

Abstract
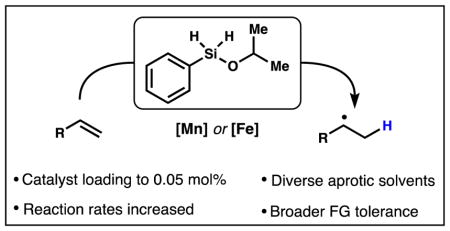
We report the discovery of an outstanding reductant for metal-catalyzed radical hydrofunctionalization reactions. Observations of unexpected silane solvolysis distributions in the HAT-initiated hydrogenation of alkenes reveal that phenylsilane is not the kinetically preferred reductant in many of these transformations. Instead, isopropoxy(phenyl)silane forms under the reactions conditions, suggesting that alcohols function as important silane ligands to promote the formation of metal hydrides. Study of its reactivity showed that isopropoxy(phenyl)silane is an exceptionally efficient stoichiometric reductant and it is now possible to significantly decrease catalyst loadings, lower reaction temperatures, broaden functional group tolerance and use diverse, aprotic solvents in iron- and manganese-catalyzed hydrofunctionalizations. As representative examples, we have improved the yields and rates of alkene reduction, hydration, hydroamination and conjugate addition. Discovery of this broadly applicable, chemoselective and solvent-versatile reagent should allow an easier interface with existing radical reactions. Finally, isotope-labeling experiments rule out the alternative hypothesis of hydrogen atom transfer from a redox-active beta-diketonate ligand in the HAT step. Instead, initial HAT from a metal-hydride to directly generate a carbon-centered radical appears to be the most reasonable hypothesis.
Graphical Abstract
INTRODUCTION
In the 1980’s, Mukaiyama reported a powerful method to functionalize electron neutral alkenes, apparently through radical intermediates.1 This groundbreaking work enabled the appendage of hydrogen and a functional group to a double bond with Markovnikov selectivity and high chemoselectivity, which subsequently allowed challenging disconnections on complex molecules.
A toolkit of synthetic methods2–8 has arisen following the seminal reports of hydration, hydroperoxidation, and hydronitrosation of alkenes by cobalt, manganese and iron beta-diketonate or salen-type complexes (Scheme 1). The intermediate carbon-centered radical or organometallic can react with oxygen in the hydration (1 to 2);2a,d with electrophilic fluorine sources in the fluorination;4c,8e with a sulfonate in the chlorination,4a azidation3e or cyanation;6c with a diazodicarboxylate3e or nitrosoarene3k in the hydroamination; or with a hydrazone in the methylation reaction.6h However, catalyst deactivation frequently occurs after a few turnovers and most of these transformations require an alcoholic solvent that competitively consumes the silane. In some cases, reducible groups like aldehydes and nitriles react competitively in the presence of alkenes, and superstoichiometric amounts of the silane or high catalyst loadings are required. Most methodologies also allow little variation in the reaction conditions (e.g. lower temperature and concentration or fewer equivalents of the radical trap), which impedes the optimization of low-yielding or excessively slow examples. These restrictions may become problematic for interfacing this direct radical generation method with existing organometallic engines. Our group has modified the Mukaiyama conditions to achieve stereoselective hydrogenation, reductive cyclization,7b alkene isomerization, diene cycloisomerization, arene annulation, and retro-cycloisomerization.6f
Scheme 1.

Alkene hydrofunctionalization toolkit.
Based on evidence from the literature and our own observations, we proposed these reactions proceed by initial hydrogen atom transfer (HAT) from a metal-hydride to directly generate a carbon-centered radical from the alkene. Early precedent for such a mechanism can be found in the stoichiometric HAT hydrogenation of anthracene and styrene with metal hydrides like HMn(CO)5. These studies suggested direct and reversible formation of a radical cage pair;9 dissociation of this pair was identified as the rate-determining step of the reduction. Similarly, in the context of our stereoselective HAT hydrogenation using PhSiH3 (3) as a stoichiometric reductant (Scheme 2), manganese catalysts were found to be superior to cobalt catalysts, which may become trapped as off-cycle organometallics via collapse of a carbon-centered radical/metal pair.6f,7b,c As observed previously,1,7a the dipivaloylmethane (dpm) ligand induced higher reaction rates than acetylacetonate (acac) at modest (10 mol %) catalyst loading for most of the examples.
Scheme 2.

Stereoselective hydrogenation of alkenes.
However, like many previous HAT-initiated reactions mediated by iron and manganese, alcohol was required as solvent. Obviously, this solvent restriction can be problematic for cationic2f,3j,10 and anionic6a,e,g,11 radical-polar crossover reactions, where nucleophilic and protic solvents lead to charge quenching. Here we demonstrate that isopropoxy-(phenyl)silane (6, Ph(i-PrO)SiH2) is a uniquely efficient reductant that permits the use of multiple solvents in manganese and iron-catalyzed reactions. In most cases, rates of reactions are increased, required reaction temperatures are decreased, and the catalyst loading of our HAT hydrogenation can be dropped as low as 0.05 mol%. Notably, isopropoxy(phenyl)silane increases the efficiency of other reactions within the growing arsenal of HAT-initiated hydrofunctionalizations. The ability to generate carbon-centered radicals from diverse unsaturated building blocks under these chemoselective conditions has led to a rapid growth of the field. Discovery of this broadly applicable, chemoselective and solvent-versatile reagent should allow easier interface with existing radical reactions.
For example, we privately disclosed this reagent to the Pronin lab who vividly illustrated its potential in a recent synthesis of emindole SB.11 A key step in this work consists of a radical-polar crossover cyclization based on radical conjugate addition followed by intramolecular aldol cyclization (Scheme 3). Reactions of 7 as well as other analogs using phenylsilane produced 8a/8b in low yield and with multiple byproducts, but the reaction was significantly improved by use of Ph(i-PrO)SiH2.
Scheme 3.

