

Abstract
Purpose
Triple-negative breast cancer (TNBC) is a highly heterogeneous disease and has the worst outcome among all subtypes of breast cancers. Although PARP inhibitors represent a promising treatment in TNBC with BRCA1/BRCA2 mutations, there is great interest in identifying drug combinations that can extend the utility of PARP inhibitors to a majority of TNBC patients with wild-type BRCA1/BRCA2. Here we explored whether mTOR inhibitors, through modulating homologous recombination (HR) repair, would provide therapeutic benefit in combination with PARP inhibitors in preclinical models of BRCA-proficient TNBC.
Experimental Design
We have studies the effects of mTOR inhibitors on HR repair following DNA double-strand breaks (DSBs). We further demonstrated the in vitro and in vivo activities of combined treatment of mTOR inhibitors with PARP inhibitors in BRCA-proficient TNBC. Moreover, microarray analysis and rescue experiments were employed to investigate the molecular mechanisms of action.
Results
We found that mTOR inhibitors significantly suppressed HR repair in two BRCA-proficient TNBC cell lines. mTOR inhibitors and PARP inhibitors in combination exhibited strong synergism against these TNBC cell lines. In TNBC xenografts, we observed enhanced efficacy of everolimus in combination with talazoparib (BMN673) compared with either drug alone. We further identified through microarray analysis and by rescue assays that mTOR inhibitors suppressed HR repair and synergized with PARP inhibitors through regulating the expression of SUV39H1 in BRCA-proficient TNBCs.
Conclusions
Collectively, these findings strongly suggest that combining mTOR inhibitors and PARP inhibitors would be an effective therapeutic approach to treat BRCA-proficient TNBC patients.
Keywords: mTOR inhibitor, PARP inhibitor, homologous recombination, synergy, SUV39H1, triple-negative breast cancer
Introduction
Triple-negative breast cancer (TNBC) is characterized by the lack of estrogen receptors (ER) and progesterone receptors (PR), in addition to the absence of overexpression of human epidermal growth factor receptor 2 (Her2). It is a highly heterogeneous disease that accounts for approximately 15% of all breast cancers (1,2). Because TNBC cannot be treated with specific hormone or anti-Her2 therapy, systematic conventional chemotherapy is usually the first-line therapy for TNBC patients. However, TNBC, especially those with advanced-stage disease, generally shows the worst prognosis among all subgroups of breast cancer patients (3), making it an extreme challenging disease in clinical oncology. Hence, one of the major focuses to direct clinical trials of TNBC is identifying targeted therapies in sensitive TNBC subsets by the presence of predictive biomarkers. Inhibitors of poly(adenosine diphosphate [ADP]-ribose) polymerase (PARP) have demonstrated activity in breast cancers with mutations in BRCA1 or BRCA2 with low toxicity profiles (4). TNBC is generally associated with significant genomic instability due to DNA-repair defects. Depending on different populations and studies, up to 10-20% of TNBC patients carry germline or sporadic mutations in BRCA1 or BRCA2 (5,6), which primarily affect the ability to repair DNA double-strand breaks (DSB) through error-free homologous recombination (HR) repair (7). PARP is one of the key players in base excision repair for repairing DNA single-strand breaks (SSB). It directly binds to DNA SSBs and is catalytically activated to mediate PARylation of itself and other proteins to recruit DNA damage repair factors (8). Failure to repair SSBs leads to DSBs during DNA replication. Thus PARP inhibition results in HR dependency for repairing DSBs. This vulnerability provides the rationale for synthetic lethal therapy with PARP inhibitors in patients with HR-incompetent cancers, such as TNBCs with BRCA1/2 mutations (9). In addition, PARP inhibitors have also been used in BRCA-proficient TNBCs (10,11), which currently have limited therapeutic options, albeit with minor evidence for activity. Therefore, discovery of agents that would cause HR defects and synergize with PARP inhibitors is urgently needed to provide preclinical evidence to direct clinical trials of rational combinations in BRCA-proficient TNBCs.
Previously, our group applied a transcriptional profiling-based approach to systematically generate an HR defect gene signature, which robustly predicts PARP inhibitor response and HR repair status in vitro (12). We then queried the Connectivity Map, which is a collection of genome-wide transcriptional expression data and shows connections among bioactive small molecules, gene expression, and diseases (13,14), to screen compounds against the HR defect gene expression profiles and identified those compounds that would potentially lead to HR defects and sensitize cancer cells to PARP inhibitors. Interestingly, Mammalian target of rapamycin (mTOR) inhibitors were among the top candidates, in terms of suppressing HR repair and possibly synergizing with PARP inhibitors. mTOR is a key downstream regulator of the phosphatidylinositide-3-kinase (PI3K) pathway, and PI3K inhibitors have been reported to compromise HR repair and sensitize breast cancer cells to PARP inhibitors (15,16). Deregulation of mTOR has been found in various human cancers (17), including TNBCs (18,19). Therefore, inhibition of mTOR signaling pathway is an attractive clinical strategy for this disease. In addition to regulating cell growth, proliferation, and metabolism in response to environmental and nutritional stimuli (20) through phosphorylating p70 ribosomal S6 kinase 1 (S6K1) and eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1), mTOR has been implicated in the DNA damage response in human cancers, possibly through modulating proteins that are essential in chromosomal integrity and DNA damage response (21-23). Recently, several studies found that mTOR regulates the DNA damage response through the NF-κB-mediated FANCD2 pathway in leukemia and rhabdomyosarcoma (24-26). However, the association between mTOR and DNA damage response in TNBCs remains largely unknown. Therefore, it was important to investigate whether mTOR inhibitors modulate HR repair and enhance the cytotoxic effects of PARP inhibitors in BRCA-proficient TNBCs.
We demonstrate herein that mTOR inhibitors suppress HR repair efficiency and synergize with PARP inhibitors in BRCA-proficient TNBCs cell lines. These observations were further confirmed in vivo in BRCA-proficient TNBC xenografts. Furthermore, this study demonstrates that mTOR inhibitors modulate HR repair through suppressing the expression of SUV39H1, a key histone methyltransferase (27). Our results thus provide evidence for the translation of rationale combination strategies with mTOR inhibitors and PARP inhibitors in BRCA-proficient TNBCs to the clinic and disclose a novel molecular mechanism by which the mTOR inhibitors modulate HR repair and synergize to PARP inhibitors.
Materials and Methods
Cell culture and chemicals
The U2OS, MDA-MB-231, BT-549 and MCF-12A cell lines were purchased from the ATCC (American Type Culture Collection) and the cell lines were authenticated by Short Tandem Repeat (STR) profiling by ATCC. U2OS cells were maintained in McCoy’s 5A medium supplemented with 10% fetal bovine serum. MDA-MB-231 and BT-549 cells were cultured in Dulbecco modified Eagle medium (DMEM) or RPMI-1640 medium supplemented with 10% fetal bovine serum, respectively. MCF-12A cells were cultured in mammary epithelial growth medium (1:1 DMEM:DMEM/F12 medium, 20% horse serum, hydrocortisone [0.5 mg/mL], insulin [10 μg/mL], recombinant epidermal growth factor [20 ng/mL], cholera toxin [100 ng/mL], and 1:100 penicillin-streptomycin [Invitrogen]). mTOR inhibitors everolimus (EVE) and KU-0063794 (KU) and PARP inhibitors olaparib (AZD2281) and talazoparib (BMN673) were purchased from Selleckchem. Myc-DDK-SUV39H1 plasmid was purchased from OriGene. Antibodies against SUV39H1, p-4EBP1 (Ser65), and Myc-Tag were purchased from Cell Signaling Technology. Vinculin antibody was purchased from Santa Cruz Biotechnology. An Annexin V FITC apoptosis detection kit was purchased from BD Biosciences and an apoptosis assay was performed following the manufacturer’s procedure. The transfection reagent FuGENE 6 and oligofectamine were purchased from Promega and Life Technologies, respectively.
