PLoS One 2012; 7: e52167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Heaphy CM, de Wilde RF, Jiao Y, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011; 333: 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schwartzentruber J, Korshunov A, Liu XY, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012; 482: 226–231. [DOI] [PubMed] [Google Scholar]
- 14. Wu G, Broniscer A, McEachron TA, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non‐brainstem glioblastomas. Nat Genet 2012; 44: 251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McDonald KL, McDonnell J, Muntoni A, et al. Presence of alternative lengthening of telomeres mechanism in patients with glioblastoma identifies a less aggressive tumor type with longer survival. J Neuropathol Exp Neurol 2010; 69: 729–736. [DOI] [PubMed] [Google Scholar]
- 16. Royds JA, Al Nadaf S, Wiles AK, et al. The CDKN2A G500 allele is more frequent in GBM patients with no defined telomere maintenance mechanism tumors and is associated with poorer survival. PLoS One 2011; 6: e26737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Crum CP, Nucci MR, Lee KR. Diagnostic gynecologic and obstetric pathology (2nd edn). Elsevier/Saunders: Philadelphia: 2011. [Google Scholar]
- 18. Yeager TR, Neumann AA, Englezou A, et al. Telomerase‐negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res 1999; 59: 4175–4179. [PubMed] [Google Scholar]
- 19. Slatter T, Gifford‐Garner J, Wiles A, et al. Pilocytic astrocytomas have telomere‐associated promyelocytic leukemia bodies without alternatively lengthened telomeres. Am J Pathol 2010; 177: 2694–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Henson JD, Reddel RR. Assaying and investigating Alternative Lengthening of Telomeres activity in human cells and cancers. FEBS Lett 2010; 584: 3800–3811. [DOI] [PubMed] [Google Scholar]
- 21. Liu L, Bailey SM, Okuka M, et al. Telomere lengthening early in development. Nat Cell Biol 2007; 9: 1436–1441. [DOI] [PubMed] [Google Scholar]
- 22. Kannan K, Inagaki A, Silber J, et al. Whole‐exome sequencing identifies ATRX mutation as a key molecular determinant in lower‐grade glioma. Oncotarget 2012; 3: 1194–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dang L, White DW, Gross S, et al. Cancer‐associated IDH1 mutations produce 2‐hydroxyglutarate. Nature 2009; 462: 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu W, Yang H, Liu Y, et al. Oncometabolite 2‐hydroxyglutarate is a competitive inhibitor of alpha‐ketoglutarate‐dependent dioxygenases. Cancer Cell 2011; 19: 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chowdhury R, Yeoh KK, Tian YM, et al. The oncometabolite 2‐hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep 2011; 12: 463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Andronesi OC, Kim GS, Gerstner E, et al. Detection of 2‐hydroxyglutarate in IDH‐mutated glioma patients by in vivo spectral‐editing and 2D correlation magnetic resonance spectroscopy. Sci Transl Med 2012; 4: 116ra114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Losman JA, Kaelin WG, Jr . What a difference a hydroxyl makes: mutant IDH, (R)‐2‐hydroxyglutarate, and cancer. Genes Dev 2013; 27: 836–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cesare AJ, Reddel RR. Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet 2010; 11: 319–330. [DOI] [PubMed] [Google Scholar]
- 29. Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008; 321: 1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009; 360: 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kerr DA, Lopez HU, Deshpande V, et al. Molecular distinction of chondrosarcoma from chondroblastic osteosarcoma through IDH1/2 mutations. Am J Surg Pathol 2013; 37: 787–795. [DOI] [PubMed] [Google Scholar]
- 32. Amary MF, Bacsi K, Maggiani F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol 2011; 224: 334–343. [DOI] [PubMed] [Google Scholar]
- 33. Atkins KA, Arronte N, Darus CJ, et al. The Use of p16 in enhancing the histologic classification of uterine smooth muscle tumors. Am J Surg Pathol 2008; 32: 98–102. [DOI] [PubMed] [Google Scholar]
- 34. Mills AM, Ly A, Balzer BL, et al. Cell cycle regulatory markers in uterine atypical leiomyoma and leiomyosarcoma: immunohistochemical study of 68 cases with clinical follow‐up. Am J Surg Pathol 2013; 37: 634–642. [DOI] [PubMed] [Google Scholar]
- 35. Stratton MR, Williams S, Fisher C, et al. Structural alterations of the RB1 gene in human soft tissue tumours. Br J Cancer 1989; 60: 202–205. [DOI] [PMC free article] [PubMed] [Google Scholar]