Key step in the total synthesis of emindole SB.
RESULTS AND DISCUSSION
Study of the intermediates formed in the HAT hydrogenation: discovery of isopropoxy(phenyl)silane
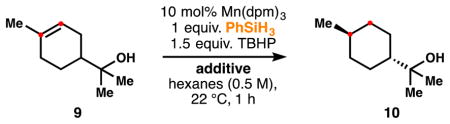
We had speculated that the alcoholic solvent in HAT-initiated hydro-functionalization (with Mn and Fe) was necessary only to increase the hydridic character of the silane reductant through formation of a pentavalent silane, as observed by Schowen and others.12 In our initial experiments, we observed that the HAT hydrogenation in hexanes reduces very little terpineol (Table 1, entry 1), whereas addition of increasing amounts of isopropanol slowly rescues the reaction (2–4), albeit at 10 mol% Mn(dpm)3.
Table 1.
Increase in reduction yield with increased i-PrOH.
| ||||
|---|---|---|---|---|
| entry | additive | conversion (%) | yield (%) | d.r. |
| 1 | None | 35 | 17 | 9.5 |
| 2 | 1 equiv. i-PrOH | 51 | 46 | 7.4 |
| 3 | 2 equiv. i-PrOH | 63 | 57 | 7.6 |
| 4 | 5 equiv. i-PrOH | 82 | 76 | 7.7 |
However, when monitoring consumption of PhSiH3 by GC, we observed a non-consistent distribution of the silane-derived products: PhSiH3 was converted to Ph(i-PrO)SiH2, Ph(i-PrO)2SiH (11), traces of PhSi(Oi-Pr)3, (Ph(i-PrO)2Si)2O, Ph(i-PrO)(t-BuO)SiH and Ph(i-PrO)(dpm)SiH among other unidentified species. Interestingly, the first solvolysis product remained at low abundance throughout the reaction (Figure 1). The observation that Ph(i-PrO)SiH2 (6) was consumed more rapidly than the other silanes led us to hypothesize that it might serve as a superior reductant. Additionally, this data suggested that alcohols do not just act as reaction media or proton sources to turn over the catalyst in radical-anion crossover reactions. Instead, alcohols function as important silane ligands in HAT-initiated reactions, likely as a result of increased Si electrophilicity and more rapid ligand exchange with the catalyst (either dpm, isopropoxy or TBHP).
Figure 1.

Consumption of phenylsilane (3) in the presence of Mn(dpm)3, TBHP and isopropanol from 1 min to 240 min.
The available literature revealed limited examples of mono-alkoxysilanes. We prepared isopropoxy(phenyl)silane (6) in 30g scale by adapting the procedure reported by Yamada et al. based on the reaction between phenylsilane and isopropanol in the presence of catalytic copper (II) hexafluoroacetylacetonate (Cu(hfac)2, Scheme 4).13 Unlike other methods, this solvolysis selectively forms (6) and only generates diisopropoxy(phenyl)silane (11) as a minor byproduct (7%). Manganese, iron, cobalt, nickel, ruthenium or platinum complexes did not selectively generate mono-solvolyzed silanes. Interestingly, attempts to isolate the solvolysis product derived from phenylsilane and ethanol, i.e. ethoxy(phenyl)silane, were not successful due to the moisture sensitivity of this adduct. In contrast, metal-catalyzed solvolysis with tert-butanol was sluggish, and therefore the isopropoxy ligand was identified as optimal for investigation. The correspondence of this procedure to the reaction conditions required for most HAT-initiated transformations is clear, which encouraged us in our new hypothesis.
Scheme 4.

Synthesis of isopropoxy(phenyl)silane.
Study of the reactivity of isopropoxy(phenyl)silane in the HAT hydrogenation
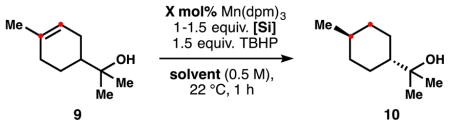
Our first generation HAT hydrogenation conditions reduce terpineol 9 in 89% yield with 10 mol% catalyst in isopropanol, but the yield drops precipitously when lowering the catalyst loading or changing solvent (Table 2, entries 1–4).
Table 2.
Effects of 6 on the catalyst loading of the HAT hydrogenation.a
| ||||
|---|---|---|---|---|
| entry | silane | conditions | conv/yield (%)b | d.r. |
| 1 | 3 | i-PrOH, 10 mol% | 94/89 | 7.5 |
| 2 | 3 | i-PrOH, 1 mol% | 61/53 | 8.3 |
| 3 | 3 | i-PrOH, 0.1 mol% | 27/28 | 9.2 |
| 4 | 3 | hexane, 10 mol% | 35/17 | 9.5 |
| 5 | 6 | i-PrOH, 10 mol% | 99/98 | 4.1 |
| 6 | 6 | i-PrOH, 1 mol% | 100/87 | 5.5 |
| 7 | 6 | hexane, 10 mol% | 94/93 | 5.2 |
| 8 | 6 | hexane, 1 mol% | 100/97 | 6.3 |
| 9 | 6 | hexane, 0.1 mol% | 100/91 | 6.6 |
| 10 | 6 | hexane, 0.05 mol%c | 100/98 | 6.6 |
| 11 | 6 | hexane, 0.02 mol%c | 48/15 | 6.2 |
| 12 | 11 | i-PrOH, 10 mol% | 98/86 | 5.3 |
| 13 | 11 | hexane, 10 mol% | 49/33 | 5.9 |
Reactions performed with 1 equiv. of PhSiH3, 1.5 equiv. of Ph(i-PrO)SiH2 or 3 equiv. of Ph(i-PrO)2SiH.
Determined by GC-FID using 1 equiv. of decane as internal standard.
Reaction time 13 h.
In contrast, use of Ph(i-PrO)SiH2 (6) leads to complete consumption of terpineol at 1 mol% catalyst (entries 6 and 8). Moreover, in hexanes at least 2,000 turnovers are reached (0.05 mol% Mn, entry 10).14 We noted that for both silanes the diastereoselectivity of the reaction degraded when the amount of isopropanol was increased, presumably due to the involvement of a larger reductant favoring an equatorial delivery in the second HAT. Interestingly, at lower catalyst loadings higher selectivities were also observed. Double solvolysis product diisopropoxy(phenyl)silane (11) was less effective both in isopropanol and hexanes (entries 12 and 13).
Reagent 6 tolerates several solvents, including non-anhydrous EtOAc, toluene, acetonitrile, THF and dichloromethane (Figure 2). The diastereoselectivity for all cases remained in a close range (see SI for further details).15 Otherwise, the reaction with phenylsilane and 10 mol% of Mn(dpm)3 was generally low yielding even when there was significant consumption of the starting material. The use of DMF as solvent leads to poor yields for both 3 and 6.
Figure 2.