Immunofluorescence staining and microscopy
To detect DNA damage-induced RAD51 foci formation, we pretreated MDA-MB-231 or MCF-12A cells with either the mTOR inhibitor everolimus (EVE, 10 μM) or KU-0063794 (KU, 10 μM) for 48 hours. To induce DSBs, we subjected the cells to ionizing radiation (IR, 10 Gy) and performed immunofluorescence staining as described previously (28). Cells were treated with cytoskeleton and stripping buffers, fixed with 4% paraformaldehyde, and subjected to permeabilization with 0.5% NP-40 and 1% Triton X-100. The cells were then incubated with primary antibody (rabbit anti-RAD51, 1:400; Abcam) for 2 hours at room temperature and were incubated with secondary antibody (Alexa Fluor 488 conjugated donkey anti-rabbit antibody, 1:400; Life Technologies) for 1 hour at room temperature. Slides were mounted in medium containing 4’,6-diamidino-2-phenylindole (DAPI, Vector Laboratories, H-1200) and were analyzed under a fluorescence microscope (Eclipse TE2000E, Nikon). We scored the percentage of cells with more than 10 RAD51 foci per cell in at least 50 cells per sample.
HR repair analysis
U2OS cells containing a single copy of the HR repair reporter substrate direct repeat green fluorescent protein (DR-GFP) in a random locus were generated as previously described (12). GFP-expressing plasmid (pEGFP-C1) was used as a transfection efficiency control, and pCBASceI plasmid (gifts from Dr. Maria Jasin; Memorial Sloan-Kettering Cancer Center, New York, NY) was used for detecting HR repair efficiency. Everolimus (EVE, 10 μM) or KU-0063794 (KU, 10 μM) was added 6 hours after the transfection. After 48 hours, GFP-positive cells were detected via flow cytometric analysis using a FACSCalibur flow cytometer with CellQuest software (Becton Dickinson). For HU (hydroxyurea)-synchronized HR repair assay (12), U2OS cells were treated with EVE (10 μM) or KU (10 μM) after I-SceI transfection and then treated with HU (2 mM) to synchronize the cell cycle for 16 hr before flow cytometry analysis of cell cycle distribution and GFP intensity. For the HR repair rescue assay, Myc-DDK-SUV39H1 plasmids were transiently transfected into U2OS cells. The next day, cells were transfected with pCBASceI plasmid. EVE (10 μM) or KU (10 μM) was added 6 hours after the transfection and GFP intensity was detected after 48 hours.
Cell-cycle analysis
Cells were suspended in staining solution (0.1% sodium citrate, 0.03% NP40, propidium iodide [0.05 mg/mL], and RNase A [0.02 mg/mL]) (29). Cell-cycle analysis was performed at The University of Texas MD Anderson Cancer Center South Campus Flow Cytometry and Cellular Imaging Core Facility.
Colony formation assay
MDA-MB-231 and BT-549 cells were seeded in 6-well plates at low density. The indicated concentrations of mTOR inhibitors and PARP inhibitors or vehicle control (dimethyl sulfoxide [DMSO]) were added the next day, and cells were left for 10-14 days to form colonies. Colonies were stained with 0.25% crystal violet and 25% methanol in phosphate-buffered saline (PBS) solution for visualization. Colonies with 50 or more cells were counted manually and digitally using ImageJ software with customized parameters that were optimized on the basis of three preliminary manual counts.
Cell proliferation assay
Cell proliferation was measured by PrestoBlue Cell Viability Reagent (Life Technologies). We seeded cells in 96-well plates in a total volume of 100 μL in triplicate in each experiment. The next day, cells were treated with DMSO or the indicated concentrations of mTOR inhibitors and PARP inhibitors. After 5 days, PrestoBlue Cell Viability Reagent (10 μL) was added to each well and incubated for optimized incubation time (4-6 hours) at 37°C. Then the fluorescence was measured by a Fluoroskan Ascent FL Microplate Fluorometer (Thermo Scientific). After background subtraction, we calculated the cell viability as the percentage change relative to the control cells.
Drug combination studies
Drug combination treatments were designed according to the Chou-Talalay equation, which accounts for both the potency (median inhibitory concentration) and the shape of the dose-effect curve, and the combination index (CI) was determined using CompuSyn software (ComboSyn, Inc.). CI < 1 indicates synergism (30).
In vivo tumorigenesis assay
All animal studies were conducted in compliance with animal protocols approved by the MD Anderson Cancer Center Institutional Animal Care and Use Committee. Six-week-old female athymic nu/nu mice (NCI, average weight 25 g) were used for all in vivo experiments. MDA-MB-231 or BT-549 cells (5 × 106) were mixed with BD Matrigel Basement Membrane Matrix (Becton Dickinson) and injected orthotopically into the lower left mammary fat pad of each mouse to establish xenografts. After the tumor volumes reached 50 mm3, the mice were randomized to groups treated daily by oral gavage with vehicle control, everolimus (EVE, 3 mg/kg) (31), BMN673 (BMN, 0.333 mg/kg) (32), or a combination of both agents. The MDA-MB-231 groups (n = 10 per group) were treated for 6 weeks, and the BT-549 groups (n = 8 per group) were treated for 8 weeks. Body weights were monitored, and the perpendicular diameters of each tumor were measured twice a week with a digital caliper. Tumor volumes were calculated with the following formula: (length×width2)/2.
Microarray analysis, cDNA synthesis, and reverse-transcription PCR
Microarray analysis was conducted as described previously (12). This analysis was used to search for genes that were differentially expressed between cells treated with DMSO and those treated with the mTOR inhibitor everolimus. Specifically, MDA-MB-231 and BT-549 cells were seeded in 6-well plates and treated with DMSO or everolimus for 48 hours. Total RNA from the cells was extracted with a mirVana miRNA isolation kit (Illumina, AM156). Complementary RNA was generated using a TotalPrep RNA amplification kit (Illumina, AMIL1791). Complementary RNA (750 ng) was loaded onto a HumanHT-12 v4 Expression BeadChip (Illumina, BD-103-0204), and hybridization and labeling of streptavidin-Cy3 was performed using the manufacturer’s procedure. The raw array data was processed using quantile normalization and log2 transformation with BRB-ArrayTools software (Biometric Research Branch, National Cancer Institute) for further analysis. A random-variance t-test was used to identify genes that were differentially expressed in the DMSO and mTOR inhibitor everolimus-treated cells. Gene expression differences were considered significant if P < 0.005. Venn diagram was created by GeneSpring GX software (Agilent Technologies, Version 12.6) and Gene set enrichment analysis was performed using the Ingenuity Pathway Analysis program (version 12710793). Raw data has been submitted to the GEO database (http://www.ncbi.nlm.nih.gov/geo/). Reverse transcription was conducted using an iScript cDNA Synthesis kit (Bio-Rad) according to the manufacturer’s instructions. Real-time PCR was conducted on the Mastercycler ep realplex (Eppendorf) using SYBR Green Real-Time PCR Master Mix (Life Technologies). Human SUV39H1 expression was quantified in real time and was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). SUV39H1 protein expression was quantified by Western blotting. Total proteins were extracted for immunoblotting as previously described (33). Vinculin was used as an internal control, and p-4EBP1 (Ser65) was used to demonstrate the inhibition of mTOR signaling.
RNA interference
For transient transfection, human SUV39H1 siRNA was purchased from Thermo Scientific (SMARTpool, ON-TARGETplus), and the SUV39H1 target sequences were CUAAGAAGCGGGUCCGUAU (J-009604-07), GGUGAAAUGGCGUGGAUAU (J-009604-08), UCGAGUACCUGUGCGAUUA (J-009604-09), and CAAAUCGUGUGGUACAGAA (J-009604-10). Human BRCA1 siRNA was purchased from Thermo Scientific (SMARTpool, ON-TARGETplus), and the BRCA1 target sequence was CAACAUGCCCACAGAUCAA (J-003461-09), CCAAAGCGAGCAAGAGAAU (J-003461-10), UGAUAAAGCUCCAGCAGGA (J-003461-11), and GAAGGAGCUUUCAUCAUUC (J-003461-12). All siRNAs were transfected with oligofectamine for 48-72 hours for further analysis.
Statistical analysis
The statistical significance of differences between groups in the in vitro experiments was determined by Student t-test. Two-way analysis of variance was used for statistical analysis for all in vivo experiments. P < 0.05 was considered as statistical significant.
