

Abstract
Clonal populations originated from benign‐looking ‘founder cells' may spread widely within pancreas instead of being localized in situ before frank pancreatic ductal adenocarcinoma (PDA) can be detected. Metachronous PDA is not common event, and we here sought to define potent origin of multiple PDAs developed in a woman using advanced genetics technologies. Curative resection of pancreatic head tumour was performed; however, ‘recurrent' lesions in the remnant pancreas were found 3.5 years later and total pancreatectomy was subsequently performed. The metachronous lesions were morphologically similar to the primary PDA. Using a next‐generation sequencing and digital PCR, all three PDAs were shown to possess rare somatic mutations in KRAS (p.T58I & p.Q61H). Curiously, identical KRAS mutations were found in low‐grade ‘intraepithelial' lesions, which localized in normal area of the pancreas and one of them possessed p53 mutation, which was also found in the PDAs. The footprint of the tumour evolution marked by mutational profiling supports a human correlate to the mouse models of ‘dissemination' occurring at the earliest stages of pancreatic neoplasia.
Keywords: metachronous pancreatic cancer, pancreatic intraepithelial neoplasia, early dissemination, next‐generation sequencing
Introduction
Pancreatic ductal adenocarcinoma (PDA) has been a devastating disease with extremely poor survival even in patients undergoing potentially curative resections. Among the long‐term survivor of PDAs, metachronous occurrence of invasive PDA in the remnant pancreas have recently been described 1, 2, 3. It has been challenging to fully discriminate metachronous PDAs from ‘intrapancreatic metastasis' 4. Because of the limited number of cases and reliable genetic markers, one could speculate if the multiple lesions were multicentric versus metastasis based on the clinical course and limited histological findings 5. Previous studies demonstrate that given metachronous PDAs were ‘metastasis' based on the sequence results showing identical KRAS mutation 1, still leaving the possibility of accidental concordance.
When multiple cancers located in the same or distant organ(s) are defined as ‘monoclonal', those are generally considered as ‘metastasis', whereas polyclonal lesions can develop from ‘field defect'. Here, we report a case with metachronous PDAs developed in remnant pancreas after the first resection of the primary tumour. In addition to precise pathological assessment, genetic approach indicated the high likelihood of ‘early dissemination' as a unique pathogenesis of intra‐pancreatic multiple cancers.
Methods
Samples and DNA extraction
PDAs as well as normal tissue (spleen) were manually dissected from 10 µm formalin‐fixed paraffin‐embedded (FFPE) sections (1–2 sections/tumour; supplementary material, Figure 1). Tumour cells from pancreatic intraepithelial neoplasia (PanIN), a precursor lesion of PDA, were microdissected using laser‐capture microdissection system (PALM MB‐III, ZEISS) to ensure that each tumour sample contained at least 90% neoplastic cells. The PanIN lesions were carefully selected to prevent contamination of the PDA cells and intraductal spreading lesions. DNA from the PanINs from each section was mixed together as pool sample (1–5 PanIN lesions per section and totally 12 PanINs; 4 PanIN‐1a and 8 PanIN‐1b lesions; see supplementary material, Figure 2 and Figure 2A). DNA was isolated using the QIAmp DNA FFPE tissue kit (Qiagen) and quantified and qualified using NanoDrop (Thermo) and Qubit2.0 (Life Technologies). Those samples were sufficient to obtain next‐generation sequencing (NGS) data (supplementary material, Table 1).
DNA Sequencing
A detailed protocol for Sanger sequencing, Targeted Amplicon Sequencing using NGS and digital PCR assay is provided in the Supplementary Methods.
Result
A 58‐year‐old woman with complaint about obstructive jaundice was referred to Asahikawa Medical University Hospital in November 2005. The patient had no family history of PDA and pancreatitis. Computed tomography (CT) revealed a tumour in the head of the pancreas, 26‐mm in diameter, directly invading the superior mesenteric vein (SMV; supplementary material, Figure 3). In December 2005, we performed a pylorus‐preserving pancreticoduodenectomy with resection of the SMV, in combination with an intraoperative radiation therapy targeting retroperitoneal cavity adjacent to the tumour (20 Gy). Pathological diagnosis was moderately differentiated adenocarcinoma (UICC; T3N1M0 Stage IIB) and no viable tumour cell was evident on the resection margin (Figure 1 and supplementary material, Figure 1). Adjuvant chemotherapy using gemcitabine was performed for 6 months.
