

Abstract
We recently reported that the transcription factor ARID3a is expressed in a subset of human hematopoietic progenitor stem cells in both healthy individuals and in patients with systemic lupus erythematosus. Numbers of ARID3a+ lupus hematopoietic stem progenitor cells were associated with increased production of autoreactive antibodies when those cells were introduced into humanized mouse models. Although ARID3a/Bright knockout mice died in utero, they exhibited decreased numbers of hematopoietic stem cells and erythrocytes, indicating ARID3a is functionally important for hematopoiesis in mice. To explore the requirement for ARID3a for normal human hematopoiesis, hematopoietic stem cell progenitors from human cord blood were subjected to both inhibition and over-expression of ARID3a in vitro. Inhibition of ARID3a resulted in decreased B lineage cell production accompanied by increases in cells with myeloid lineage markers. Over-expression of ARID3a inhibited both myeloid and erythroid differentiation. In addition, inhibition of ARID3a in hematopoietic stem cells resulted in altered expression of transcription factors associated with hematopoietic lineage decisions. These results suggest that appropriate regulation of ARID3a is critical for normal development of both myeloid and B lineage pathways.
Introduction
ARID3a is a member of a large family of ARID (A+T Rich Interaction Domain) proteins that bind to A+T rich DNA sequences. Members of this evolutionarily conserved family have been implicated in the control of a variety of processes, including embryonic development, chromatin remodeling, and cell cycle regulation (reviewed in (1–4)). Human ARID3a and the mouse orthologue, Bright (B cell regulator of immunoglobulin heavy chain transcription) bind to sequences 5′ of some IgH promoters and to the matrix attachment regions (MARs) that flank the intronic IgH enhancer (5–9), where, in association with Bruton’s tyrosine kinase (Btk) and the transcription factor II-I (TFII-I), they upregulate IgH transcription in activated B cells (10, 11). Additional studies with transgenic mice that over-expressed Bright/ARID3a indicated roles for this protein in marginal zone versus follicular B cell fate decisions, and as a contributing factor for production of autoantibodies (12, 13).
Although ARID3a expression in adults was originally thought to be limited to B lymphocyte lineage cells (reviewed in 14), it is clearly expressed in multiple fetal and embryonic tissues, as well as in adult hematopoietic stem cells (15–17). Knockouts of the Xenopus, Drosophila and mouse ARID3a orthologues resulted in embryonic lethality, suggesting critical roles for ARID3a during early development (17–20). In the mouse, lethality resulted from failed erythropoietic events between days 9 and 12 of fetal development (17). Furthermore hematopoietic stem cells were reduced by >90% in those mice, suggesting an important role for Bright/ARID3a in early hematopoiesis (17). Although we recently showed that ARID3a was variably expressed in multiple human hematopoietic subsets in healthy individuals and in lupus patients (16, 21), the functions of ARID3a during normal human hematopoiesis have not been studied.
To address the role of ARID3a in human hematopoiesis, we used lentiviral and retroviral constructs to inhibit, or constitutively over-express, ARID3a in lineage negative, CD34+ hematopoietic stem progenitor cells (HSPCs) in several in vitro systems that allow hematopoietic differentiation. Our data indicate that ARID3a promotes early B lineage decisions and that constitutive expression of ARID3a in early human HPSCs negatively impacts differentiation of myeloid lineage cells.
Methods and Materials
Cloning and expression of ARID3a
Full length expression constructs of human native and dominant-negative (DN) ARID3a were derived identically to the described mouse vectors (11, 22) and ligated into the polylinker site of the retroviral plasmid LZRSpBMN-linker-IRES-EGFP (23) (kind gift from Linda Thompson, OMRF) using T4 ligase (Invitrogen) following the manufacturer’s protocol. All constructs were verified by sequencing at the OMRF sequence facility. Viral vectors (native or wild type (WT) ARID3a, DN ARID3a, or a control GFP-only vector) were transfected individually into amyotrophic Phoenix viral packaging cells, as previously described (24). After 48 hours, viral supernatants were harvested.
Progenitor cells and cell lines
All cytokines were purchased from R&D Systems. MS-5 murine stromal cells were maintained in α-MEM (Cellgro) supplemented with 10% FCS, 10 units/ml penicillin-streptomycin, and 2 mM L-Glutamine (Invitrogen) (25, 26). Human cord blood was graciously provided with informed consent according to institutional Investigation Review Board protocols (IRB# 02-29) by Dr. Teresa Folger (Women’s Hospital, OK) or was purchased as mononuclear cells from Precision Bioservices or Stem Cell Technologies. Mononuclear cells were isolated by Ficoll-Hypaque gradient and enriched for CD34+ cells using magnetic column separation as per manufacturer’s directions (Miltenyi Biotech). CD34 enriched cells were used immediately or stored in RPMI based freezing medium containing 20% FCS and 5% DMSO at −80° C until further use. In some cases, cells were sorted for CD34+, lin− (CD19, CD33, CD13, CD7, CD56, and CD10) cells using a MOFLO flow cytometer (Becton Dickenson), cultured for 48 hours with Stem Pro media (Invitrogen) supplemented with 100 U penicillin and 100 μg streptomycin and a cytokine cocktail including stem cell factor (SCF) (100ng/ml), FLT3 ligand (50ng/ml), and thrombopoietin (TPO) (10ng/ml) (R&D Systems) to allow for maximal proliferation before viral transduction.
Viral transductions
For knockdown of ARID3a, Lin− CD34+ cells (HSPCs) or HSCs were treated with lentivirus containing shRNA for ARID3a or an unrelated shRNA scrambled control as previously described (27), using a multiplicity of infection (MOI) of 0.5 and 6μg/ml polybrene for 24 hours. Half of the medium was replaced with fresh medium containing lentivirus at an MOI of 0.5 for another 24 hours. Following virus treatment, HSPCs were seeded into stromal-free cultures for differentiation of B lineage cells. Over-expression of dominant negative (DN) human ARID3a or native (wild type, WT) ARID3a was accomplished using retroviral vectors that co-expressed GFP with ARID3a as described (10). Briefly, Retronectin (Takara) coated, 96-well EIA/RIA plates (Corning) were blocked with PBS containing 2% BSA overnight at 4° C and incubated with viral supernatants for 3 hours. HSPCs were transduced by addition to virus-coated plates, followed by incubation for 48 hours at 37° C with 5% CO2. Transduced cells were harvested by gentle pipetting, and sorted for GFP+ cells using a MOFLO flow cytometer. Cells were either placed onto a confluent layer of MS-5 stromal cells, or were plated in semi-solid medium.