Comparison of the solvent tolerance between silanes 3 (first column, 10 mol%) and 6 (second column, 1 mol%) in the HAT hydrogenation of terpineol.
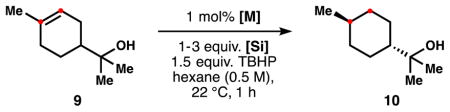
The concentration of the HAT hydrogenation when using isopropoxy(phenyl)silane can be decreased or increased with little effect on the reactivity (Table 3, entries 1–3). Interestingly, the transformation was slower under diluted conditions but led to a slight increase in diastereoselectivity. The reaction can also be run at low temperatures, which progressively increases the d.r. to 11.3 (entries 4–5).11 Fortunately, the 25-fold less-expensive catalyst Mn(acac)3 (Sigma) performs reasonably well in the reaction with 6, forming 10 in 89 % yield, d.r. 7.2 (Table 4, entry 2), whereas this catalyst performs poorly with PhSiH3. Co and Fe-catalysts are also less effective (3–4). The rest of the silanes screened were inactive under the optimized reaction conditions (entries 5–9).
Table 3.
Versatility of the reaction conditions using 6 in the HAT hydrogenation.

| ||||
|---|---|---|---|---|
| entry | Temp. (°C) | conditions | conv/yield (%)a | d.r. |
| 1 | 22 | 0.1 mol%, 1 M | 100/92 | 6.3 |
| 2 | 22 | 0.1 mol%, 0.25 M | 99/97 | 6.6 |
| 3 | 22 | 0.1 mol%, 0.1 Mb | 100/97 | 7.6 |
| 4 | 0 | 1 mol%, 0.5 M | 91/91 | 7.5 |
| 5 | −30 | 1 mol%, 0.5 M | 96/71 | 11.3 |
Determined by GC-FID using 1 equiv. of decane as internal standard.
Reaction time 13 h.
Table 4.
Reactivity of alternative catalyst/silanes in the HAT hydrogenation.a
| ||||
|---|---|---|---|---|
| entry | silane | catalyst | conv/yield (%)b | d.r. |
| 1c | 3 | Mn(acac)3 | 31/25 | 10.5 |
| 2 | 6 | Mn(acac)3 | 96/89 | 7.2 |
| 3 | 6 | Fe(dpm)3 | 53/17 | 5.3 |
| 4 | 6 | Co(dpm)2 | 39/35 | 6.3 |
| 5 | Ph2SiH2 | Mn(dpm)3 | 19/0 | – |
| 6 | Ph3SiH | Mn(dpm)3 | 3/0 | – |
| 7 | PMHS | Mn(dpm)3 | 26/0 | – |
| 8 | Et3SiH | Mn(dpm)3 | 32/0 | – |
| 9 | DEMS | Mn(dpm)3 | 33/3 | – |
Reactions performed with 1 equiv. of PhSiH3 or 1.5 equiv. for the rest.
Determined by GC-FID using 1 equiv. of decane as internal standard.
Use of 10 mol% in isopropanol.
Finally, attempts to reduce terpineol in the absence of any silane or any metal complex led to no formation of the desired product 10 (Scheme 5). However, excluding silane from the reaction led to partial consumption of 9 towards an unidentified mixture of products, implying competitive background reactivity in the HAT hydrogenation. Interestingly, 7% of reduction (d.r. increased to 8.0) was observed in the absence of TBHP but very rapid catalyst deactivation occurred.
Scheme 5.

Study of the background reactivity in the HAT hydrogenation.
Discovery of a competing hydrosilylation reaction: different behaviors between phenylsilane and isopropoxy(phenyl)silane
Finally, we also noted that in the absence of TBHP, certain substrates give rise to a competing, occasionally dominant hydrosilylation pathway using phenylsilane (Scheme 6), probably through addition of a silane radical to the alkene, and radical chain propagation via Si-H abstraction by the tertiary carbon radical.16 However, we have not found silane 6 to cause hydrosilylation. The industrial importance of hydrosilylation and the natural abundance of manganese may make this transformation also valuable.
Scheme 6.

A competing hydrosilylation reaction.
Silane 13 is unreactive under the HAT hydrogenation conditions and is therefore unlikely to be involved in the reduction mechanism. These results suggest multiple roles for TBHP including re-oxidation of the metal-complex and suppression of competing side-reactions. Moreover, TBHP might also be involved in the ligand exchange with the catalyst, which is much more efficient with silane 6 presumably as a result of increased Si electrophilicity.
Kinetic profiles of the HAT hydrogenation
As shown in Figure 3, we observed silane 6 to exhibit much higher rates of hydrogenation than PhSiH3. At 1 mol% catalyst, PhSiH3 is ineffective in hexane (0.25 M) for the HAT hydrogenation of terpineol 9, but its performance in i-PrOH is still poor.
Figure 3.

Reduction of terpineol monitored by GC-FID. Reaction performed with Ph(i-PrO)SiH2 in hexanes (
 ) or isopropanol (
) or isopropanol (
 ) and with PhSiH3 in hexanes (●) or isopropanol (
) and with PhSiH3 in hexanes (●) or isopropanol (
 ).
).
Interestingly, the optimum performance of 6 occurs in hexane, whereas i-PrOH decreases the rate – although even here 6 far surpasses 3. We noted that the reduction rates of 9 significantly dropped over time independent of the silane or the solvent used. Conversely, we also observed a variation in the diastereoselectivity, which increased over time in the cases studied. For instance, silane 6 in hexane led to an increase in d.r. from 5.6 to 6.7 in 60 min whereas silane 3 in isopropanol, gave an increase of 7.0 to 10.1 (see SI for further details). These results suggested a complex mechanistic scenario for the alkene reduction, in which the source of hydrogen may change over time, in line with solvolysis affecting rates of hydrogen transfer.
Study of the applicability of isopropoxy(phenyl)silane in the HAT hydrogenation
These second generation conditions lead to higher yields at lower catalyst, reductant and oxidant loadings than our first generation conditions for a subset of previously poorly performing alkenes.
In some cases the gains are modest, in others profound, for example, in aldehydes and ketones like 14 and 15. In our first generation system, those substrates proved problematic due to competitive reduction,17 whereas alkenes can be reduced preferentially with 6 – 929% (3 mol%) and 168% increase (1 mol%), respectively (Figure 4). Aldehyde 14 is highly sensitive under the reaction conditions and good efficiency was only achieved at 0 ºC. Reduction of the alkene of nitrile 16 was also drastically more effective with 6 and 1 mol% of catalyst (309% increase) as well as unsaturated carbonyl 17 (118% increase). All the examples performed with phenylsilane required the use of isopropanol as solvent whereas silane 6 allowed a solvent screen to further increase the yields.
Figure 4.