Figure 1.
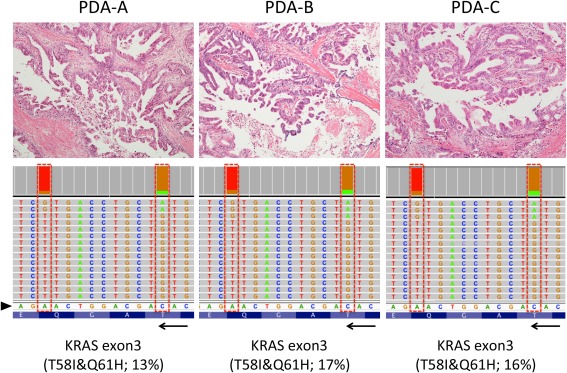
Representative histology and KRAS mutation pattern in three metachronous PDAs. Histologies of the three PDAs are shown (top panels; original magnifications, ×10 objective). All three tumours were moderately differentiated adenocarcinoma with papillary proliferation of cancer cells. Each sample shows the representation of the reads aligned to the reference genome in the KRAS genes (displayed as reverse‐complement of Exon 3; bottom panels). Arrowhead and arrow indicate of forward sequences of KRAS gene and its direction (3′ < 5′). Those histologically similar lesions possessed unique haplotype in KRAS (p.T58I&p.Q61H) analysed separately using NGS.
In May 2009, CT scan demonstrated ‘recurrent' lesions in the body and tail of the remnant pancreas (supplementary material, Figure 4). Retrospectively, these tumours could be pointed out by the serial CT scans performed after the surgery; emergence of the mass in the body of the pancreas in August 2008 was preceded by tumour in the pancreatic tail, which was first recognized in May 2008.
Total pancreatectomy was then performed in June 2009 without radiographic evidence of distant metastasis. The second (in the pancreatic tail) and third lesion (in the body of the pancreas) were histologically similar to the primary lesion developed in the head of the pancreas (Figure 2 and supplementary material, Figures 1 and 2). S‐1 was administrated for the second adjuvant chemotherapy; however, lung metastasis and peritoneal dissemination were evident 17 months after the second resection. The patient died from the metastatic disease in 66 months and 24 months after initial and second resection, respectively.
Figure 2.
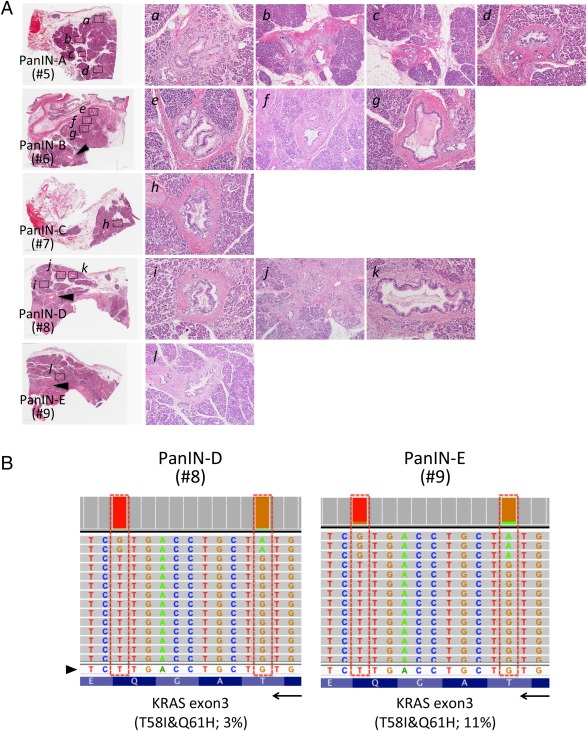
Assessment of intraductal atypical lesions pathologically identified in the ‘normal' pancreas surrounding first primary tumour. (A) Normal areas of the resected specimen apart from PDA were used to seek low‐grade PanINs surrounding the invasive PDA in the head of the pancreas and five sections were selected (A–E; given numbers indicate sample IDs for the NGS‐targeted sequencing; see supplementary material, Table 1 and supplementary material, Figure 2). Pool DNA samples were isolated using laser‐captured microdissected PanIN lesions from five sections (PanIN‐A–E) covering normal pancreas. Sufficient amount of DNA could be isolated from 1 to 5 PanIN lesions per slide (a∼l; 4 PanIN‐1a and 8 PanIN‐1b lesions). Arrowheads in H&E staining (PanIN‐D and E) indicate main‐pancreatic duct. Original magnifications are ×10 objective for a, c, e, g, h, i, k and ×4 for b, f, j, l. (B) Representative of the reads aligned to the reference genome in the KRAS genes (displayed as reverse‐complement of Exon 3) are shown. Arrowhead and arrow indicate of forward sequences of KRAS gene and its direction (3′ < 5′). Pool DNA samples from PanIN‐D and ‐E possessed the double mutation in KRAS (p.T58I&p.Q61H).