Results
mTOR inhibitors suppress HR repair
Previously, we used an established HR-defect gene signature and the Connectivity Map to predict compounds that would potentially inhibit HR repair (12). We proceeded to investigate the effects of mTOR inhibitor, one of the top compounds screened in terms of inducing HR-defect gene expression profiles, in regulating HR in BRCA-proficient TNBC. It is well known that ionizing radiation (IR)-induced DSBs result in the formation of foci of RAD51, a key DNA recombinase that promotes homologous pairing and strand exchange in HR repair (34), and that RAD51 foci formation has been commonly recognized as a marker of HR competence (35). To determine whether mTOR inhibitors affect HR repair in BRCA-proficient TNBC, we treated MDA-MB-231 cells, a BRCA1/2 wild-type TNBC cell line, with two mTOR inhibitors with distinct mechanisms of action, including allosteric mTOR inhibitor everolimus (EVE) (36) and catalytic mTOR inhibitor KU-0063794 (KU) (37), and quantified formation of IR-induced RAD51 foci. We found that both EVE and KU significantly decreased IR-induced RAD51 foci formation (Fig. 1A and B), suggesting impaired HR repair. It has been reported that mTOR inhibition is associated with G1 cell cycle arrest (38). As HR repair occurs predominantly in S/G2 phase, when a sister chromatid is available as the template (7), it is important to clarify whether the observed HR defect in response to mTOR inhibitors is cell-cycle dependent. We observed that the cell-cycle distribution in EVE- or KU-treated cells did not change dramatically (Fig. 1B, lower panel), which indicated that mTOR inhibitors directly suppressed HR repair in BRCA-proficient TNBC cells. Interestingly, we found no apparent inhibition of IR-induced RAD51 foci formation or altered cell-cycle distribution in a non-transformed breast epithelial cell line MCF-12A cells (Supplementary Fig. S1A and B), indicating that HR repair efficiency in normal cells is left intact after mTOR inhibitors treatment.
Figure 1.
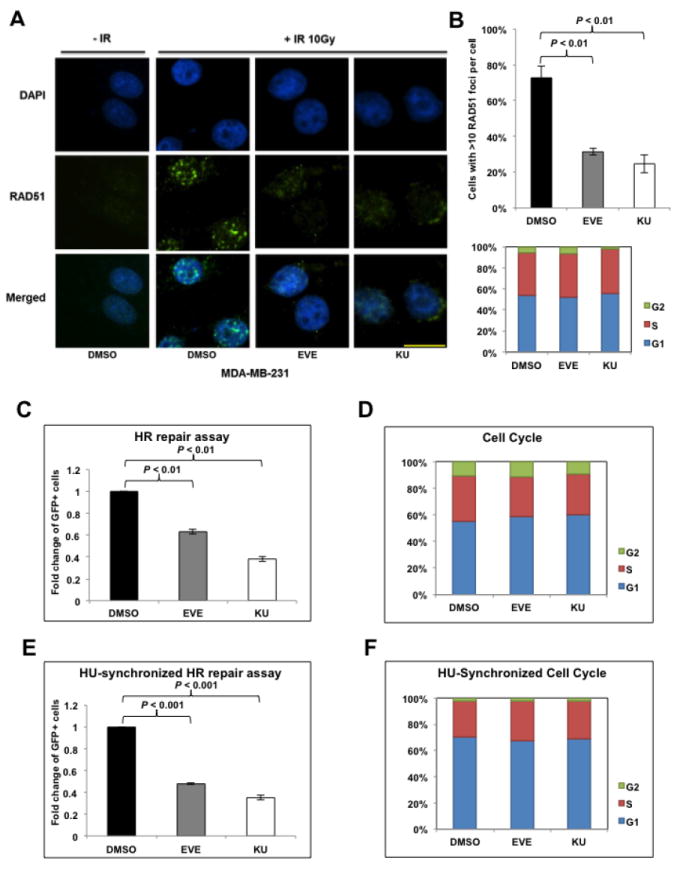
mTOR inhibitors suppress homologous recombination (HR) repair.
A. Microscopic analysis of MDA-MB-231 cells pretreated with dimethyl sulfoxide (DMSO, control) or mTOR inhibitors everolimus (EVE, 10 μM) or KU-0063794 (KU, 10 μM). DAPI was used for visualization of the nucleus. Images are representative of three independent experiments. Scale bar, 10 μm. B. Percentage of MDA-MB-231 cells with more than 10 RAD51 foci per cell in at least 50 cells for each treatment (upper panel). Means±SD (error bars) of three experiments are shown. P < 0.01 (EVE vs DMSO; KU vs DMSO). Lower panel demonstrates the cell-cycle distribution in MDA-MB-231 cells pretreated with EVE or KU. C. U2OS-DR-GFP cells treated with EVE (10 μM) or KU (10 μM) were subjected to HR repair assay. Each value is relative to the percentage of GFP-positive cells in pCBASceI-transfected control cells. Results are shown as means±SD from three independent experiments. P < 0.01 (EVE vs DMSO; KU vs DMSO). D. Cell-cycle analysis results for Figure 1C. E, U2OS-DR-GFP cells were treated with EVE (10 μM) or KU (10 μM) after pCBASceI transfection and were then treated with hydroxyurea (HU, 2 mM) to synchronize the cell cycle for 16 hours before the HR repair analysis. Results are shown as means±SD from three independent experiments. P < 0.01 (EVE vs DMSO; KU vs DMSO). F. Cell-cycle analysis results for Figure 1E.
To further confirm this observation, we analyzed the effect of mTOR inhibitors on HR repair using a direct HR repair assay, which has been previously described (39). In this system, a single copy of the HR reporter DR-GFP is randomly incorporated into U2OS cells, a human osteosarcoma cell line that is frequently used in studies of DNA damage response and repair. Transient expression of pCBASceI plasmid leads to a DSB in the upstream GFP gene and the ability of cells to repair these DSBs through HR can be quantified by the percentage of GFP-positive U2OS cells through flow cytometry. Our results showed that the mTOR inhibitors EVE or KU significantly reduced HR repair efficiency, as shown by the decreased percentage of GFP-positive U2OS cells (Fig. 1C). Additionally, we found no apparent alteration of the cell-cycle distribution (Fig. 1D) after mTOR inhibitors treatment in these HR repair model cells. Indeed, EVE or KU substantially decreased HR repair (Fig. 1E) even after hydroxyurea (HU) treatment, which is commonly used for cell-cycle synchronization (Fig. 1F) (40). Therefore, consistent with the prediction by our HR-defect gene signature and the Connectivity Map, our results demonstrated that mTOR inhibitors suppressed HR repair in BRCA-proficient TNBC cells.
mTOR inhibitors synergize with PARP inhibitors in vitro
It is well recognized that in the absence of HR repair via BRCA-dependent mechanisms, the loss of BER through PARP inhibition will lead to catastrophic genomic instability and cell death (41). Since mTOR inhibitors suppressed HR repair, we hypothesized that mTOR inhibitors would subsequently enhance the cytotoxicity of PARP inhibitors in BRCA-proficient TNBCs. We then evaluated the in vitro activity of two mTOR inhibitors, EVE and KU, in BRCA-proficient TNBC cells treated with one of two PARP inhibitors in clinical trials for breast cancer therapy, talazoparib (BMN673, BMN, BioMarin) and olaparib (AZD2281, AZD, Astrazeneca). Colony formation assay results showed that mTOR inhibitors significantly increased the sensitivity of both MDA-MB-231 (Fig. 2A, upper panel and B, upper panel) and BT-549 cells to PARP inhibitors (Fig. 2A, middle panel and B, lower panel). More importantly, compared with the single agent treatment, combinations of mTOR inhibitors and PARP inhibitors did not further inhibit colony formation in MCF-12A cells (Fig. 2A, lower panel), suggesting that mTOR/PARP inhibitor combination therapy may specifically target BRCA-proficient TNBC cells and spare normal cells.
Figure 2.