In vitro Cultures
For stromal-cell free B lineage cultures, HSPCs were plated in triplicate at 10,000 cells per well as described (28, 29). Briefly, cells were cultured in QBSF®60 (Quality Biological, Inc.) supplemented with 10% FBS (Atlanta Biologicals), 100U Penicillin/Streptomycin (Gibco), 10% human mesenchymal stem cell (hMSC) conditioned media (LaCell LLC) and containing 10ng/ml SCF, 10ng/ml granulocyte stimulating factor (G-CSF), 5ng/ml FLT3 ligand, and 5ng/ml IL-7 (R&D Systems). Cells were cultured for 4 weeks, fed weekly with half volumes of fresh, cytokine supplemented media, harvested, counted and assessed by flow cytometry.
Stromal cell co-cultures were performed as previously described (30, 31) with slight modification. Briefly, 1×104 MS-5 murine stromal cells/well were plated in 96-well plates for 24 hours, prior to seeding with 200 GFP+, virally transduced HSPCs. Cells were grown in α-MEM supplemented with 10% FCS, 100 ng/ml SCF, 10 ng/ml G-CSF, and 1×10−7 M Dup697 (Cayman Chemical). To assess NK cell development, cells were grown with 5 ng/ml IL-15 and 10ng/ml FLT3 ligand (25). Cultures were fed weekly by removal of half of the medium and replacement with fresh cytokine-containing medium for a period of 3 to 4 weeks.
Monocytic, granulocytic and erythrocytic lineage cells were assessed using a methylcellulose colony assay. Assays were initiated similarly with 1000 GFP+ virally transduced or control HSPCs using Methocult GF (Stemcell Technologies), according to the manufacturer’s directions. Cultures were incubated for 12 days, after which time colonies were analyzed visually and counted using an Axiovert 25 inverted microscope. Pictures were acquired with an AxioCam HRc (Zeiss).
Flow Cytometry
Cord blood cells were harvested, stained, and confirmed to be lineage negative by flow cytometry using human hematopoietic lineage markers (CD2, CD3, CD14, CD16, CD19, CD56, CD235a) allophycocyanin (APC) (eBioscience). Other fluorescent antibodies used were: CD19 BV510, streptavidin (SA) PerCP-Cy5.5 (BD Biosciences), CD10 Biotin (Caltag), CD34 PE, CD38 Alexa Fluor 700, CD7 PE-Cy5, CD135 (FLT-3) PE-Cy5, CD10 PE-Cy7, CD127 (IL7Rα) PE-Cy7, CD49f PerCP-Cy5.5, c-Kit Brilliant Violet 421, CD45RA Brilliant Violet 570, CD135 Biotin, SA APC-Cy7 (BioLegend), CD34 PE, CD10 Pacific Blue, CD19 PE-Cy5, CD19 PE-Cy7 (BioLegend), CD56 FITC and CD33 APC (BD Biosciences), CD19 APC, CD13 APC, CD7 APC and CD14 APC, CD106 Biotin (Caltag). SA PE-Texas Red, and the 7AAD viability marker were purchased from Ebiosciences. Following surface marker staining, cells were fixed with 2% paraformaldehyde, permeabilized with 0.1% Tween-20 and stained with goat anti-human ARID3a antibody (15), followed by rabbit anti-goat FITC (Invitrogen). Appropriate isotype controls (BD Biosciences, eBiosciences, BioLegend) were used for determining negative staining of hematopoietic progenitor subsets as described (32, 33). Doublet exclusion was used to ensure analyses of single cells prior to forward/side scatter gating. Murine CD106 was used to gate out murine stromal cells from stromal cell culture analyses, and 7AAD or propidium iodide were used to gate and measure cell viability in cultures. Data were collected using an LSRII (BD Biogenics) and FACSDiva (BD Biosciences) software version 4.1, and were analyzed using FlowJo (Tree Star) software version 10.
Microarray Analyses
RNA was isolated from sorted GFP positive cells using Trireagent (MRC, Cincinnati, OH), was quantified on a Nanodrop scanning spectrophotometer, and cDNA preparation and Microarray analyses were performed by the Oklahoma Medical Research Foundation Microarray Core Facility by hybridizing cDNA to a human microarray library containing 21,329 genes (34). Four independent microarrays were analyzed from four independent transfection experiments and allowed detection of expression of 6000 genes. Data were normalized and differentially expressed genes were identified as described in detail previously (35). Data are available at the Gene Expression Omnibus (GEO) under series number GSE33777 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE33777).
HSPC Transcript Analyses
CD34-enriched cells were sorted for HSCs (CD34+CD38−CD45RA−CD49f+) using a FACSAria II (BD Biosciences). Viral infections were performed as described above, and RNA was isolated from sorted GFP positive cells using Trireagent (MRC) after 48 hours. RNA concentration and quality were assessed using the Agilent Total RNA Pico Chips on the 2100 Bioanalyzer (Agilent, Böblingen, Germany). Primers were generated using the DeltaGene Assay designer (Fluidigm) optimized for DeltaGene assays on the BiomarkHD system, and primer pair specificity was determined via melting curve analyses for use at 400 nM. Primer sequences and gene names are shown in Supplementary Table S1. cDNA was prepared with Fluidigm PreAmp Master Mix (PM100-5580), and preamplification of cDNA was performed with the DeltaGene Assay Kit (Fluidigm) and used at a final 1:10 dilution in DNA suspension buffer (TEKnova, PN: T0221). Quantitative PCR was conducted on the BioMark™ HD system and transcript expression was assessed using the Gene Expression (GE) 96.96 IFC chip (Fluidigm) using the standard DeltaGene Assay. Thermocycling parameters included an initial phase of 98°C for 40 s followed by 40 cycles, consisting of 95°C for 10 s and 60°C for 40 s. Raw Cq (Ct) values were obtained from the Fluidgm Biomark software, along with quality control calls. Ct values that failed quality control were dropped from subsequent analyses. GAPDH was used as a housekeeping standard, and ΔΔCt values were calculated from averaged duplicates as previously described (27).
Statistics
Data were statistically evaluated using the student T test to compare distribution of variables between pairs of groups. The non-parametric ANOVA (Kruskal-Wallis) followed by Dunn post hoc tests were used for 3 group comparisons. Statistical analysis was performed with Prism (Graphpad) software version 6.03. P values of less than 0.05 were considered significant.
Results
ARID3a is expressed in the majority of hematopoietic stem cells in human cord blood
We previously showed that ARID3a was differentially expressed in multiple subsets of adult peripheral blood HSPCs in lupus patients versus healthy controls (16). To better define the normal roles for ARID3a in early hematopoiesis, we assessed expression in multiple hematopoietic subsets of lineage negative, CD34+ cord blood HSPCS as defined by Notta et. al. (33). Using flow cytometry, HSPCs were gated to allow identification of hematopoietic stem cells (HSCs), multi-myeloid progenitors (MMPs), multi-potent progenitors (MPP), and multi-lymphoid progenitors (MLP) (Fig. 1A) (16). Analysis of these subsets for numbers of ARID3a+ cells revealed that 74% of HSCs and MMPs expressed ARID3a, while only 43% of MLPs and 20% of MPPs were ARID3a positive (Fig. 1B). Thus, ARID3a expression is highest in the earliest HSC progenitors, but is not limited to a specific HSPC subset.