We sought to characterize the three PDAs developed in the current case, since metachronous PDA is not a common event and only limited cases were previously reported 1, 2. First, Sanger sequencing was performed for genotyping KRAS, a major driver for PanIN/PDA and intraductal papillary mucinous neoplasm (IPMN) of the pancreas, and GNAS, which is frequently mutated in IPMNs 6. Neither mutations in KRAS (codon 12&13) nor GNAS (codon 201&227) was identified in the tumours (data not shown). To further define the genetic profile of larger number of tumour‐associated mutations, NGS was then used. Sequencing of PCR‐amplified fragments‐containing multiple mutation sites on known tumour‐related genes was conducted to detect and quantitate mutated alleles using a multigene NGS panel (Supplementary Table 1).
All three PDAs possessed exactly identical KRAS mutations in codon 58 (COSM87288; p.T58I) and codon 61 (COSM554; p.Q61H) (Figure 1 and supplementary material, Table 2). The double mutation in KRAS was also demonstrated in the primary PDA by Sanger sequence (supplementary material, Figure 5). Curiously, the double mutations in KRAS p.T58I and p.Q61H were found in a single allele and considered as haplotype. The presence of mutant KRAS allele was detected by digital PCR using specific probe targeting p.T58I and p.Q61H (supplementary material, Figure 6), confirming that a single allele possessed both mutations.
Additional somatic mutation in p53 at codon 175 (COSM10648; p.R175H) was shared by all PDAs and secondly developed lesion in the pancreatic tail was shown to possess mutation in VHL and Smad4 albeit at low frequency. Other unique mutations including tumour suppressor genes associated with PDA/IPMN such as SMARCA4 7 and RNF43 8 were not identified (data not shown).
Finally, we thought to determine if the precursor lesions pathologically identified in the ‘normal' pancreas surrounding primary PDA resected in 2005 also have the unique mutations in KRAS (Figure 2 and supplementary material, Figure 2). As summarized in supplementary material, Table 2, the double KRAS mutation in codon 58&61 was found in PanIN samples from 2 of 5 sections analyzed (PanIN‐D and ‐E). Although their variant frequency was very small (3–11%), the haplotype in KRAS was verified by digital PCR (supplementary material, Figure 6). One of the two samples also have mutation in p53 (p.R175H), whereas others were shown to possess wild‐type p53.
Discussion
It has been reported that intraglandular metastasis of PDA was found in about 5% of cases in the study using over 300 cases and overall survival and disease‐free survival for patients with the ‘synchronous' PDA were significantly shorter than for those with single lesion 9. Despite of the histological imitation observed in the three PDAs in the current case, we hesitated to consider that the recurrent lesions directly metastasized from the first primary tumour because of the following reasons; recurrent lesions were evident more than 3 years after the resection of the primary tumour, surgical margin on the pancreas was negative for viable cancer cells at the time of first resection, and the second tumour developed in a geographically distant site from the primary tumour. Therefore, ‘local recurrence' due to microscopic residual tumour cells during the first resection was unlikely. Actually, larger number of mutations was found in the second lesion; however, newer third lesion possessed no more mutation relative to the primary tumour implying that these recurrent lesions independently developed. In addition, careful pathological assessment identified the multiple PanINs in the ‘normal' pancreas adjacent to the primary tumour (Figure 1), suggesting that multiple precursors could be wildly distributed to the entire pancreas.