Reduction of alkenes bearing susceptible functional groups. Reactions performed with 1.5 equiv. of [Si]/TBHP at 22 ºC for 15–60 min. The solvent used was optimized for each substrate with 6 and isopropanol was required for 3 (see SI for further information). aReaction performed at 0 ºC.
Besides increasing yields, isopropoxy(phenyl)silane obviated the need for superstoichiometric reductant and oxidant (e.g. 6 equiv. PhSiH3, 4 equiv. TBHP) or high catalyst loading (e.g. 20 mol%).7b,c This is the case of enol ether 18 and haloalkenes 19, 20 and 21, which were reduced significantly more efficiently than our first generation conditions (Figure 5). Interestingly, although good yields were obtained for trisubstituted haloalkenes (79% and 70%), an impurity in the reaction tended to inhibit turnover, hampering complete conversion – related protocols required stoichiometric amounts of catalyst for similar scaffolds.7c
Figure 5.

Reduction of enol ethers and haloalkenes. Reactions performed with 2 equiv. of [Si]/TBHP at 22 ºC for 4 h. The solvent used was optimized for each substrate with 6 and isopropanol was required for 3 (see SI for further information). aAddition of 1 equiv. of CaCO3.
Treatment of dienes like 22 with a silane in the presence of Mn(dpm)3 leads to the HAT-initiated reductive cyclization (Scheme 7).7b In this example, when the metal hydride-generated carbon-centered radical is trapped by a pendant alkene to form a subsequent radical that is reduced. The cyclization step competitively forms the 5- and 6-membered rings independent of the silane or solvent used in the reaction (23a and 23b, respectively). This intrinsic behavior is consistent with a radical transformation.15 With 1 mol% of catalyst the first-generation conditions lead to the reductive cyclization in 28% isolated yield whereas isopropoxy(phenyl)silane increases this yield to 87%.
Scheme 7.

Comparison between silanes 3 and 6 in the HAT-initiated reductive cyclization.
Styrenes continue to be poorly performing substrates (Figure 6), although even 24 improved with 6 (styrenes tend to dimerize through a persistent benzyl radical, which could be minimized by performing the reduction under more dilute conditions). t-Bu-methylenecyclohexane 25 on the other hand is reduced quantitatively with 6 in only 15 min, but rather inefficiently with 3.
Figure 6.

Comparison of the reduction of a styrene and an exocyclic alkene with silane 3 and 6 (see SI for further information).
Hydrogenation of hindered Δ5,6 steroids like cholesterol (26) can be modestly improved with 6 (Figure 7). An important, if esoteric, showcase example for the continuing utility of the generally thermodynamic preference of alkene reduction is shown in example 27. Here a Δ4,5 steroid is reduced to the trans-A/B ring junction (confirmed by x-ray crystallography, Figure 8),18 whereas all literature precedent shows, to the best of our knowledge, preference for cis- upon catalytic hydrogenation (with the exception of dissolving metal reduction, as discussed previously).7b
Figure 7.

Reduction of hindered alkenes in steroid-derivatives. Reactions performed with 2 equiv. of [Si]/TBHP at 22 ºC for 0.5–2 h. The solvent used was optimized for each substrate with 6 and isopropanol was required for 3 (see SI for further information).
Figure 8.

Trans-A/B ring junction of 27 confirmed by x-ray crystallography.
Isotope labelling experiments of the HAT hydrogenation
As a mechanistic aid to the eventual design of an asymmetric variant of HAT hydrogenation, we sought to identify the source of the second hydrogen addition, which in most cases forms the sole stereogenic center from a prochiral alkene.19 Many prior studies have examined the source of the first hydrogen using deuterium labeling.2e,f,3e,4a,6b,g,d,7a,8a–c,e Only Herzon has explored double radical hydrogen incorporation under Mukaiyama-type conditions,4d and these studies did not identify the second source of hydrogen incorporation.20 Furthermore, a significant question arose only recently in studies by Norton who demonstrated that Co(dmgBF2)2-H complexes 28 and 29 may exist as equilibria of Co-H and O-H tautomers, either of which could undergo HAT (Scheme 8).21 We wondered whether either hydrogen in our Mn-catalyzed system could derive from a non-innocent ligand, or whether hydrogen scrambling between a metal center and its ligand could occur. The analogous structure for metal hydride β-diketonate 30 would involve C-H tautomers 31 or 32. Since silane 6 gave us the ability to hydrogenate alkenes in aprotic solvent that cannot exchange with free- or bound-diketonate, we interrogated these competing sources of hydrogen/deuterium.
Scheme 8.

Tautomerization of metal complexes.
Hydrogenation of substrate 33 with PhSiD3 and Mn(dpm)3 showed complete incorporation at C4 (100%, i.e. 66% H integration by 1H NMR). These results are consistent with repeated observations of complete initial (C4) deuterium incorporation22 using PhSiD3 or NaBD4 and a non-deuterated ligand.2e,f,3e,4a,6b,g,d,7a,8a–c,e However, this prior data alone does not exclude hydrogen scrambling with the ligand due to the inverse kinetic isotope effect associated with metal-hydride HAT; i.e. the Mn-D might be preferentially transferred to the alkene even as a small population in equilibrium.23 Moreover, only 63% D was observed at C2, suggesting a competing non-silane-derived source for the second hydrogen (Scheme 9). However, deuterated catalyst Mn(d1-dpm)3 (25 mol%) in hexanes leads to no deuterium incorporation at either C2 or C4. The absence of deuteration could not be due to a high kH/kD via C-H HAT24 in light of the previous PhSiD3 incorporation experiments. Based on this data, there is no evidence that HAT results from a redox-active (C-H bond forming) non-innocent beta-diketonate ligand.
Scheme 9.