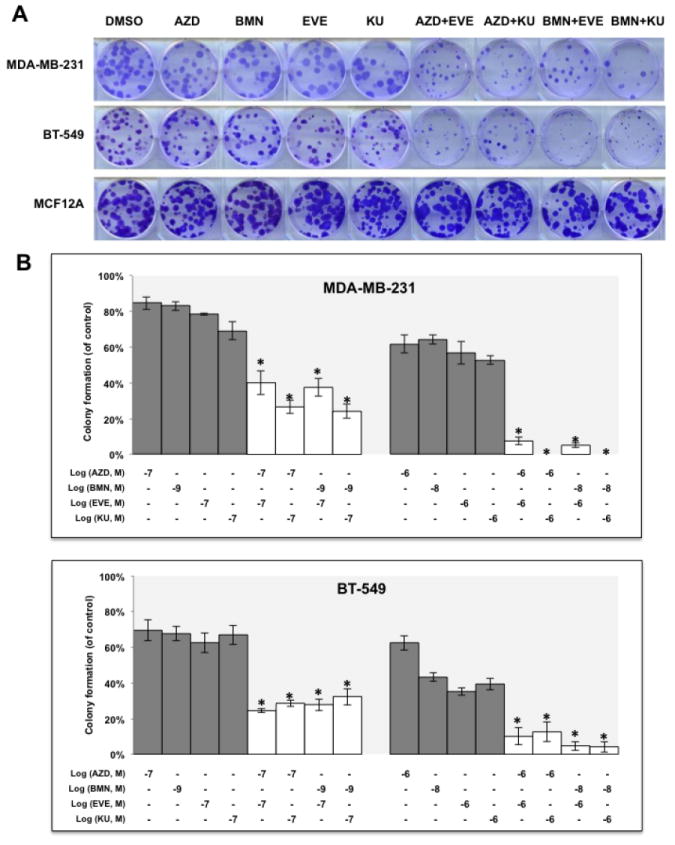
mTOR inhibitors synergize with PARP inhibitors in BRCA-proficient TNBCs in vitro.
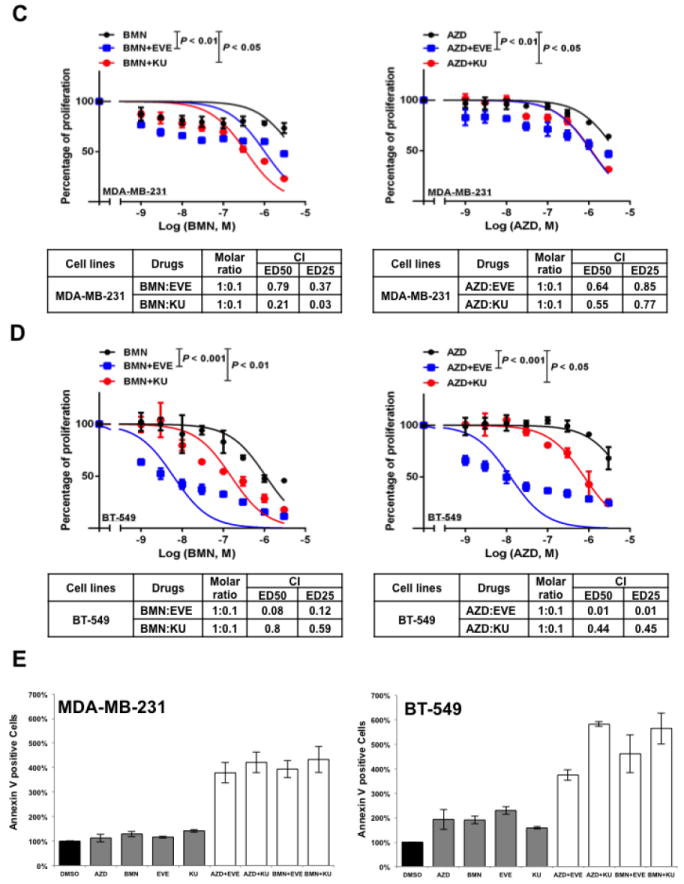
A. Colony formation assay results for MDA-MB-231 and BT-549 cells treated with DMSO, mTOR inhibitor alone (EVE, 10-7 M; KU, 10-7 M), PARP inhibitor alone (AZD, 10-7 M; BMN, 10-9 M) or the indicated combinations of agents. Images are representative of at least three independent experiments. B. Representative results of the quantification of the colonies treated with the indicated concentrations of mTOR inhibitors and PARP inhibitors as shown in Fig 2A. Values are relative to those of DMSO-treated cells and represent the mean±SD from three independent experiments. *P < 0.05 (combination vs single-agent treatment). C. Cell proliferation assay of MDA-MB-231 cells treated with single agents or combinations of PARP inhibitor BMN (left panel) or AZD (right panel) with mTOR inhibitors EVE or KU. Each value is relative to the value of cells treated with DMSO (control). Results are shown as mean±SD from three independent experiments. The combination index (CI) values are listed at the bottom. ED25 and ED50 represent 25% and 50% effective dose, respectively. D. Cell proliferation assay of BT-549 cells treated with single agents or combinations of BMN (left panel) or AZD (right panel) with EVE or KU. Each value is relative to the value of cells treated with DMSO (control). Results are shown as means±SD from three independent experiments. The CI values are listed at the bottom. E. Apoptosis analysis of MDA-MB-231 (left panel) and BT-549 cells (right panel) treated with the indicated single agent (AZD, 2 μM; BMN, 2 μM; EVE, 2 μM; KU, 2 μM) or different combinations of PARP inhibitors and mTOR inhibitors. Results are shown as means±SD from three independent experiments.
Next, we performed a proliferation assay to further determine whether mTOR inhibitors synergize with PARP inhibitors in vitro. In MDA-MB-231 cells, combined treatment of mTOR inhibitors significantly decreased the IC50 for PARP inhibitors and a marked synergy was observed between mTOR inhibitors and PARP inhibitors based on the calculated CI values (30) (Fig. 2C, left panel, mTOR inhibitors combined with BMN; right panel, mTOR inhibitors combined with AZD). Similar synergistic effect was observed in additional BRCA-proficient TNBC cells, BT-549 (Fig. 2D, left panel, mTOR inhibitors combined with BMN; right panel, mTOR inhibitors combined with AZD). Additionally, combinations of mTOR inhibitors and PARP inhibitors markedly increased the percentage of apoptotic cells in both MDA-MB-231 (Fig. 2E, left panel) and BT-549 cells (Fig. 2E, right panel) when compared with cells treated with single agent alone.
mTOR inhibitor sensitizes PARP inhibitor in TNBC xenografts
We further assessed the effects of everolimus and BMN673 and their combination on tumor growth in BRCA-proficient TNBC in vivo. EVE or BMN as a single agent delayed tumor growth in MDA-MB-231 xenografts. Nonetheless, the BMN+EVE combination was superior to BMN (P = 0.0143) or EVE alone (P < 0.001) in reducing tumor growth (Fig. 3A and B). Additionally, in BT-549 xenografts, the tumor volume in the BMN+EVE combination group was also significantly decreased compared to that in the BMN (P < 0.001) or EVE (P < 0.001) single agent group (Fig. 3D and E). Most importantly, the combination induced tumor regressions in MDA-MB-231 model. No body weight loss was detected during the 6 weeks (MDA-MB-231 xenografts, Fig. 3C) or 8 weeks (BT-549 xenografts, Fig. 3F) of treatment, indicating that consistent with in vitro findings (Fig. 2A, lower panel), combination therapy of mTOR inhibitor and PARP inhibitor may particularly target cancer cells. Taken together, the approach of mTOR inhibitors and PARP inhibitors combination therapy may specifically increase the therapeutic efficacy in BRCA-proficient TNBC without increasing the toxicity to normal cells.
Figure 3.
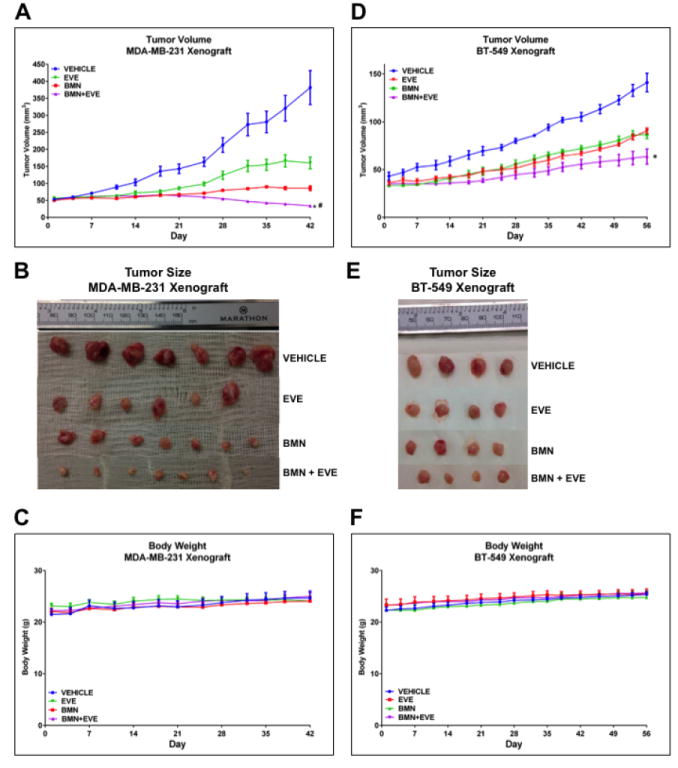
mTOR inhibitors sensitize BRCA-proficient TNBC xenografts to PARP inhibitors.