KRAS mutation is a major ‘driver' for PanIN/PDA and IPMN 10. Different pattern of KRAS mutation would strongly support multicentric occurrence of the three PDAs. However, we found that all PDAs developed in the current case has identical mutations in KRAS (p.T58I&p.Q61H), both of which have been shown to act as driver and play a role in drug resistance 11, 12. Because of extreme rarity of identical combination of double KRAS mutations in a single allele, it is reasonable to speculate that the metachronous PDAs may be derived from a common ‘founder lesion' 13 rather than independently initiated within a ‘field defect'. In addition to the intrapancreatic lesions, metastatic disease in other organs developed in the current case and targeted sequencing of these samples could determine if the rare KRAS alleles (or other mutations) are present and support the model of clonal relationship to a founder cell 14, 15.
Among the genetically heterogeneous PanIN populations marked by NGS‐assay in the current case, we specifically focused on the ‘precursors' with unique mutation in KRAS (p.T58I&p.Q61H) (Figure 2). One of the PanINs also possessed p53 mutation (p.R175H), which is again shared by the PDAs. p53R175H provides tumour cells gain‐of‐function that inactivates ATM‐dependent DNA damage responses, which facilitates to overcome KRAS‐induced senescence and ultimately promote metastasis 16. It should be noted that those PanINs were apart from the invasive front of the primary PDA and intraductal extension was excluded because of their low‐grade of atypia (PanIN1a‐1b). Therefore, the PanIN with the unique KRAS mutations and p53R175H could be the ‘founder lesion' of given metachronous PDAs (proposed model is shown in Figure 3). The progression model supports the notion that the three PDAs potentially originated from the precursors disseminated during earlier stages rather than metastasized from the established PDA.
Figure 3.
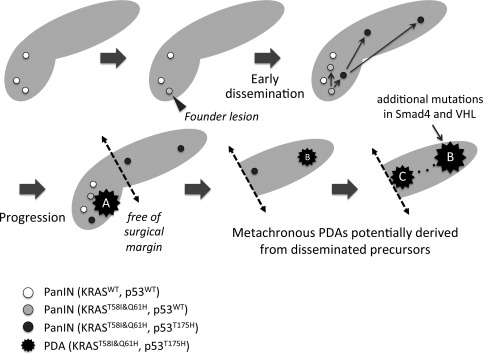
Proposed progression model from multiple PanINs. Early precursor lesions (PanINs) with wild‐type KRAS emerged to create field of cancer, and some of them acquired oncogenic KRAS, p.T58I and p.Q61H (Founder lesion). Additional somatic mutation in p53 p.T175H occurred in part of the PanINs with the unique KRAS haplotype, which may in turn spread into other area of the pancreas (‘early dissemination' indicated by arrows). One of them progressed into invasive PDA (PDA‐A), which was resected by the Whipple's procedure with negative surgical margin. However, other disseminated precursors independently progressed to form other PDAs in the tail (PDA‐B) and body (PDA‐C) of the pancreas, finally connecting each other through main pancreatic duct (dotted line). In addition to the typical mutations in KRAS and p53, PDA‐B has other somatic mutations in Smad4 and VHL.
A model of ‘early dissemination' in which epithelial‐to‐mesenchymal transition (EMT) and dissemination can precede frank tumour formation was demonstrated by lineage‐labelling system using genetically engineered mouse 17. Earlier study showed that tumour cells can leave the primary tumour before mutations of the p53 gene occur, supporting haematogenous dissemination of neoplastic cells can occur even without losing tumour suppressors 18. Additionally, recent human study also demonstrated that naive pancreatic tumour cells, which do not have invasive property, can spread into circulation 19. In the current report, we presented precise pathological and genetic assessments strongly implying migration/dissemination of such ‘benign‐looking' PDA precursor cells within pancreas in human. Focal immunostaining of EMT factors such as Snail and integrin‐linked kinase was demonstrated in low‐grade PanINs albeit at low frequency relative to PDA 20, supporting the ability of these precursors to migrate into circulation and ultimately form metastatic foci. In addition, a longitudinal monitoring of EMT and mesenchymal‐to‐epithelial transition (MET) features has been demonstrated in circulating tumor cells (CTCs) 21. Therefore, naive circulating ‘precursor' cells may represent MET during migration process from circulation to the duct epithelial lining.