Involvement of the ligand in the hydrogen atom transfer.
When the hydrogenation was performed in d8-i-PrOD or using t-BuOOD, we observed no D incorporation at either C2 or C4,7c despite the low bond dissocation energy of hydroperoxy O-H bonds, ruling out these alternative sources of H or D (see SI for further details). But more surprisingly, hydrogenation of 33 with PhSiD3 and t-BuOOD in d8-i-PrOD led to very similar results, i.e. only partial deuteration at the internal carbon C2. We observed that reduction of 33 under more dilute conditions led to very little improvement (0.1 M, 66% D at C2), nor did excess of PhSiD3 (6 equiv, 72% D) or modified catalyst loading (5 mol% Mn, 49% D; and 100 mol% Mn, 68% D). Where was the internal hydrogen coming from?
Deuteration of substrate 33 in the benzylic positions gave us a clue. Reduction of d4-33 with PhSiH3 delivered 9% deuterium incorporation at C2 during the hydrogenation (Scheme 10), indicating that the mysterious internal hydrogen is derived from the substrate itself. This level of deuterium incorporation is lower than the reverse isotope labeling (37% H, Scheme 9) likely reflecting a normal KIE of hydrogen transfer. This isomerization could either be mediated by a Mn(II) reverse HAT or inter-substrate transfer. Unless there is a very high inverse KIE from the metal hydride, this scrambling must occur between the intermediate substrate radical and substrate 33 since we see 100% D incorporation at C4 (Scheme 9) and a metal hydride formed by reverse HAT would label C4 with H. Abstraction of the benzylic hydrogen from 33 would not be surprising since its BDE should lie somewhere between diphenylmethane (81.4 kcal/mol) and 1,4-pentadienes (76.4 kcal/mol), similar to the BDE of 1,4-cyclohexadiene (76.9 kcal/mol), used previously as a stoichiometric reductant.25
Scheme 10.

Isotope scrambling depending on the substrate.
Reduction of substrates 35 and 37 with PhSiD3 neatly showed 100% deuterium incorporation at C4, but 76% and 17% of hydrogen, respectively, were present at C2. Incorporation of additional deuterium (129% D) at C4 of 36 confirms the equilibrium intermediacy of an alkene subsequent to the initial HAT. Substrate 37, in which the C-H bonds adjacent to the newly formed radical possess higher BDEs than the benzylic positions of 33, demonstrates much lower levels of scrambling, i.e. higher levels of deuterium incorporation. Therefore the amount of competitive isomerization is highly dependent on the structure of the substrate, and is not intrinsic to all reductions.
Finally, we also observed that the hydrogenation of 33 produced 70% of 34 in the absence of TBHP when 1 equiv. of the manganese complex was used. This result suggests that the reduction of the radical intermediate does not occur exclusively from the metal-hydride since the Mn(II) product cannot reform a Mn(III)-H (see Scheme 5). Based on this stoichiometric reaction and on the labeling results above, the main second reductant appears to be the silane itself. The resulting silyl radical can add to alkenes in the absence of TBHP to effect hydrosilylation (see Scheme 6). In the context of hydrogenation, the fate of the resulting silyl radical in the catalytic cycle or alternate radical chain is uncertain–it might form a silylmanganese (III) reservoir–but TBHP appears to play a role in its consumption to prevent hydrosilylation.26 This data also illustrates obstacles to the design of an asymmetric variant of HAT hydrogenation using the Mukaiyama reaction manifold, especially since a metal complex may only be partially involved in the stereogenic second hydrogen donation.
An intermolecular competition experiment between PhSiH3 and PhSiD3 (1:1) in the reduction of 33 revealed major incorporation of deuterium in the terminal C4 position (Scheme 11), whereas no preference was observed in C2 after considering the background source of hydrogen (isomerization). This preference for D-incorporation demonstrates an inverse KIE present in the catalytic cycle, either at the initial HAT step or in formation of the metal hydride/deuteride from the metal(III) precatalyst.27
Scheme 11.