Figure 1.

Human cord blood HSPC subsets express variable levels of ARID3a. A. Cord blood HSPCs were stained and analyzed by flow cytometry to show representative proportions of the following HSPC subpopulations: HSC (Lin− CD34+ CD38− CD49f+ CD45RA−), MPP (Lin− CD34+ CD38− CD49f− CD45RA−), MLP (Lin− CD34+ CD38− CD10+ CD7−) and MMP (Lin− CD34+ CD38+ CD135+ CD7−). Gates used for each population are shown. Dashed box indicates isotype controls. B. Percentages of cells in each progenitor subset and percentages of cells within each subset that co-stained for intracellular ARID3a are indicated, and representative flow cytometry data are shown. Means and standard error bars are shown from two independent experiments.
ARID3a-expressing HSPCs express hematopoietic cytokine receptors
We noted that ARID3a expression was highest in a small subset of HSPCs (26%) that expressed high levels of CD34 (Fig 2A). Further analyses for hematopoietic cytokine receptors commonly observed on HSCs revealed that the majority of ARID3a+ HSPCs expressed the IL7 receptor α (IL7Rα), FLT3 and c-kit (Fig. 2B–D). These data suggest that ARID3a+ HSPCs may contribute to early hematopoietic events.
Figure 2.
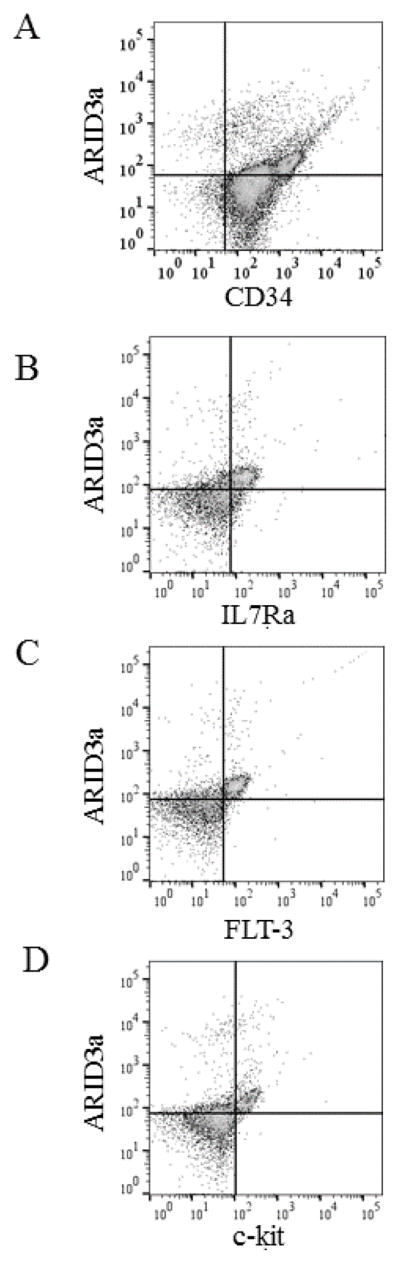
A subset of ARID3a+ HSPCs exhibit increased expression of CD34 and cytokine receptors. A. Flow cytometric analyses of HSPCs for CD34 and ARID3a expression indicate a subset of cells express increased levels of CD34. Lin−CD34+ HSPCs were also assessed for expression of IL7Ra (B), FLT-3 (C) and c-kit (D) with ARID3a. Quadrants are based on isotype controls.
Normal B lymphopoiesis requires ARID3a
Because ARID3a was originally described as a B lineage-specific protein (reviewed in 3), we determined if ARID3a contributed to B lineage development. Therefore, we assessed the ability of HSPCs transduced with shRNA against ARID3a or an unrelated control shRNA to develop into B lineage cells in vitro under conditions that allow B lineage development. ARID3a was effectively knocked down after two days, while scramble control shRNAs did not affect ARID3a expression (Fig. 3A). Although ARID3a-specific shRNA and control shRNA-treated cultures developed pre-pro, pro, pre and immature B lineage cells as defined by the surface markers indicated in Fig. 3B, cell cultures derived from ARID3a-deficient HSPCs resulted in a 75% reduction in total B lineage cells compared to scramble shRNA treated cultures (Fig. 3C). Cultures of ARID3a-deficient cells showed increased proportions of pro-B cells versus pre-B cells compared to ratios of these subsets in the untreated and scramble-control treated cultures. Numbers of pre-B cells in the ARID3a-deficient cultures were decreased by 86%, suggesting ARID3a may contribute to maturation of cells from the pro-B to pre-B cell stage, or to expansion of the pre-B cell stage itself. Together, these data suggest ARID3a is important for human B lineage commitment and maturation.
Figure 3.

Loss of ARID3a impairs B lineage development from human cord blood HSPCs. A. HSPCs were seeded into HSC culture media supplemented with SCF, FLT3 and TPO (triplicates of 10,000 cells/well) for 48 hours, prior to treatment with polybrene only, or lentivirus containing either ARID3a-specific, or scramble-control shRNA. A portion of cells were assessed for ARID3a knockdown after 48 hours. B. Following virus treatment, cells were maintained for one month in media supplemented with conditioned feeder media, G-CSF, IL7, SCF, and FLT3 for B lineage development. Representative flow cytometry data indicate the gating strategy used for B lymphocyte subsets and monocytes from virus treated samples. C. Percentage of B lineage pre-pro B cells (CD34+ CD33− CD10+ CD19−), pro-B cells (CD34+ CD33− CD10+ CD19+), pre-B cells (CD34− CD33− CD10+ CD19+) and B cells (CD34− CD33− CD10− CD19+)) for no virus control, ARID3a shRNA, and scramble control cultures are shown. Means and SE for 4 independent experiments are shown. Statistical significance was determined by unpaired t test: **, p=0.0032; ***, p=0.0002; ****, p<0.0001.
ARID3a inhibition results in increased numbers of myeloid lineage cells
Although B lineage cells were decreased in ARID3a shRNA treated cultures, total numbers of cells were not reduced compared to the shRNA control virally treated cultures (not shown). Rather, numbers of CD33+ myeloid lineage cells were dramatically increased in the ARID3a-deficient cultures (Fig. 4A–D). Cultures of ARID3a-deficient cells contained nearly 80% CD33+ cells versus the >80% B lineage development observed in both the untreated and control shRNA-treated cultures (Fig. 4A, 3C) indicating that ARID3a-deficiency increases myeloid development at the expense of B lineage development. Because retention of CD34+ cells was also increased in ARID3a-deficient cultures relative to scramble control treated cultures, (Fig. 4B) we hypothesized that some of the CD33+ cells we observed might also co-express CD34. Indeed, the majority of the CD33+ cells retained expression of CD34 in this culture (Fig. 4C). These results are consistent with the hypothesis that inhibition of ARID3a may allow cells to preferentially initiate development into myeloid lineage cells.