A-C. Female nude mice (10 per group) with orthotopically implanted MDA-MB-231 xenografts were treated by oral gavage with vehicle control, everolimus (EVE, 3 mg/kg), BMN 673 (BMN, 0.333 mg/kg) or a combination of both agents daily for 6 weeks. A. Tumor volumes were determined on the indicated days of treatment. Results are shown as means±SEM. Two-way analysis of variance was used to determine statistical significance of differences between groups: *P < 0.001 (BMN+EVE vs EVE); #P = 0.0143 (BMN+EVE vs BMN). B. Representative pictures of orthotopically implanted tumor tissue from each group at the time of study termination (day 42). C. Body weight-time curve in MDA-MB-231 xenograft is shown. D–F. Female nude mice (8 per group) with orthotopically implanted BT-549 xenografts were treated by oral gavage with vehicle control, EVE (3 mg/kg), BMN (0.333 mg/kg), or a combination of both agents daily for 8 weeks. D. Tumor volumes were determined on the indicated days of treatment. Results are shown as means±SEM. Two-way analysis of variance was used to determine statistical significance of differences between groups: *P < 0.001 (BMN+EVE vs EVE; BMN+EVE vs BMN). E. Pictures of orthotopically implanted tumor tissue from each group at the time of study termination (day 56). F. Body weight-time curve in BT-549 xenograft is shown.
mTOR inhibitors suppress SUV39H1 expression
Next, we searched for the underlying mechanisms that are responsible for mTOR inhibitors-induced suppression of HR repair and consequent sensitization to PARP inhibitors in BRCA-proficient TNBC. We found that mTOR inhibitors did not affect the mRNA or protein expression of key HR-related factors, including BRCA1, RAD51 and RPA2 (Supplementary Fig. 2A and B). To obtain a broad molecular understanding of this process, we applied a genome-wide expression profiling approach to systematically analyze cellular transcriptome alterations induced by mTOR inhibitor. Among the altered genes in response to mTOR inhibitor that are implicated in DNA replication, recombination, and repair by Ingenuity Pathway Analysis (IPA), we found that SUV39H1 was most significantly decreased in both MDA-MB-231 and BT-549 cells (Supplementary Fig. 3). SUV39H1 is a specific H3K9 histone methyltransferase (42) and has been implicated in DSB repair (43,44). Loss of SUV39H1 decreases DSB repair mediated by HR and increases cell sensitivity to PARP inhibition (45). Hence, SUV39H1 might be a key molecule involved in the suppression of HR repair by mTOR inhibitors. Consistent with our microarray data, we found that MDA-MB-231 (Fig. 4A) and BT-549 (Fig. 4B) cells exposed to EVE or KU had significantly decreased SUV39H1 mRNA than DMSO-treated control cells (P < 0.001). In addition, markedly reduced SUV39H1 protein levels were observed in response to EVE or KU treatment when compared to DMSO treatment (Fig. 4C). The level of p-4EBP1 (Ser65) served as a marker for mTOR signaling activity. More importantly, cells lacking SUV39H1 demonstrated impaired HR repair (Fig. 4D) without changes in cell-cycle distribution (Fig. 4E). In the same experiment, BRCA1 knockdown was used as a positive control for defect in HR repair. Furthermore, recruitment of RAD51 to DSBs was significantly reduced following SUV39H1 loss in BRCA-proficient TNBC MDA-MB-231 cells (Fig. 4F). Consistently, depletion of SUV39H1 increased sensitivity of MDA-MB-231 cells to PARP inhibitors (Fig. 4G, left panel, AZD; right panel, BMN).
Figure 4.
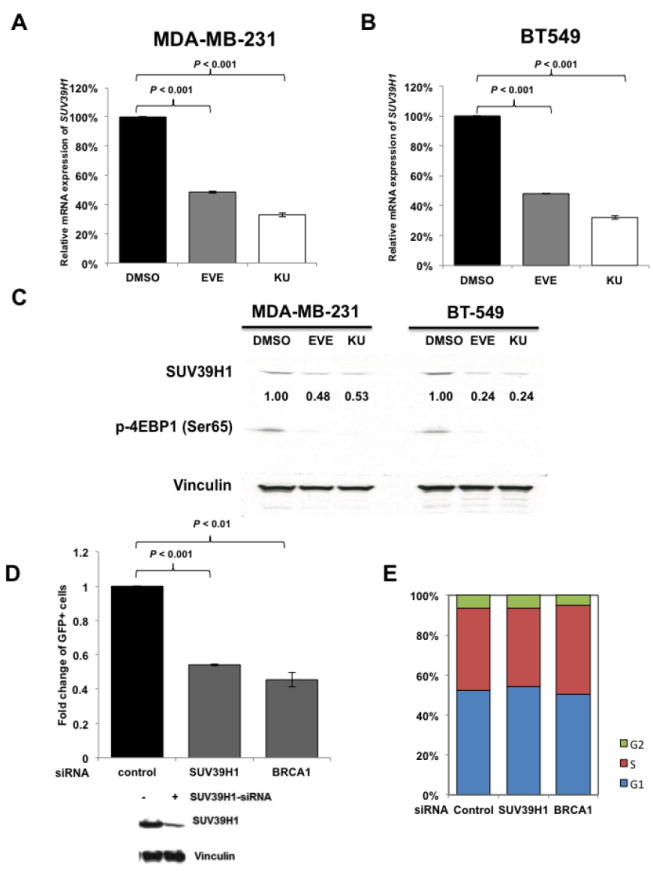
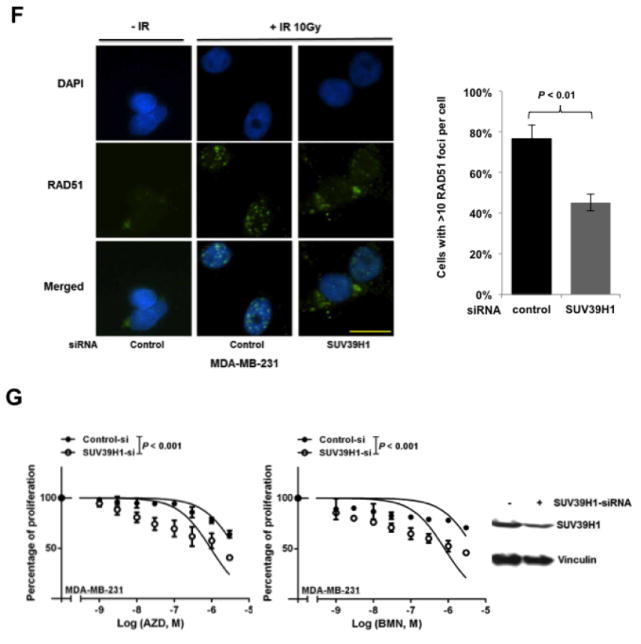
mTOR inhibitors suppress SUV39H1 expression.
A–B. MDA-MB-231 (A) and BT-549 (B) cells were treated with EVE (10 μM) or KU (10 μM) for 48 hours. The total RNA was extracted to detect SUV39H1 mRNA by real-time reverse transcription PCR (RT-PCR) with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an internal control. Fold change of SUV39H1 mRNA was plotted. Results are shown as means±SD from three independent experiments. P values are shown in the figures. C. MDA-MB-231 and BT-549 cells were treated with EVE (10 μM) or KU (10 μM) for 48 hours. Total proteins were extracted for immunoblotting. Vinculin was used as an internal control and p-4EBP1 (Ser65) was used to demonstrate mTOR pathway inhibition. The ratio of SUV39H1 protein expression relative to vinculin is listed below the blot. D. U2OS-DR-GFP cells were transfected with control siRNA, BRCA1 siRNA or SUV39H1 siRNA and were subjected to HR repair assay. Each value is relative to the percentage of GFP-positive cells in pCBASceI-transfected control cells. Results are shown as means±SD from three independent experiments. P < 0.001 (SUV39H1 siRNA vs control siRNA) and P < 0.01 (BRCA1 siRNA vs control siRNA). Western blots to demonstrate SUV39H1 knockdown are presented below. E. Cell-cycle analysis results for Figure 4D. F. (left panel) Microscopy analysis of MDA-MB-231 cells transiently transfected with control siRNA, or SUV39H1 siRNA for 48 hours. DAPI was used for visualization of the nucleus. Images are representative of three independent experiments. Scale bar, 10 μm. (Right panel) Percentage of MDA-MB-231 cells with more than 10 RAD51 foci per cell in at least 50 cells for each treatment. Means±SD (error bars) of three experiments are shown. P < 0.01 (SUV39H1 siRNA vs control siRNA). G. Control or SUV39H1 siRNA was transiently transfected into MDA-MB-231 cells. PARP inhibitors AZD or BMN was added the next day, and cell proliferation was evaluated with PrestoBlue Cell Viability Reagent 72 hours later. Western blots to demonstrate SUV39H1 knockdown are presented to the right.