H/D intermolecular competition experiment.
Similarly, comparison of a kinetic profile of PhSiD3 against PhSiH3 in the hydrogenation of terpineol 9 in isopropanol showed a small inverse kinetic isotope effect on the overall rate (Figure 9). Such inverse KIEs have been suggested as hallmarks of an HAT mechanism in stoichiometric reductions mediated by metal hydrides.9 The relatively small size of the observed KIE may suggest a tempering effect of normal KIEs at other points in the catalytic cycle, but we are cautious to over-interpret this observation since pre-catalyst formation might also lead to an inverse KIE. Nevertheless, this is the first inverse KIE observed in the Mukaiyama literature, and establishes an important point of departure for subsequent mechanistic study.28
Figure 9.
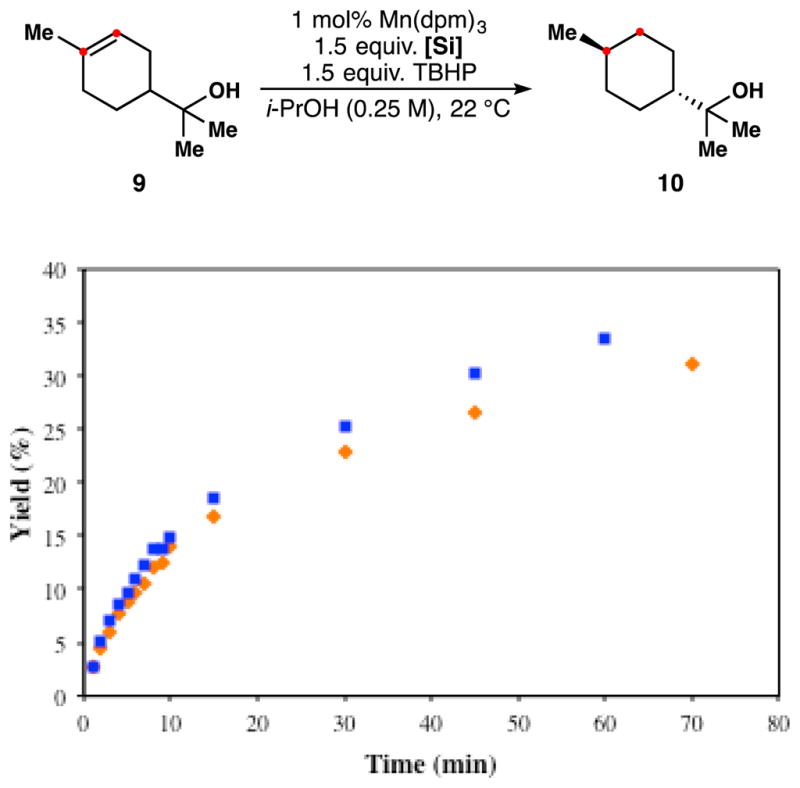
Reduction of terpineol monitored by GC-FID. Reaction performed in isopropanol with PhSiH3 (
) or PhSiD3 (
).
Combined with work from our lab showing that organocobalt complexes appear to be parasitic species in HAT isomerization,6f and that AIBN or alcohol2a can replace silane reductant using cobalt catalysis, these results support the hypothesis that these hydrofunctionalizations proceed by metal-hydride HAT to an alkene and exclude the competing alternatives of hydrometallation and ligand HAT. Obviously, reductive elimination pathways are not available to the planar tetradentate porphyrin or salen metal complexes frequently used in this chemistry.29 Given the high energy of carbon-centered radicals and the low bond dissociation enthalpy of C-H bonds adjacent to carbon radicals, these M-H HAT reactions must rely on a surprisingly high reactivity of the intermediate metal-hydrides.9
Study of the applicability of isopropoxy(phenyl)silane in other HAT-initiated hydrofunctionalizations: hydration of alkenes
In his seminal publications, Mukaiyama reported the hydration of alkenes using a silane in an alcoholic solvent in the presence of a cobalt complex and oxygen (1 atm).30 Later, Magnus reported the use of manganese catalysts in the umpolung hydration of α,β-unsaturated compounds.2d Silane 6 increases the performance of Mn-catalyzed hydration of 1 in a non-alcoholic solvent (Figure 10). The reaction affords 2 using 5 mol% of either with Mn(dpm)3 or Mn(acac)3 in THF under O2 atmosphere, but PhSiH3 performs poorly in the absence of isopropanol. Interestingly, Co(acac)2 was a competent catalyst with both 3 and 6 in THF.
Figure 10.

Hydration of phenylbutene 1 with O2 monitored by GC-FID. Reaction performed with Mn(acac)3/Ph(i-PrO)SiH2 in THF (
), Mn(dpm)3/Ph(i-PrO)SiH2 in THF (
) and Mn(acac)3/PhSiH3 in THF (●) or isopropanol (
). Co(acac)2/PhSiH3 and Ph(i-PrO)SiH2 in THF are both efficient.
Study of the reductive olefin coupling
In addition to improving the yields, catalyst loadings and solvent breadth of HAT hydrogenation, isopropoxy(phenyl)silane (6) allows Baran’s iron-catalyzed conjugate addition to be run at low catalyst loadings and ambient temperature.6e As shown in Scheme 12, PhSiH3 (3) does not perform well towards the formation of 41 or 43 under those conditions.
Scheme 12.

Comparison of silane 3 and 6 in radical conjugate addition.
In contrast, silane 6 far outstrips silane 3 and gives yields comparable to, or better than, the original conditions, which used high catalyst loading and excess of enone or alkene (30–40 mol% [Fe], 3 equiv. of 39 or 42, EtOH, 60 °C). Interestingly, no turn-over was observed in the absence of an alcoholic solvent also with silane 6 – isopropanol was used as a cosolvent of EtOAc or DCM, as a proton source is mechanistically required for this transformation. Thus, the radical intermediate attacks the α,β-unsaturated carbonyl, which presumably forms an iron enolate that gets protonated to afford the final product and re-oxidize the catalyst.
Study of the hydroamination of alkenes
Similarly, silane 6 allows the recently reported HAT-initiated iron-catalyzed hydroamination of alkenes to be run at low catalyst loading (1 mol%) and ambient temperature (Scheme 13).3k
Scheme 13.