Figure 4.

Loss of ARID3a in HSPCs generates cells expressing myeloid lineage markers. A. Flow cytometric analyses of cells as shown in Fig. 3 was used to evaluate expression of CD33 in no virus control, shRNA, and scramble shRNA cultures. B. Cultures were further evaluated for retention of CD34 on total cells and without other lineage markers (CD34 only: CD34+ CD33− CD10− CD19−) in each set of cultures. C. Percentages of cells expressing the myeloid lineage marker CD33 with CD34 (CD34+ CD33+ CD19−) and without CD34 (CD34− CD33+ CD19−) are indicated. D. Representative flow cytometry data show increased CD33 expression in ARID3a shRNA cultures. All data indicate means and SE for 4 experiments. Statistical significance was determined by unpaired t test: *, p<0.014; **, p=0.0032; ***, p=0.0002; ****, p<0.0001.
Over-expression of ARID3a resulted in decreased numbers of myeloid lineage cells
We next asked if over-expression of native (wild type, WT) ARID3a, and a dominant negative (DN) form of ARID3a containing two point mutations in the DNA-binding domain (22), adversely affected myelopoiesis. ARID3a binds DNA as a dimer and hence over-expression of the mutant ARID3a dimerizes with endogenous ARID3a and blocks its DNA-binding function (22). Both retroviral vectors encoding these proteins allowed simultaneous expression of ARID3a and the fluorescence tracker, GFP. HSPCs were transduced with either the GFP-only expressing control virus, or virus expressing WT or DN ARID3a with equivalent transduction efficiencies (Fig. 5A). GFP+ HSPCs from each set of transductions were sorted to allow culture initiation with equivalent numbers of transduced cells. Cells were then allowed to differentiate on murine MS-5 stromal cell layers for three weeks using standard conditions designed to support development of both myeloid and B lineage cells (25, 26). Although total numbers of native (WT) ARID3a-expressing cells were only slightly lower than those expressing DN ARID3a or the control viral vector, all three sets of cells expanded more than 10-fold (Fig. 5B). Numbers of B lineage cells obtained under these culture conditions in all cultures was low compared to those obtained in liquid cultures and did not allow consistent evaluation (not shown). Strikingly, expression of WT ARID3a led to reduced numbers of both immature (60% fewer) and mature myeloid lineage cells (nearly 80% fewer) compared to control vector cultures, suggesting ARID3a expression inhibited development of myeloid lineage cells (Fig. 5C and D). As predicted from the shRNA knockdown data in Fig. 3, cultures expressing DN ARID3a showed 2–3 times more myeloid cells compared to control vector cultures, despite the presence of equivalent cell numbers at the end of culture (Fig. 5B-D). Numbers of CD14+ cells appeared to be even more dramatically affected by ARID3a over-expression than did those analyzed using CD33/13 surface expression with increases of 3.4-fold (Fig. 5D). These data indicate that over-expression of ARID3a inhibits myeloid lineage cell expansion in vitro while blocking endogenous ARID3a expands numbers of myeloid lineage cells.
Figure 5.

Over-expression of ARID3a in HSPCs inhibits myeloid lineage development. A. HSPCs were transduced with GFP-only (CON), dominant negative (DN) or wild type (WT) ARID3A expressing virus and were sorted for GFP expression 48 hours after transduction using the indicated. Representative percentages of GFP+ cells FACS sorted for in vitro assays are shown. B. GFP+ cells (200 cells/well) were cultured for three weeks under conditions that support myelopoiesis. Total cell numbers were counted via flow cytometric analyses and data for each culture are expressed as fold expansions over the starting numbers of cells. C. Numbers of CD33/13+cells from control (Ctrl), DN and native (WT) ARID3a-expressing cultures were enumerated by flow cytometry and are graphed as the averages of triplicate wells. D. Numbers of CD14+ cells mature myeloid lineage cells were similarly evaluated from the same cultures. Standard error bars are shown. Data are representative of eight independent experiments. Kruskal-Wallis evaluation for three-group analyses of CD33/13+ and CD14+ populations were P=0.0063 and p<0.0001 respectively, with significances of DN compared with WT (**, p=0.0087) for CD33/13+, and significance of Ctrl compared with DN and WT (***, p=0.001 and ****, p<0.0001), and DN compared with WT (****, p<0.0001).
Liquid co-cultures initiated with the addition of IL-15 (5ng/ml) and FLT3 ligand (10ng/ml) to allow the development of NK cells were assessed for the presence of the NK marker, CD56, after 4 weeks (25). Although only small numbers of NK cells developed (10–14%), there was no apparent difference in the numbers among control, DN or WT ARID3a expressing cultures (data not shown). These data indicate that NK cell development in this culture system was not affected by ARID3a levels.
ARID3a over-expression inhibits development of multiple mature cell types derived from the myeloid lineage
The data above indicate ARID3a has a negative impact on myeloid cells in liquid co-cultures that can differentiate down non-lymphoid pathways. To further assess the effects of ARID3a expression on myeloid lineage cell development, an alternate semi-solid assay that supports the development of myeloid, granulocytic and erythroid lineage cells was used (36). Cultures were initiated with 1000 virally infected GFP+ HSPCs cells, and were analyzed after 12 days for numbers and types of colonies formed. Although both control virus and DN ARID3a transduced cultures typically yielded 100 colonies per culture (Fig. 6A), cultures that over-expressed ARID3a gave rise to fewer than 20 colonies per dish, a more dramatic effect than we had observed in liquid cultures (Fig. 5B). Therefore, ARID3a over-expression appeared to affect colony maturation of monocyte lineage cells.
Figure 6.

ARID3a over-expression inhibits differentiation of multiple myeloid lineage colony types. A. Total numbers of colonies Control expressing Sorted HSPCs expressing control (CON), DN and WT ARID3a virus were plated (1000 cells per dish) in methylcellulose cultures and analyzed after twelve days for total numbers of myeloid lineage colony forming units. Data indicate means from triplicate dishes and aregraphed with standard error bars. B. Individual colony phenotypes were identified microscopically and average numbers of erythroid (E), monocyte (M), granulocyte/monocyte (GM), granulocyte/erythroid/megakaryocyte/monocyte (GEMM) and erythroid burst blast (BFU-E) colony forming units from triplicate dishes are presented graphically with standard error bars. Examples of the phenotypes of three prominent colony types identified are shown at the right at 50x magnification. Data are representative of two independent experiments.