SUV39H1 overexpression partially rescues HR repair suppressed by mTOR inhibitors and blocks synergism between mTOR inhibitors and PARP inhibitors
To confirm that SUV39H1 plays an essential role in the suppression of HR repair by mTOR inhibitors, we performed rescue assays to evaluate HR repair function and sensitivity to PARP inhibitors after restoration of SUV39H1. We found that in U2OS-DR-GFP cells, overexpression of SUV39H1 significantly increased HR repair efficiency that had been reduced by mTOR inhibitors EVE or KU (Fig. 5A). Furthermore, overexpression of SUV39H1 in MDA-MB-231 and BT-549 cells treated with BMN alone did not change cell proliferation; however, when these cells were treated with the combination of BMN and KU (Fig. 5B) or BMN and EVE (Fig. 5C), cells with SUV39H1 overexpression were less sensitive to the drug treatments than the vector-transfected cells. This indicated that SUV39H1 overexpression suppressed, at least partially, the synergy between PARP inhibitors and mTOR inhibitors in BRCA-proficient TNBC cells. Taken together, these data strongly support the hypothesis that mTOR inhibitors suppress HR repair and synergize with PARP inhibitors in BRCA-proficient TNBCs by regulating SUV39H1 expression.
Figure 5.
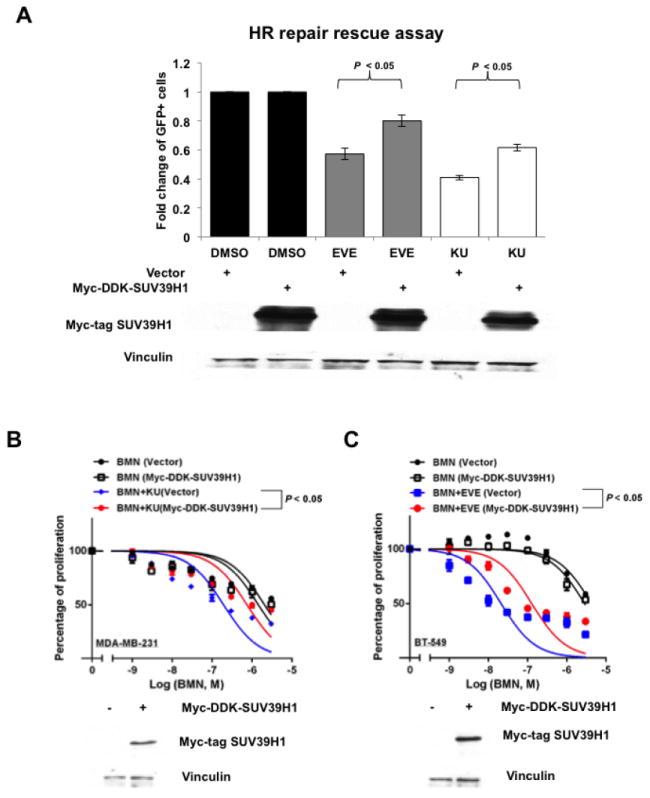
SUV39H1 overexpression rescues HR repair suppressed by mTOR inhibitors and blocks the synergy between PARP inhibitors and mTOR inhibitors.
A. Myc-DDK-SUV39H1 plasmid was transiently transfected into U2OS-DR-GFP cells to induce SUV39H1 overexpression. Six hours after transfection of pCBASceI plasmid, mTOR inhibitor EVE (10 μM) or KU (10 μM) was added. The HR repair assay was performed after 48 hours. (Upper panel) Each value is relative to the percentage of GFP-positive cells in pCBASceI-transfected control cells. Results are shown as means±SD from three independent experiments. Lower panel shows the overexpression of SUV39H1 proteins (Myc-Tag mAb) in U2OS-DR-GFP cells. B–C. Myc-DDK-SUV39H1 plasmid was transiently transfected into MDA-MB-231 (B) or BT-549 (C) cells for SUV39H1 overexpression. BMN 673 (BMN) or combinations of BMN and mTOR inhibitors (EVE or KU) were added the next day, and cell proliferation was evaluated with PrestoBlue Cell Viability Reagent 72 hours later. Western blots to demonstrate overexpression of SUV39H1 (Myc-Tag mAb) are at the bottom of each panel.
Discussion
The search for combination therapies that would result in impaired HR with subsequent sensitization to PARP inhibitors in cells with competent HR repair has been an area of great research interest. To identify more precise biomarkers to predict patients’ response to PARP inhibitors, our group previously identified an HR defect gene signature to predict HR repair status and sensitivity to PARP inhibitors (12). Using the Connectivity Map as an effective drug discovery platform, we were able to predict small molecules that could potentially suppress HR repair and sensitize cells to PARP inhibitors. We found that mTOR inhibitors were at the top of the list in terms of inducing HR-defect gene expression profiles. Our current study demonstrated that, as the Connectivity Map predicted, mTOR inhibitors significantly impaired RAD51 foci formation in response to DSBs in BRCA-proficient TNBC cells. Additionally, using U2OS cells with stable DR-GFP as an HR repair model, we found that mTOR inhibitors directly decreased HR repair efficiency, even after cell-cycle synchronization. Notably, our results also showed that mTOR inhibitors synergized with PARP inhibitors in BRCA-proficient TNBC cells in vitro and also significantly enhanced the cytotoxicity of PARP inhibitors in BRCA-proficient TNBC xenografts in vivo. Our findings suggest that mTOR inhibitors can suppress HR repair in BRCA-proficient TNBCs, therefore making these cancers vulnerable to PARP inhibitors. Importantly, although comparatively high concentrations of mTOR inhibitors are used in the current study, our observations are biological relevant, since treatment of mTOR inhibitors do not affect HR repair (Supplementary Fig. 1) and their combinations with PARP inhibitors do not further reduce cell viability (Fig. 2A) in non-transformed breast epithelial cells. In addition, mTOR inhibitor and PARP inhibitor combination is well tolerated in mice (Fig. 3C and F). All these preclinical evidences provide rationales for application of dual mTOR and PARP inhibitions in BRCA-proficient TNBC cells. Further, mTOR inhibitors could potentially extend the clinical application of PARP inhibitors to a wider spectrum of cancers. Of note, PI3K inhibitors, histone deacetylase (HDAC) inhibitors, and Hsp90 inhibitors were also on our list with regard to impairing HR repair (12). This is indeed consistent with several recent studies. For example, two research groups independently reported (15,16) that PI3K inhibition compromised HR repair, which led to down-regulation of BRCA1, decreased RAD51 foci formation, and decreased sensitization of BRCA1/2-wild-type TNBC cells or TNBC patient-derived primary tumor xenografts to PARP inhibitors. Additionally, PI3K-mediated down-regulation of BRCA1 has been found to be ERK-dependent, through the involvement of the ETS1 transcription factor (15). It is also reported that Hsp90 inhibition suppressed BRCA1-dependent HR repair (46) and altered the sensitivity of BRCA1-mutant cells to PARP inhibitors (47). Another study found that HDAC inhibitor treatment depleted BRCA1 and other HR proteins and synergized with PARP inhibitors in human TNBC cells (48). Together, our studies hold great promise to expand the clinical treatment options for TNBCs in the near future.