Prevalence of high‐grade PanINs in patients underwent surgical resection of PDA is considered as risk for the post‐operative recurrence 22, and patients underwent pancreatectomy for chronic pancreatitis sometimes developed disseminated PDA although only PanINs and no tumours were found on histologic analysis 23. Therefore, the incidental PanINs can generally be observed in classic PDA; however, the significance on their outcome is still controversial 24 and further studies are required. In the current case, we identified ‘low‐grade PanIN' with KRAS and p53 mutation, and presence of such a microscopic lesion with two genetic hits may potentially be a factor predicting recurrence caused by development of metachronous cancer(s).
In conclusion, unique KRAS mutations (p.T58I&p.Q61H) were identified in the metachronous PDAs in the current case. Surprisingly, the identical mutations were also found in independent low‐grade ‘intraepithelial' lesions with and without additional mutation in p53, highlighting an alternative mode of tumour progression through ‘early disseminations'. Since PDAs are occasionally accompanied with multiple microscopic lesions in radiographically normal pancreas, precise genetic profiling of these incidental lesions will be demanded.
Grant support
Supported by grants from the Ministry of Education, Science, Sports, and Culture of Japan (25461029 to Y.M.), Pancreas Research Foundation in Japan (to Y.M.), Akiyama Life Science Foundation (to Y.M.) and Andrew Warshaw Institute for Pancreatic Cancer Research (to Y.M.).
Author contributions
KI, acquisition of data; HK, study concept and design, and drafting of the manuscript; YO, analysis and interpretation of data; JS, critical revision of the manuscript for important intellectual content; SC, acquisition of data; HF, study supervision; MM, acquisition of data; HH, administrative, technical, or material support; TF, study supervision; HF, study supervision; TK, analysis and interpretation of data; KN, study supervision; YM, study concept and design, and critical revision of the manuscript for important intellectual content.
Supporting information
Supplementary Figure 1. Gross appearance of the resected pancreas.
Supplementary Figure 2. Assessment of intraductal atypical lesions pathologically identified in the “normal” pancreas surrounding first primary tumour resected.
Supplementary Figure 3. Imaging diagnosis of the primary pancreatic ductal adenocarcinoma (PDA).
Supplementary Figure 4. Serial CT images during post‐operative follow up.
Supplementary Figure 5. DNA Sanger Sequencing confirming KRAS mutations in codon 58 and 61.
Supplementary Figure 6. Droplet digital PCR confirming haplotype mutation in KRAS p.T58I and p.Q61H.
Supporting Information
Supporting Information
Supporting Information
Acknowledgements
The authors are thankful to the late Dr. Yoshihiko Tokusashi who was outstanding pathologist in Asahikawa Medical University and supported precise pathological assessment of the resected specimens. The authors also thank Eiko Aoyanagi and Yuji Fukuda for tissue section preparation; Munehiko Ogata for nucleic acid purification; and Manish K. Gala and Tomoki Yokochi for helpful discussion.
Conflict of interest: Several authors (MM, HH and TF) on this manuscript are employees of Life Technologies Japan, Inc., whose technology is used in this study.