Each colony was individually evaluated microscopically by its morphology and color to allow identification of mature colony subtypes as described previously (37). Erythroid (E), monocyte (M), granulocyte/monocyte (GM), mixed granulocyte/erythroid/megakaryocyte/monocyte (GEMM) and erythroid burst-forming unit (BFU-E) colonies were present in similar numbers in control and DN ARID3a transduced cultures (Fig. 6B). As expected from the stromal cell liquid cultures used in Fig. 5C, monocyte colonies in DN ARID3a cultures were slightly increased relative to control cultures with the use of a DN protein. However, all types of colonies were decreased in cultures initiated from ARID3a over-expressing cells. Mature erythroid colonies were present at such low levels in all cultures that the effect of ARID3a on those cells was difficult to evaluate. Nonetheless, numbers of erythroid burst forming units were significantly decreased in ARID3a over-expressing cultures suggesting that ARID3a levels may influence both erythroid and mature myeloid cell development. Colonies were also evaluated under a fluorescent microscope to assess GFP expression. While >95% of the vector control and DN ARID3a colonies were GFP+, only 6% of the total WT ARID3a colonies were GFP+. This suggests that most of the colonies that were present in the WT ARID3a transduced cultures had lost GFP, and presumably, ARID3a, expression. Thus, these semi-solid assays not only verified the previous co-culture data, but also indicated that over-expression of WT ARID3a inhibited colony formation and maturation of myeloid lineage cells. These data are consistent with the idea that loss of ARID3a is important for normal myeloid lineage development.
Expression of ARID3a in a mature B cell line identifies potential ARID3a gene targets in B cells
We asked if expression of ARID3a in a human B cell line (Raji) that lacked endogenous ARID3a activity might identify potential genes affected by ARID3a levels. RNA was isolated from control transfected and ARID3a-expressing cells, and microarray analyses were performed from four independent cultures. Only 15 new genes were expressed above background control levels and 37 more were upregulated by ARID3a. ARID3a expressing cells had 491 down-regulated genes compared to control cells, and an additional 217 genes which were no longer expressed above background. IgM heavy chain RNA was induced 5-fold, consistent with previous in vitro reporter experiments (10, 11), while SOX2, a stem cell-associated gene, was down-regulated by ARID3a expression, consistent with our previous findings that inhibition of ARID3a increased SOX2 expression (27). Semi-quantitative RT-PCR of several differentially regulated genes, including TCEB2 (elongin B) and TLR6, experimentally validated the microarray data (not shown). Differentially expressed genes also included TLR6, TLR7, IL5RA, FCGR3B, TNFRSF13C (BAFFR), TNFRSF17 (BCMA) and TNFSF13 (APRIL) (Table 1), all of which have been associated with B lymphocyte and/or myeloid lineage functions. Further analyses of these data for relevant signaling pathways affected by ARID3a expression revealed a high percentage of differentially expressed genes were associated with TGF-β signaling pathways, consistent with studies of the Xenopus orthologue of ARID3a (18), and data suggesting roles for this pathway in HSPC maintenance, proliferation, and differentiation (38). These data show that ARID3a expression leads to both enhanced and decreased expression of multiple immunoregulatory genes in human cells, and provide a new source for identifying gene targets that contribute to the effects we observed when ARID3a levels are dysregulated during hematopoiesis.
Table I.
ARID3a expression in a mature B cell line results in differential expression of multiple genes with immunomodulatory functions.
| Gene Symbol | Description | GenBank | Fold Change | p-value |
|---|---|---|---|---|
| HUMIGHMO30* | Immunoglobulin μ chain antibody MO30 | AY039026 | 5.82 | 0.04497 |
| IL5RA | Interleukin 5 receptor, alpha | NM_000564 | 1.66 | 0.037165 |
| TLR6* | Toll-like Receptor 6 | NM_006068 | −6.5 | 0.042789 |
| TLR7* | Toll-like Receptor 7 | NM_016562 | −3.83 | 0.004648 |
| FCBR3B | Fc fragment of IgG, low affinity IIIb, receptor; CD16b | J04162 | −3.43 | 0.005577 |
| TNFSF13* | Tumor necrosis factor (ligand) superfamily, member 13; APRIL | NM_003808 | −2.16 | 0.035478 |
| THY1 | Thy-1 cell surface antigen | AK057865 | −2.13 | 0.033825 |
| IL2RA* | Interleukin 2 receptor, alpha | NM_000417 | −2.07 | 0.036808 |
| IRF3 | Interferon regulatory factor 3 | NM_001571 | −2.05 | 0.044526 |
| TNFRSF17 | Tumor necrosis factor receptor superfamily, member 17; BCMA | NM_001192 | −1.88 | 0.011811 |
| CCR5 | Chemokine (C-C motif) receptor 5 | NM_000579 | −1.86 | 0.021458 |
| ABIN-1* | A20-binding inhibitor of NF-κB activation-2 | NM_024309 | −1.81 | 0.030763 |
| POU2AF1 | POU domain, class 2, associating factor 1; BOB-1; OCA-B | NM_006235 | −1.77 | 0.011926 |
| IGL@ | Immunoglubulin lambda locus | AK057065 | −1.7 | 0.043435 |
| CD79B | CD79B antigen (immunoglobulin-associated beta) | NM_000626 | −1.61 | 0.036085 |
| TNFRSF13C | Tumor necrosis factor receptor superfamily, member 13C, BAFFR | NM_052945 | −1.61 | 0.025292 |
Lower signal indistinguishable from background; Student’s T-test p-value
ARID3a inhibition in cord blood HSCs increases myeloid lineage gene expression transcripts
To directly assess the effects of ARID3a expression on genes important for lymphocyte versus myeloid lineage development (reviewed in (32, 39, 40)), we performed qRT-PCR on isolated HSCs treated with control or ARID3a-specific shRNAs. A total of 36 genes, in addition to several housekeeping genes were successfully evaluated and the results of those genes with the most significant changes in expression are shown in Fig. 7. Knockdown was effective as shown by downregulation of ARID3a in shRNA treated samples (Fig. 7). As expected from our previously published data (27, 41), the stem cell-associated genes LIN28A and NANOG, were increased in ARID3a inhibited versus control cells. ARID3a inhibition resulted in upregulation of CEBPB, ID2, RUNX1 and CSF1, all genes associated with myelopoiesis compared to cells treated with control shRNAa (Fig, 7). STAT3, an intracellular signaling factor important in myelopoiesis (42), was also upregulated in ARID3a inhibited cells. Conversely, TCF3 (E2A) and PBX1, genes associated with B lineage development (43, 44), were downregulated in ARID3a-inhibited HSCs. Interestingly, TAL1 (also called SCL), a gene for which expression is reported to be important for megakaryocyte and erythrocyte development (45), was also downregulated. Together, these data support conclusions from our in vitro culture data suggesting that lower levels of ARID3a expression in HSCs affect gene expression patterns correlated with promotion of myelopoiesis and suppression of lymphopoiesis.