It has been reported that PI3K inhibition down-regulates BRCA1 in TNBCs (15). In our study, mTOR inhibitors everolimus and KU-0063794 did not affect the expression of major HR-related factors, including BRCA1, RAD51 and RPA2 (Supplementary Fig. 2). Our exploration of the molecular mechanisms underlying suppression of HR by mTOR inhibitors through systematic analysis of cellular transcriptome alterations induced by these agents revealed down-regulation of SUV39H1 expression in two BRCA-proficient TNBC cell lines. SUV39H1 is the most studied methyltransferase of lysine 9 of histone 3 (H3K9) (27). It forms a multimeric complex with other histone H3K9 methyltransferases, and is likely involved in pericentric heterochromatin formation and regulation of gene expression. SUV39H1 is rapidly loaded onto the chromatin at DSBs and methylates H3K9. The resulting H3K9me3 activates Tip60 histone acetyltransferase, which in turn acetylates ataxia telangiectasia mutated kinase (ATM) and promotes ATM-dependent phosphorylation of DSB repair proteins (45). Inhibition of SUV39H1 dramatically decreases HR repair efficiency and the recruitment of BRCA1 and replication protein A to DSBs (45). In this study, we found that SUV39H1 inhibition impaired RAD51 foci formation in MDA-MB-231 cells (Fig. 4F) and reduced HR repair in the U2OS-DR-GFP HR repair model (Fig. 4D). Importantly, we showed that the overexpression of SUV39H1 partially restored HR repair efficiency that had been suppressed by mTOR inhibitors, indicating that the regulation of SUV39H1 by the mTOR pathway contributes to HR repair. Additionally, exogenous expression of SUV39H1 suppressed the synergy between PARP inhibitors and mTOR inhibitors, which provides a rational explanation of how mTOR inhibitors modulate HR repair in BRCA-proficient TNBCs. SUV39H1 is known to be specifically methylated by SET domain-containing protein 7 (SET7/9), and this methylation subsequently facilitates genome instability and eventually inhibits cell proliferation (49). Interestingly, we showed that the decrease of SUV39H1 protein expression was comparable to the decrease of SUV39H1 mRNA expression in both MDA-MB-231 and BT-549 cells (Fig 4A-C), indicating that mTOR signaling might regulate SUV39H1 predominantly at the transcriptional level, which need to be further investigated for a thorough understanding in our future work. Also, whether these observations apply to TNBC with BRCA1/2 mutations or other breast cancer subgroups requires further investigations.
It is well known that TNBC is a highly heterogeneous disease with variable prognosis, which has added increased complexity to the development of personalized therapeutics for TNBCs. In our current study, we have focused on exploring combination therapies that would expand the spectrum of TNBC patients who would benefit from PARP inhibitors, especially those with intact HR. Therefore, we have used two TNBC cell line models, MDA-MB-231 and BT-549, to represent specific groups of TNBC that are BRCA1/2 wild-type (BRCA-proficient) and are categorized as HR-intact through our HR-defect gene signature. However, it remains unknown whether or how heterogeneous HR activity would exist in individual TNBC tumors, which may be answered in the future by single cell transcriptome analysis. In addition, we have demonstrated the therapeutic benefit from the combination of mTOR inhibitors and PARP inhibitors on BRCA-proficient TNBC using both in vitro cultured cells and in vivo mouse xenograft model, which are two standard models in cancer biology. In the future, we will further verify these intriguing results on patient derived xenograft (PDX) model, which more closely resemble the original tumors (50), in order to promote novel and effective personalized clinical trials for TNBC patients.
In summary, our results demonstrate that mTOR inhibitors suppress HR repair, and further show that mTOR inhibitors dramatically enhance the cytotoxicity of PARP inhibitors in BRCA-proficient TNBCs in vitro and in vivo, without affecting HR repair or the proliferation of non-transformed breast epithelial cells. We have also revealed a novel mechanism of how mTOR signaling regulates HR repair and PARP inhibitor sensitivity in BRCA-proficient TNBC. These results present an effective approach for therapeutic combination of mTOR inhibitors with PARP inhibitors in BRCA-proficient TNBCs and might also benefit patients with other types of cancer.
Supplementary Material
Translational Relevance.
Novel targeted therapies are urgently needed for TNBC that have limited therapeutic options and poor prognosis. PARP inhibitors provide benefit for TNBC with BRCA1/BRCA2 mutations. In order to expand the spectrum of TNBC patients who would benefit from PARP inhibitors, especially those that are BRCA-proficient, rational combination therapies that lead to HR defects and consequently synergize with PARP inhibitors should be evaluated. Here, we report that mTOR inhibitors are highly effective in suppressing HR repair. In addition, combination treatment of BRCA-proficient TNBCs with mTOR inhibitors and PARP inhibitors synergistically inhibits proliferation in vitro and exhibits enhanced efficacy in vivo. Finally, we found that mTOR inhibitors suppress SUV39H1, a novel mechanism by which they regulate HR repair and synergize with PARP inhibitors. These findings provide rationales for translation of the therapeutic combination of mTOR inhibitors with PARP inhibitors into clinical trials targeting BRCA-proficient TNBC patients, and potentially patients with tumors of other lineages.
Acknowledgments
Financial support:
This work was supported by The Department of Defense (DOD) Breast Cancer Research Program (BCRP) Era of Hope Scholar Award (W81XWH-10-1-0558; S.-Y. Lin).
Footnotes
Potential conflicts of interest:
No potential conflict of interest was disclosed.
References
- 1.Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer Registry. Cancer. 2007;109(9):1721–8. doi: 10.1002/cncr.22618. [DOI] [PubMed] [Google Scholar]
- 2.Crown J, O’Shaughnessy J, Gullo G. Emerging targeted therapies in triple-negative breast cancer. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2012;23(Suppl 6):vi56–65. doi: 10.1093/annonc/mds196. [DOI] [PubMed] [Google Scholar]
- 3.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. The New England journal of medicine. 2010;363(20):1938–48. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 4.Yap TA, Sandhu SK, Carden CP, de Bono JS. Poly(ADP-ribose) polymerase (PARP) inhibitors: Exploiting a synthetic lethal strategy in the clinic. CA: a cancer journal for clinicians. 2011;61(1):31–49. doi: 10.3322/caac.20095. [DOI] [PubMed] [Google Scholar]
- 5.Hartman AR, Kaldate RR, Sailer LM, Painter L, Grier CE, Endsley RR, et al. Prevalence of BRCA mutations in an unselected population of triple-negative breast cancer. Cancer. 2012;118(11):2787–95. doi: 10.1002/cncr.26576. [DOI] [PubMed] [Google Scholar]
- 6.Wong-Brown MW, Meldrum CJ, Carpenter JE, Clarke CL, Narod SA, Jakubowska A, et al. Prevalence of BRCA1 and BRCA2 germline mutations in patients with triple-negative breast cancer. Breast cancer research and treatment. 2015;150(1):71–80. doi: 10.1007/s10549-015-3293-7. [DOI] [PubMed] [Google Scholar]
- 7.Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell research. 2008;18(1):99–113. doi: 10.1038/cr.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dantzer F, Schreiber V, Niedergang C, Trucco C, Flatter E, De La Rubia G, et al. Involvement of poly(ADP-ribose) polymerase in base excision repair. Biochimie. 1999;81(1-2):69–75. doi: 10.1016/s0300-9084(99)80040-6. [DOI] [PubMed] [Google Scholar]
- 9.Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26(22):3785–90. doi: 10.1200/JCO.2008.16.0812. [DOI] [PubMed] [Google Scholar]
- 10.Johnson N, Li YC, Walton ZE, Cheng KA, Li D, Rodig SJ, et al. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nature medicine. 2011;17(7):875–82. doi: 10.1038/nm.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mayer IA, Abramson VG, Lehmann BD, Pietenpol JA. New strategies for triple-negative breast cancer--deciphering the heterogeneity. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20(4):782–90. doi: 10.1158/1078-0432.CCR-13-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peng G, Chun-Jen Lin C, Mo W, Dai H, Park YY, Kim SM, et al. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nat Commun. 2014;5:3361. doi: 10.1038/ncomms4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313(5795):1929–35. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 14.Qu XA, Rajpal DK. Applications of Connectivity Map in drug discovery and development. Drug discovery today. 2012;17(23-24):1289–98. doi: 10.1016/j.drudis.2012.07.017. [DOI] [PubMed] [Google Scholar]