References
- 1. Wada K, Takada T, Yasuda H, et al. A repeated pancreatectomy in the remnant pancreas 22 months after pylorus‐preserving pancreatoduodenectomy for pancreatic adenocarcinoma. J Hepatobiliary Pancreat Surg 2001; 8: 174–178. [DOI] [PubMed] [Google Scholar]
- 2. Kleeff J, Reiser C, Hinz U, et al. Surgery for recurrent pancreatic ductal adenocarcinoma. Ann Surg 2007; 245: 566–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miyazaki M, Yoshitomi H, Shimizu H, et al. Repeat pancreatectomy for pancreatic ductal cancer recurrence in the remnant pancreas after initial pancreatectomy: is it worthwhile? Surgery 2014; 155: 58–66. [DOI] [PubMed] [Google Scholar]
- 4. Izumi S, Nakamura S, Mano S, et al. Resection of four synchronous invasive ductal carcinomas in the pancreas head and body associated with pancreatic intraepithelial neoplasia: report of a case. Surg Today 2009; 39: 1091–1097. [DOI] [PubMed] [Google Scholar]
- 5. Dalla Valle R, Mancini C, Crafa P, et al. Pancreatic carcinoma recurrence in the remnant pancreas after a pancreaticoduodenectomy. JOP 2006; 7: 473–477. [PubMed] [Google Scholar]
- 6. Wu J, Matthaei H, Maitra A, et al. Recurrent GNAS Mutations Define an Unexpected Pathway for Pancreatic Cyst Development. Sci Transl Med 2011; 3: 92ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shain AH, Giacomini CP, Matsukuma K, et al. Convergent structural alterations define SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proc Natl Acad Sci USA 2012; 109: E252–E259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu J, Jiao Y, Dal Molin M, et al. Whole‐exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin‐dependent pathways. Proc Natl Acad Sci USA 2011; 108: 21188–21193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oguro S, Shimada K, Ino Y, et al. Pancreatic intraglandular metastasis predicts poorer outcome in postoperative patients with pancreatic ductal carcinoma. Am J Surg Pathol 2013; 37: 1030–1038. [DOI] [PubMed] [Google Scholar]
- 10. Izawa T, Obara T, Tanno S, et al. Clonality and field cancerization in intraductal papillary‐mucinous tumors of the pancreas. Cancer 2001; 92: 1807–1817. [DOI] [PubMed] [Google Scholar]
- 11. Agarwal A, Eide CA, Harlow A, et al. An activating KRAS mutation in imatinib‐resistant chronic myeloid leukemia. Leukemia 2008; 22: 2269–2272. [DOI] [PubMed] [Google Scholar]
- 12. Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early‐ and late‐stage human malignancies. Sci Transl Med 2014; 6: 224ra224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yachida S, Jones S, Bozic I, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010; 467: 1114–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Forshew T, Murtaza M, Parkinson C, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med 2012; 4: 136ra168. [DOI] [PubMed] [Google Scholar]
- 15. Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012; 366: 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu DP, Song H, Xu Y. A common gain of function of p53 cancer mutants in inducing genetic instability. Oncogene 2010; 29: 949–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rhim AD, Mirek ET, Aiello NM, et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012; 148: 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Offner S, Schmaus W, Witter K, et al. p53 gene mutations are not required for early dissemination of cancer cells. Proc Natl Acad Sci USA 1999; 96: 6942–6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rhim AD, Thege FI, Santana SM, et al. Detection of circulating pancreas epithelial cells in patients with pancreatic cystic lesions. Gastroenterology 2014; 146: 647–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schaeffer DF, Assi K, Chan K, et al. Tumor expression of integrin‐linked kinase (ILK) correlates with the expression of the E‐cadherin repressor snail: an immunohistochemical study in ductal pancreatic adenocarcinoma. Virchows Arch 2010; 456: 261–268. [DOI] [PubMed] [Google Scholar]
- 21. Yu M, Bardia A, Wittner BS, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013; 339: 580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Frankel TL, LaFemina J, Bamboat ZM, et al. Dysplasia at the surgical margin is associated with recurrence after resection of non‐invasive intraductal papillary mucinous neoplasms. HPB 2013; 15: 814–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sakorafas GH, Sarr MG. Pancreatic cancer after surgery for chronic pancreatitis. Dig Liver Dis 2003; 35: 482–485. [DOI] [PubMed] [Google Scholar]
- 24. Hassid BG, Lucas AL, Salomao M, et al. Absence of pancreatic intraepithelial neoplasia predicts poor survival after resection of pancreatic cancer. Pancreas 2014; 43: 1073–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Gross appearance of the resected pancreas.
Supplementary Figure 2. Assessment of intraductal atypical lesions pathologically identified in the “normal” pancreas surrounding first primary tumour resected.
Supplementary Figure 3. Imaging diagnosis of the primary pancreatic ductal adenocarcinoma (PDA).
Supplementary Figure 4. Serial CT images during post‐operative follow up.
Supplementary Figure 5. DNA Sanger Sequencing confirming KRAS mutations in codon 58 and 61.
Supplementary Figure 6. Droplet digital PCR confirming haplotype mutation in KRAS p.T58I and p.Q61H.
Supporting Information
Supporting Information
Supporting Information