Figure 7.

Inhibition of ARID3a expression in CB HSCs affects expression of transcription factors implicated in myeloid versus lymphoid lineage development. RNA from CB HSCs seeded into HSC culture media supplemented with SCF, FLT3 and TPO and treated with ARID3a-specific or scramble-control shRNA for 48 hours was assessed for expression of 36 genes. Data for genes showing changes > 2-fold are indicated with standard error bars. Genes with increased expression but not shown here were: IFI44, IFIT3, OAS3, IFNA2, IFNB1, IRF1 and Oct4. Genes assessed with no changes in expression were IFI6, PLSCR1, CXCL13, ICOS, IFNRA1, MYD88, PTGS2, TNFA, CDKN1A, SH2B3, BAK1, BCL2, IL10RA, and TLR9. Data are representative of 4 replicates. Statistical significance was determined by unpaired t test: *, p<0.04
Discussion
The data presented here suggest new roles for ARID3a function in early human hematopoiesis, and particularly in B lymphocyte lineage commitment and development. ARID3a inhibition suggests that this transcription factor is important for B lineage commitment, and the differentiation of pro-B lineage cells to pre-B cells. Conversely, our data indicate that down-regulation of ARID3a is important for development of mature myeloid lineage cells in vitro, because over-expression of native ARID3a resulted in inhibition of myeloid lineage differentiation. Together, these data imply that ARID3a plays important functions at several independent stages of human hematopoiesis.
As indicated in the schematic diagram in Fig. 8, ARID3a was variably expressed in multiple subtypes of HSPCs, with the highest expression in the multipotent, self-renewing HSC subset. This finding is consistent with data from fetal liver cells obtained from ARID3a/Bright knockout mice which showed 90% reductions in the HSC subset suggesting the majority of those cells required ARID3a for development (17). MPPs, which maintain multipotency but are no longer self-renewing also showed some ARID3a-expressing cells, but were greatly reduced relative to HSCs (Fig. 8). However, ARID3a expression was more abundant in both MLPs, which are lymphoid lineage committed and myeloid lineage committed MMPs, (reviewed in (32, 46–48). These data suggest that ARID3a functions may be important at multiple points during early hematopoiesis.
Figure 8.
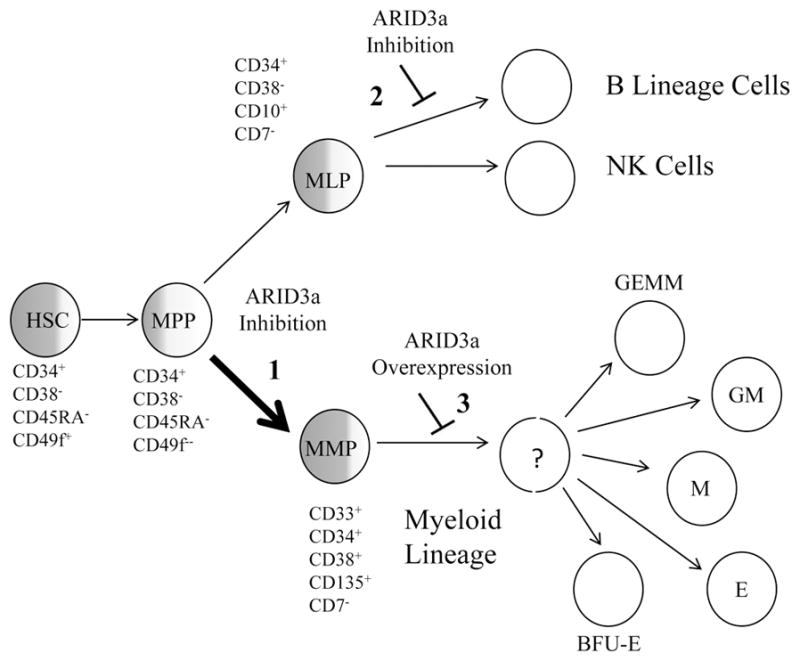
Schematic diagram for ARID3a functions during human hematopoiesis. Degrees of shading represent the proportion of cells initially expressing ARID3a in HSPC subsets HSC, hematopoietic stem cell; MPP, multipotent progenitor, MLP, multi-lymphoid progenitor and MMP, multi-myeloid progenitor. ARID3a inhibition resulted in increased development of myeloid lineage cells (1, thicker arrow) versus lymphocyte lineages, and inhibited B lymphocyte lineage development without affecting natural killer (NK) cells (2). Conversely, ARID3a over-expression blocked myeloid lineage development at unidentified stages resulting in decreased maturation of erythroid (E), monocyte (M), granulocyte/monocyte (GM), granulocyte/erythroid/megakaryocyte/monocyte (GEMM) and erythroid burst blast (BFU-E) colonies (3).
Interestingly, ARID3a expression in HSPCs was associated with expression of FLT3, c-kit and the IL7Rα (Fig. 2). Human, but not murine, hematopoietic progenitor cells express FLT3, where it may be a potent survival signal (reviewed in 49). FLT3 is expressed on 40–80% of early hematopoietic progenitors, from HSCs to lymphoid and myeloid progenitors, and it has been reported to function as a survival signal for all stages (46, 49, 50). SCF, which binds to c-kit, has also been implicated in early progenitor survival ((reviewed in 51)). Although ARID3a, FLT3 and c-kit were co-expressed at the initiation of culture (Fig. 2), ARID3a inhibition did not affect cell expansion in vitro, suggesting that ARID3a is not itself involved in survival signals. Our qRT-PCR data also suggest ARID3a levels do not affect survival since CDKN1A, BAK1 and BCL2 were not changed in cells without ARID3a (Fig. 7, legend). While the role of IL-7 signaling on early human hematopoietic progenitors and B lineage development remains controversial, our previous data link IL-7Rα expression with proliferation of HSPCs in vitro (16). Furthermore, the IL7Rα gene shows putative ARID3a binding by ChIP-seq analysis (our unpublished data and data available from the ENCODE group), indicating a potential role for ARID3a in IL7Rα expression.