- 15.Ibrahim YH, Garcia-Garcia C, Serra V, He L, Torres-Lockhart K, Prat A, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer discovery. 2012;2(11):1036–47. doi: 10.1158/2159-8290.CD-11-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Juvekar A, Burga LN, Hu H, Lunsford EP, Ibrahim YH, Balmana J, et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer discovery. 2012;2(11):1048–63. doi: 10.1158/2159-8290.CD-11-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Populo H, Lopes JM, Soares P. The mTOR signalling pathway in human cancer. International journal of molecular sciences. 2012;13(2):1886–918. doi: 10.3390/ijms13021886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walsh S, Flanagan L, Quinn C, Evoy D, McDermott EW, Pierce A, et al. mTOR in breast cancer: differential expression in triple-negative and non-triple-negative tumors. Breast. 2012;21(2):178–82. doi: 10.1016/j.breast.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 19.Montero JC, Esparis-Ogando A, Re-Louhau MF, Seoane S, Abad M, Calero R, et al. Active kinase profiling, genetic and pharmacological data define mTOR as an important common target in triple-negative breast cancer. Oncogene. 2014;33(2):148–56. doi: 10.1038/onc.2012.572. [DOI] [PubMed] [Google Scholar]
- 20.Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nature reviews Cancer. 2004;4(5):335–48. doi: 10.1038/nrc1362. [DOI] [PubMed] [Google Scholar]
- 21.Bandhakavi S, Kim YM, Ro SH, Xie H, Onsongo G, Jun CB, et al. Quantitative nuclear proteomics identifies mTOR regulation of DNA damage response. Molecular & cellular proteomics : MCP. 2010;9(2):403–14. doi: 10.1074/mcp.M900326-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen H, Ma Z, Vanderwaal RP, Feng Z, Gonzalez-Suarez I, Wang S, et al. The mTOR inhibitor rapamycin suppresses DNA double-strand break repair. Radiation research. 2011;175(2):214–24. doi: 10.1667/rr2323.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen C, Lancaster CS, Shi B, Guo H, Thimmaiah P, Bjornsti MA. TOR signaling is a determinant of cell survival in response to DNA damage. Molecular and cellular biology. 2007;27(20):7007–17. doi: 10.1128/MCB.00290-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo F, Li J, Du W, Zhang S, O’Connor M, Thomas G, et al. mTOR regulates DNA damage response through NF-kappaB-mediated FANCD2 pathway in hematopoietic cells. Leukemia. 2013;27(10):2040–6. doi: 10.1038/leu.2013.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo F, Li J, Zhang S, Du W, Amarachintha S, Sipple J, et al. mTOR kinase inhibitor sensitizes T-cell lymphoblastic leukemia for chemotherapy-induced DNA damage via suppressing FANCD2 expression. Leukemia. 2014;28(1):203–6. doi: 10.1038/leu.2013.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen C, Oswald D, Phelps D, Cam H, Pelloski CE, Pang Q, et al. Regulation of FANCD2 by the mTOR pathway contributes to the resistance of cancer cells to DNA double-strand breaks. Cancer research. 2013;73(11):3393–401. doi: 10.1158/0008-5472.CAN-12-4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fritsch L, Robin P, Mathieu JR, Souidi M, Hinaux H, Rougeulle C, et al. A subset of the histone H3 lysine 9 methyltransferases Suv39h1, G9a, GLP, and SETDB1 participate in a multimeric complex. Molecular cell. 2010;37(1):46–56. doi: 10.1016/j.molcel.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 28.Hu R, Wang E, Peng G, Dai H, Lin SY. Zinc finger protein 668 interacts with Tip60 to promote H2AX acetylation after DNA damage. Cell Cycle. 2013;12(13):2033–41. doi: 10.4161/cc.25064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krishan A. Rapid flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J Cell Biol. 1975;66(1):188–93. doi: 10.1083/jcb.66.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58(3):621–81. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 31.Lane HA, Wood JM, McSheehy PM, Allegrini PR, Boulay A, Brueggen J, et al. mTOR inhibitor RAD001 (everolimus) has antiangiogenic/vascular properties distinct from a VEGFR tyrosine kinase inhibitor. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15(5):1612–22. doi: 10.1158/1078-0432.CCR-08-2057. [DOI] [PubMed] [Google Scholar]
- 32.Shen Y, Rehman FL, Feng Y, Boshuizen J, Bajrami I, Elliott R, et al. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(18):5003–15. doi: 10.1158/1078-0432.CCR-13-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peng Y, Dai H, Wang E, Lin CC, Mo W, Peng G, et al. TUSC4 functions as a tumor suppressor by regulating BRCA1 stability. Cancer research. 2015;75(2):378–86. doi: 10.1158/0008-5472.CAN-14-2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baumann P, West SC. Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends in biochemical sciences. 1998;23(7):247–51. doi: 10.1016/s0968-0004(98)01232-8. [DOI] [PubMed] [Google Scholar]
- 35.Graeser M, McCarthy A, Lord CJ, Savage K, Hills M, Salter J, et al. A marker of homologous recombination predicts pathologic complete response to neoadjuvant chemotherapy in primary breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16(24):6159–68. doi: 10.1158/1078-0432.CCR-10-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baselga J, Campone M, Piccart M, Burris HA, 3rd, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. The New England journal of medicine. 2012;366(6):520–9. doi: 10.1056/NEJMoa1109653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garcia-Martinez JM, Moran J, Clarke RG, Gray A, Cosulich SC, Chresta CM, et al. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR) The Biochemical journal. 2009;421(1):29–42. doi: 10.1042/BJ20090489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C, Blenis J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Molecular and cellular biology. 2004;24(1):200–16. doi: 10.1128/MCB.24.1.200-216.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peng G, Yim EK, Dai H, Jackson AP, Burgt I, Pan MR, et al. BRIT1/MCPH1 links chromatin remodelling to DNA damage response. Nature cell biology. 2009;11(7):865–72. doi: 10.1038/ncb1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosner M, Schipany K, Hengstschlager M. Merging high-quality biochemical fractionation with a refined flow cytometry approach to monitor nucleocytoplasmic protein expression throughout the unperturbed mammalian cell cycle. Nature protocols. 2013;8(3):602–26. doi: 10.1038/nprot.2013.011. [DOI] [PubMed] [Google Scholar]
- 41.Anders CK, Winer EP, Ford JM, Dent R, Silver DP, Sledge GW, et al. Poly(ADP-Ribose) polymerase inhibition: “targeted” therapy for triple-negative breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16(19):4702–10. doi: 10.1158/1078-0432.CCR-10-0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fuks F, Hurd PJ, Deplus R, Kouzarides T. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic acids research. 2003;31(9):2305–12. doi: 10.1093/nar/gkg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peters AH, O’Carroll D, Scherthan H, Mechtler K, Sauer S, Schofer C, et al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell. 2001;107(3):323–37. doi: 10.1016/s0092-8674(01)00542-6. [DOI] [PubMed] [Google Scholar]
- 44.Peng JC, Karpen GH. Heterochromatic genome stability requires regulators of histone H3 K9 methylation. PLoS genetics. 2009;5(3):e1000435. doi: 10.1371/journal.pgen.1000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ayrapetov MK, Gursoy-Yuzugullu O, Xu C, Xu Y, Price BD. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(25):9169–74. doi: 10.1073/pnas.1403565111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stecklein SR, Kumaraswamy E, Behbod F, Wang W, Chaguturu V, Harlan-Williams LM, et al. BRCA1 and HSP90 cooperate in homologous and non-homologous DNA double-strand-break repair and G2/M checkpoint activation. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(34):13650–5. doi: 10.1073/pnas.1203326109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson N, Johnson SF, Yao W, Li YC, Choi YE, Bernhardy AJ, et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(42):17041–6. doi: 10.1073/pnas.1305170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ha K, Fiskus W, Choi DS, Bhaskara S, Cerchietti L, Devaraj SG, et al. Histone deacetylase inhibitor treatment induces ‘BRCAness’ and synergistic lethality with PARP inhibitor and cisplatin against human triple negative breast cancer cells. Oncotarget. 2014;5(14):5637–50. doi: 10.18632/oncotarget.2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang D, Zhou J, Liu X, Lu D, Shen C, Du Y, et al. Methylation of SUV39H1 by SET7/9 results in heterochromatin relaxation and genome instability. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(14):5516–21. doi: 10.1073/pnas.1216596110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang X, Lewis MT. Establishment of Patient-Derived Xenograft (PDX) Models of Human Breast Cancer. Current protocols in mouse biology. 2013;3(1):21–9. doi: 10.1002/9780470942390.mo120140. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.