Comparison of silane 3 and 6 in the hydroamination of alkenes.
The reaction leads from good to excellent yields, whereas the efficiency was highly reduced with PhSiH3 in isopropanol. In this case, the results suggested that the radical intermediate attacked a nitroso group that would be subsequently reduced and protonated, therefore a minimum of 3 equiv. of an alcohol were also required for the formation of 46 (see SI for further details).
CONCLUSIONS
We have shown that the required alcohol solvent used in Mn and Fe-catalyzed HAT-initiated hydrofunctionalizations partly serves as an accelerating substituent on the silane reductant. This solvent restriction posed a significant limitation to the field, which hampered the refinement of the established protocols, as we have shown here. The intermediate reductant derived from isopropanol, Ph(i-PrO)SiH2 (6), greatly improves many of these reactions, including our 1st generation HAT hydrogenation, hydration and hydroamination of alkenes as well as the reductive olefin coupling. Isopropoxy(phenyl)-silane can accelerate and improve HAT-mediated transformations that are non-optimal under standard conditions. Outstanding results have been obtained: decreased catalyst loadings down to 0.05 mol%, lowered reaction temperatures and use of diverse, aprotic solvents, which preclude superstoichiometric amounts of reagents, and broader functional group tolerance. The solvent versatility also allowed the novel radical-polar crossover methods as the Pronin lab has already shown in a recent synthesis of emindole SB.11 We also include data to suggest that HAT does not occur from the ligand, which is an alternative hypothesis for HAT reactions involving the dimethylglyoxime (dmg) ligand. Further isotope labeling experiments revealed preferential deuterium incorporation during the HAT along with hydrogen scrambling from the substrate. Moreover, discovery of a competing hydrosilylation reaction revealed multiple roles for TBHP besides re-oxidation of the catalyst. The mechanistic hypothesis that currently best fits the data is M-H HAT to the alkene forming a carbon-centered radical. Thus, the structural and energetic implications from this HAT hypothesis, along with the versatility provided by Ph(i-PrO)SiH2, should stimulate many discoveries and facilitate the invention of new chemical reactions. New methods and applications in chemical synthesis are currently under development in our group.
Supplementary Material
Acknowledgments
Funding Sources
Financial support for this work was provided by the NIH (GM104180 and F31 GM111050 to R.M.). Additional support was generously provided by Eli Lilly, Novartis, Bristol-Myers Squibb, Amgen, Boehringer-Ingelheim, the Sloan Foundation and the Baxter Foundation.
We thank Dr. Curtis Moore and Prof. Arnold L. Rheingold for x-ray crystallographic analysis. We thank Professor Dale Boger for valuable discussions.
Footnotes
Detailed experimental procedures, spectral data and chromatograms are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For a review and references to early work, see: Mukaiyama T, Yamada T. Bull Chem Soc Jpn. 1995;68:17.Hoffmann RW. Chem Soc Rev. 2016;45:577. doi: 10.1039/c5cs00423c.
- 2.C–O bond formation: Mukaiyama T, Isayama S, Inoki S, Kato K, Yamada T, Takai R. Chem Lett. 1989;18:449.Isayama S, Mukaiyama T. Chem Lett. 1989;18:573.Isayama S, Mukaiyama T. Chem Lett. 1989;18:1071.Magnus P, Payne AH, Waring MJ, Scott DA, Lynch V. Tetrahedron Lett. 2000;41:9725.Tokuyasu T, Kunikawa S, McCullough KJ, Masuyama A, Nojima M. J Org Chem. 2005;70:251. doi: 10.1021/jo048359j.Shigehisa H, Aoki T, Yamaguchi S, Shimizu N, Hiroya K. J Am Chem Soc. 2013;135:10306. doi: 10.1021/ja405219f.Hu X, Maimone TJ. J Am Chem Soc. 2014;136:5287. doi: 10.1021/ja502208z.
- 3.C–N bond formation: Kato K, Mukaiyama T. Chem Lett. 1990;19:1395.Kato K, Mukaiyama T. Chem Lett. 1990;19:1917.Hata E, Kato K, Yamada T, Mukaiyama T. J Synth Org Chem Jpn. 1996;54:728.Gaspar B, Waser J, Carreira EM. Synthesis. 2007:3839.Waser J, Gaspar B, Nambu H, Carreira EM. J Am Chem Soc. 2006;128:11693. doi: 10.1021/ja062355+.Waser J, González-Gómez JC, Nambu H, Huber P, Carreira EM. Org Lett. 2005;7:4249. doi: 10.1021/ol0517473.Waser J, Nambu H, Carreira EM. J Am Chem Soc. 2005;127:8294. doi: 10.1021/ja052164r.Waser J, Carreira EM. Angew Chem Int Ed. 2004;43:4099. doi: 10.1002/anie.200460811.Waser J, Carreira EM. J Am Chem Soc. 2004;126:5676. doi: 10.1021/ja048698u.Shigehisa H, Koseki N, Shimizu N, Fujisawa M, Niitsu M, Hiroya K. J Am Chem Soc. 2014;136:13534. doi: 10.1021/ja507295u.Gui JH, Pan CM, Jin Y, Qin T, Lo JC, Lee BJ, Spergel SH, Mertzman ME, Pitts WJ, La Cruz TE, Schmidt MA, Darvatkar N, Natarajan SR, Baran PS. Science. 2015;348:886. doi: 10.1126/science.aab0245.
- 4.C-X (halide) bond formation: Gaspar B, Carreira EM. Angew Chem Int Ed. 2008;47:5758. doi: 10.1002/anie.200801760.Gaspar B, Waser J, Carreira EM. Org Syn. 2010;87:88.Shigehisa H, Nishi E, Fujisawa M, Hiroya K. Org Lett. 2013;15:5158. doi: 10.1021/ol402696h.Ma X, Herzon SB. Chem Sci. 2015;6:6250. doi: 10.1039/c5sc02476e.
- 5.C–S/Se bond formation: Girijavallabhan V, Alvarez C, Njoroge FG. J Org Chem. 2011;76:6442. doi: 10.1021/jo201016z.; (b) Ref 4d.