Although the role of IL-7 in human versus mouse B lymphopoiesis has been controversial (52–54), IL-7 signaling may be particularly important at the pro-B cell stage (54). We found that inhibition of ARID3a in total HSPCs resulted in a 75% decrease in B lineage cells, and cells that did commit to the B lineage showed impairment of progression from the proB to pre-B cell stage when compared to both the non-transfected controls and the scramble lentiviral controls (Fig. 3C). This is consistent with our previous findings indicating tight regulation of ARID3a transcription during B lineage differentiation and increased expression of ARID3a in pre-B cells (15). Interestingly, the microRNA miR-125b is differentially expressed during B lineage differentiation and its expression affects B cell differentiation (55). In other studies (56), ARID3a was identified as a target for miR-125 in progenitor B cells, although it is unclear if the effects of miR-125 on pre-B cell development are solely mediated through ARID3a. Together, these data indicate functionally important roles for ARID3a during human B lineage differentiation.
Inhibition of ARID3a function in HSPCs under stromal free B lineage culture conditions substantially decreased numbers of B lineage cells with a concomitant increase in cells expressing the myeloid lineage marker CD33 (Fig. 4). However, NK cell numbers were not affected implying effects on B lineage development may be most prominent after the MLP developmental stage (Fig. 8). The majority of myeloid lineage cells maintained expression of the early lineage marker CD34, suggesting they were not fully committed myeloid lineage cells. However, in stromal cell cultures, ARID3a inhibition with DN ARID3a also led to increases in myeloid cell numbers, including those that expressed the mature myeloid lineage marker CD14 (Fig. 5). Conversely, elevation of ARID3a function by over-expression was detrimental for maturation of multiple types of mature myeloid lineage cells (Figs. 5 and 6). These two independent experimental approaches suggest that down-regulation of ARID3a is an important event for maturation of multiple myeloid lineage cell types.
It is now possible to evaluate lineage potential of HSPCs using humanized mouse models (16, 57). However, HSPCs are a heterogeneous population of progenitors that express variable levels of ARID3a within those subpopulations. Our previous studies revealed that some patients with systemic lupus erythematosus (SLE) had large numbers of ARID3a+ HSPCs compared to ARID3a expression levels in HSPCs from healthy controls (16). Therefore, we sought to determine if high levels of ARID3a expression in progenitor cells might be associated with differences in engraftment and hematopoietic development. Although patient cells with increased expression of ARID3a exhibited abnormal features in in vitro cultures, SLE HSPCs with high ARID3a expression did not differ in hematopoietic development of B cells or myeloid populations from HSPCs with normal levels of ARID3a (16). Differences between effects observed in vitro and in vivo could result from homeostatic events over the 16–20 week engraftment period, and/or to the mixed nature of the progenitor populations. ARID3a/Bright knockout mice showed defects in early hematopoiesis without a total loss of all B lineage cells (17). Therefore, ARID3a levels may contribute to lineage choices without being required for development of all B lineage cells.
Identifying the genetic pathways by which ARID3a affects myeloid versus B lineage development will require more detailed analyses. However, introduction of ARID3a into the ARID3a negative human B cell line Raji revealed multiple genes with immunomodulatory functions that are affected by ARID3a levels (Table I). Mature B lineage markers that are differentially expressed during B cell development, including TNFRSF17 (BCMA) and BAFFR (58), were identified as potential targets for ARID3a consistent with our findings that ARID3a is important for B cell maturation. Conversely, FcBR3B and the M-CSF receptor (CSF1R), genes often associated with the myeloid lineage, were down-regulated. Expression of CSF1, was upregulated in ARID3a-inhibited HSCs (Fig. 7), and was observed by others to be more highly expressed in progenitors in the myeloid versus lymphoid lineage pathway (59). Furthermore, studies in humanized mice indicated CSF1 is required for appropriate development and function of macrophages and monocytes following adoptive transfer of human HSPCs (60). Intriguingly, ARID3a levels affected the transcription of a number of TLRs (Table I). Expression of TLR2 was increased in ARID3a-inhibited HSCs (Fig. 7). Others showed that TLR stimulation through TLR1/2 (often co-expressed with TLR6), biased lineage commitment of human HSCs toward the myeloid lineage at the expense of B lymphocyte lineage cells (61, 62).
While over-expression of ARID3a in Raji cells resulted in down regulation of CEBPB, inhibition of ARID3a in HSCs led to increased CEBPB expression (Fig. 7). CEBPB is required for macrophage differentiation and myeloid versus lymphoid development in mice, and has also been implicated as a transcription factor important for maintenance of human myeloid lineage cells (63–65). RUNX1 has been reported to be required for megakaryocyte and platelet development (reviewed in 66), and was increased in ARID3a-inhibited HSCs, while our in vitro data showed decreased megakaryocyte development in vitro with ARID3a over-expression. Further, we found a marked decrease in TCF3 expression following loss of ARID3a expression. TCF3 has been shown to be necessary for development of B lineage committed cell types (reviewed in 44). Based on these data, ARID3a expression likely plays a role in the regulation of multiple genes important for hematopoiesis. Because our data indicate that ARID3a can act to suppress, as well as enhance transcription (27), the functions of ARID3a may differ in various cell types. Additional experiments will be required to assess the role ARID3a has in the regulation of each of these genes.
Although hematopoietic development schemes in the mouse have been described in detail due to the ease with which those cells can be grown in culture, human systems have lagged behind. Furthermore, while murine model systems provide important new information about many disease systems, there are increasing examples suggesting human hematopoietic events do not always mimic mouse models. Therefore, studies to better understand early hematopoietic events in human HSPCs and the factors that contribute to lineage decisions in human cells are important, particularly for manipulation of this important system in both regenerative medicine and transplantation. The data presented in this report indicate an important role for ARID3a in hematopoiesis lineage commitment and differentiation events, likely with differing functions depending on the developmental stage of the subsets. Studies of the mechanisms by which ARID3a expression functions in fate choice and differentiation, as well as consequences of aberrant expression of ARID3a in early hematopoietic progenitors are currently underway.
Supplementary Material
Acknowledgments
The authors wish to thank Dr. T. Folger for cord blood samples, B. Hurt and S. Wasson for manuscript preparation, M. Coggeshall, S. Kovats, S. Ferrell, J. Ward and K. Rose for technical assistance and helpful discussions. We also thank D. Hamilton and the OMRF Flow Cytometry Core, and N. Dominguez, K. Bean and J. Te of the OMRF Arthritis & Clinical Immunology Oklahoma Rheumatic Disease Resources and Cores Center and OMRF Translational Informatics & Core Resources center.
Footnotes
Funding: This work was supported by the March of Dimes, NIH AI45864 and AI044215 to CFW, P30 AR053483, Phenotyping Core support (JMG), and U19-AI062629-10 (K. Mark Coggeshall, PI), pilot (CFW).