- 6.C–C bond formation: Isayama S, Mukaiyama T. Chem Lett. 1989;18:2005.Wang LC, Jang HY, Roh Y, Lynch V, Schultz AJ, Wang X, Krische MJ. J Am Chem Soc. 2002;124:9448. doi: 10.1021/ja020223k.Gaspar B, Carreira EM. Angew Chem Int Ed. 2007;46:4519. doi: 10.1002/anie.200700575.Gaspar B, Carreira EM. J Am Chem Soc. 2009;131:13214. doi: 10.1021/ja904856k.Lo JC, Yabe Y, Baran PS. J Am Chem Soc. 2014;136:1304. doi: 10.1021/ja4117632.Crossley SWM, Barabé F, Shenvi RA. J Am Chem Soc. 2014;136:16788. doi: 10.1021/ja5105602.Lo JC, Gui J, Yabe Y, Pan CM, Baran PS. Nature. 2014;512:343. doi: 10.1038/nature14006.Dao HT, Li C, Michaudel Q, Maxwell BD, Baran PS. J Am Chem Soc. 2015;137:8046. doi: 10.1021/jacs.5b05144.Zheng J, Wang D, Cui S. Org Lett. 2015;17:4572. doi: 10.1021/acs.orglett.5b02294.
- 7.C=C reduction: Magnus P, Waring MJ, Scott DA. Tetrahedron Lett. 2000;41:9731.Iwasaki K, Wan KK, Oppedisano A, Crossley SWM, Shenvi RA. J Am Chem Soc. 2014;136:1300. doi: 10.1021/ja412342g.King SM, Ma X, Herzon SB. J Am Chem Soc. 2014;136:6884. doi: 10.1021/ja502885c.
- 8.For a related set of radical hydrofunctionalizations, discovered independent of Ref. 1–7 and based on iron complexes/borohydrides, see: Ishikawa H, Colby DA, Boger DL. J Am Chem Soc. 2008;130:420. doi: 10.1021/ja078192m.Ishikawa H, Colby DA, Seto S, Va P, Tam A, Kakei H, Rayl TJ, Hwang I, Boger DL. J Am Chem Soc. 2009;131:4904. doi: 10.1021/ja809842b.Leggans EK, Barker TJ, Duncan KK, Boger DL. Org Lett. 2012;14:1428. doi: 10.1021/ol300173v.Gotoh H, Sears JE, Eschenmoser A, Boger DL. J Am Chem Soc. 2012;134:13240. doi: 10.1021/ja306229x.Barker TJ, Boger DL. J Am Chem Soc. 2012;134:13588. doi: 10.1021/ja3063716.
- 9.(a) Feder HM, Halpern J. J Am Chem Soc. 1975;97:7186. [Google Scholar]; (b) Sweany RL, Halpern J. J Am Chem Soc. 1977;99:8335. [Google Scholar]; (c) Eisenberg DC, Norton JR. Isr J Chem. 1991;31:55. [Google Scholar]; (d) Choi J, Tang L, Norton JR. J Am Chem Soc. 2007;129:234. doi: 10.1021/ja066325i. [DOI] [PubMed] [Google Scholar]
- 10.(a) Abley P, Dockal ER, Halpern J. J Am Chem Soc. 1972;94:659. doi: 10.1021/ja00791a016. [DOI] [PubMed] [Google Scholar]; (b) Magnuson RH, Halpern J, Levitin IY, Vol’pin ME. J Chem Soc Chem Comm. 1978:44. [Google Scholar]
- 11.George DT, Kuenstner EJ, Pronin SV. J Am Chem Soc. 2015;137:15413. doi: 10.1021/jacs.5b11129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) O’Donnell K, Bacon R, Chellappa KL, Schowen RL, Lee JK. J Am Chem Soc. 1972;94:2500. [Google Scholar]; (b) Howie CR, Lee JK, Schowen RL. J Am Chem Soc. 1973;95:5286. [Google Scholar]; (c) Revunova K, Nikonov GI. Chem Eur J. 2014;20:839. doi: 10.1002/chem.201302728. [DOI] [PubMed] [Google Scholar]
- 13.Gunji Y, Yamashita Y, Ikeno T, Yamada T. Chem Lett. 2006;35:714. [Google Scholar]
- 14.The stark drop in conversion between entries 10 and 11 probably indicates inhibition from an impurity near 0.0002 equiv abundance. However, neither the impurity nor its source could be identified.
- 15.Beckwith ALJ, Bowry VW, Ingold KU. J Am Chem Soc. 1992;114:4983. [Google Scholar]
- 16.(a) Chatgilialoglu C. Acc Chem Res. 1992;25:188. [Google Scholar]; (b) Chen C, Hecht MB, Kavara A, Brennessel WW, Mercado BQ, Weix DJ, Holland PL. J Am Chem Soc. 2015;137:13244. doi: 10.1021/jacs.5b08611. [DOI] [PubMed] [Google Scholar]
- 17.(a) Magnus P, Fielding MR. Tetrahedron Lett. 2001;42:6633. [Google Scholar]; (b) Yamada T, Ikeno T, Ohtsuka Y, Kezuka S, Sato M, Iwakura I. STAM. 2006;7:184. and references cited therein. [Google Scholar]
- 18.CCDC 1445790 contains the supplementary crystallographic data for this paper (25 x-ray). This data is provided free of charge by the Cambridge Crystallographic Data Centre.
- 19.Sibi MP, Porter NA. Acc Chem Res. 1999;32:163. [Google Scholar]
- 20.In Ref 8b, treatment of anhydrovinblastine with superstoichiometric amounts of Fe2(ox)3 and NaBD4 in the absence of oxygen led to alkene reduction with complete deuterium incorporation in both carbons, suggesting differences in the reaction paths between this and our work.
- 21.Estes DP, Grills DC, Norton JR. J Am Chem Soc. 2014;136:17362. doi: 10.1021/ja508200g. [DOI] [PubMed] [Google Scholar]
- 22.In Ref 7c, partial deuterium incorporation is observed, but many sources of hydrogen are present.
- 23.Anslyn EV, Dougherty DA. Modern Physical Organic Chemistry. University Science Books; 2006. [Google Scholar]
- 24.Mayer JM. Acc Chem Res. 2011;44:36. doi: 10.1021/ar100093z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.(a) Cherkason A, Jonsson M. J Chem Inf Comput Sci. 2000;40:1222. doi: 10.1021/ci000387p. [DOI] [PubMed] [Google Scholar]; (b) Pratt DA, Mills JH, Porter NA. J Am Chem Soc. 2003;125:5801. doi: 10.1021/ja034182j. [DOI] [PubMed] [Google Scholar]; (c) Gao Y, DeYonker NJ, Garrett EC, III, Wilson AK, Cundari TR, Marshall P. J Phys Chem A. 2009;113:6955. doi: 10.1021/jp901314y. [DOI] [PubMed] [Google Scholar]
- 26.Chatgilialoglu C. Chem Rev. 1995;95:1229. [Google Scholar]
- 27.Simmons EM, Hartwig JF. Angew Chem Int Ed. 2012;51:3066. doi: 10.1002/anie.201107334. [DOI] [PubMed] [Google Scholar]
- 28.In Ref 3e, the cobalt(III)-catalyzed hydrohydrazination and hydro-azidation of olefins showed normal kinetic isotope effects (2.2 and 1.65, respectively), demonstrating differences in rate determining steps between these and the hydrogenation reactions.
- 29.(a) Gridnev AA, Ittel SD. Chem Rev. 2001;101:3611. doi: 10.1021/cr9901236. [DOI] [PubMed] [Google Scholar]; (b) De Bruin B, Dzik WI, Li S, Wayland BB. Chem Eur J. 2009;15:4312. doi: 10.1002/chem.200802022. [DOI] [PubMed] [Google Scholar]; (c) Sorokin AB. Chem Rev. 2013;113:8152. doi: 10.1021/cr4000072. [DOI] [PubMed] [Google Scholar]
- 30.Inoki S, Kato K, Isayama S, Mukaiyama T. Chem Lett. 1990:1869. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.