References
- 1.Wilsker D, Patsialou A, Dallas PB, Moran E. ARID proteins: a diverse family of DNA binding proteins implicated in the control of cell growth, differentiation, and development. Cell Growth Differ. 2002;13:95–106. [PubMed] [Google Scholar]
- 2.Patsialou A, Wilsker D, Moran E. DNA-binding properties of ARID family proteins. Nucleic Acids Res. 2005;33:66–80. doi: 10.1093/nar/gki145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ratliff ML, Templeton TD, Ward JM, Webb CF. The Bright Side of Hematopoiesis: Regulatory Roles of ARID3a/Bright in Human and Mouse Hematopoiesis. Front Immunol. 2014;5:113. doi: 10.3389/fimmu.2014.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilsker D, Probst L, Wain HM, Maltais L, Tucker PW, Moran E. Nomenclature of the ARID family of DNA-binding proteins. Genomics. 2005;86:242–251. doi: 10.1016/j.ygeno.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 5.Goebel P, Montalbano A, Ayers N, Kompfner E, Dickinson L, Webb CF, Feeney AJ. High frequency of matrix attachment regions and cut-like protein x/CCAAT-displacement protein and B cell regulator of IgH transcription binding sites flanking Ig V region genes. J Immunol. 2002;169:2477–2487. doi: 10.4049/jimmunol.169.5.2477. [DOI] [PubMed] [Google Scholar]
- 6.Webb CF, Das C, Eneff KL, Tucker PW. Identification of a matrix-associated region 5′ of an immunoglobulin heavy chain variable region gene. Mol Cell Biol. 1991;11:5206–5211. doi: 10.1128/mcb.11.10.5206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnston CM, Wood AL, Bolland DJ, Corcoran AE. Complete sequence assembly and characterization of the C57BL/6 mouse Ig heavy chain V region. J Immunol. 2006;176:4221–4234. doi: 10.4049/jimmunol.176.7.4221. [DOI] [PubMed] [Google Scholar]
- 8.Webb C, Zong RT, Lin D, Wang Z, Kaplan M, Paulin Y, Smith E, Probst L, Bryant J, Goldstein A, Scheuermann R, Tucker P. Differential regulation of immunoglobulin gene transcription via nuclear matrix-associated regions. Cold Spring Harb Symp Quant Biol. 1999;64:109–118. doi: 10.1101/sqb.1999.64.109. [DOI] [PubMed] [Google Scholar]
- 9.Herrscher RF, Kaplan MH, Lelsz DL, Das C, Scheuermann R, Tucker PW. The immunoglobulin heavy-chain matrix-associating regions are bound by Bright: a B cell-specific trans-activator that describes a new DNA-binding protein family. Genes Dev. 1995;9:3067–3082. doi: 10.1101/gad.9.24.3067. [DOI] [PubMed] [Google Scholar]
- 10.Rajaiya J, Hatfield M, Nixon JC, Rawlings DJ, Webb CF. Bruton’s tyrosine kinase regulates immunoglobulin promoter activation in association with the transcription factor Bright. Mol Cell Biol. 2005;25:2073–2084. doi: 10.1128/MCB.25.6.2073-2084.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rajaiya J, Nixon JC, Ayers N, Desgranges ZP, Roy AL, Webb CF. Induction of immunoglobulin heavy-chain transcription through the transcription factor Bright requires TFII-I. Mol Cell Biol. 2006;26:4758–4768. doi: 10.1128/MCB.02009-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shankar M, Nixon JC, Maier S, Workman J, Farris AD, Webb CF. Anti-nuclear antibody production and autoimmunity in transgenic mice that overexpress the transcription factor Bright. J Immunol. 2007;178:2996–3006. doi: 10.4049/jimmunol.178.5.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oldham AL, Miner CA, Wang HC, Webb CF. The transcription factor Bright plays a role in marginal zone B lymphocyte development and autoantibody production. Mol Immunol. 2011;49:367–379. doi: 10.1016/j.molimm.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Webb CF. The transcription factor, Bright, and immunoglobulin heavy chain expression. Immunol Res. 2001;24:149–161. doi: 10.1385/IR:24:2:149. [DOI] [PubMed] [Google Scholar]
- 15.Nixon JC, Rajaiya JB, Ayers N, Evetts S, Webb CF. The transcription factor, Bright, is not expressed in all human B lymphocyte subpopulations. Cell Immunol. 2004;228:42–53. doi: 10.1016/j.cellimm.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 16.Ratliff ML, Ward JM, Merrill JT, James JA, Webb CF. Differential expression of the transcription factor ARID3a in lupus patient hematopoietic progenitor cells. J Immunol. 2015;194:940–949. doi: 10.4049/jimmunol.1401941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Webb CF, Bryant J, Popowski M, Allred L, Kim D, Harriss J, Schmidt C, Miner CA, Rose K, Cheng HL, Griffin C, Tucker PW. The ARID family transcription factor bright is required for both hematopoietic stem cell and B lineage development. Mol Cell Biol. 2011;31:1041–1053. doi: 10.1128/MCB.01448-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Callery EM, Smith JC, Thomsen GH. The ARID domain protein dril1 is necessary for TGF(beta) signaling in Xenopus embryos. Dev Biol. 2005;278:542–559. doi: 10.1016/j.ydbio.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 19.Shandala T, Kortschak RD, Gregory S, Saint R. The Drosophila dead ringer gene is required for early embryonic patterning through regulation of argos and buttonhead expression. Development. 1999;126:4341–4349. doi: 10.1242/dev.126.19.4341. [DOI] [PubMed] [Google Scholar]
- 20.Shandala T, Kortschak RD, Saint R. The Drosophila retained/dead ringer gene and ARID gene family function during development. Int J Dev Biol. 2002;46:423–430. [PubMed] [Google Scholar]
- 21.Ward JM, Rose K, Montgomery C, Adrianto I, James JA, Merrill JT, Webb CF. Disease activity in systemic lupus erythematosus correlates with expression of the transcription factor AT-rich-interactive domain 3A. Arthritis Rheumatol. 2014;66:3404–3412. doi: 10.1002/art.38857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nixon JC, Rajaiya J, Webb CF. Mutations in the DNA-binding domain of the transcription factor Bright act as dominant negative proteins and interfere with immunoglobulin transactivation. J Biol Chem. 2004;279:52465–52472. doi: 10.1074/jbc.M403028200. [DOI] [PubMed] [Google Scholar]
- 23.Heemskerk MH, Hooijberg E, Ruizendaal JJ, van der Weide MM, Kueter E, Bakker AQ, Schumacher TN, Spits H. Enrichment of an antigen-specific T cell response by retrovirally transduced human dendritic cells. Cell Immunol. 1999;195:10–17. doi: 10.1006/cimm.1999.1520. [DOI] [PubMed] [Google Scholar]
- 24.Kinsella TM, Nolan GP. Episomal vectors rapidly and stably produce high-titer recombinant retrovirus. Hum Gene Ther. 1996;7:1405–1413. doi: 10.1089/hum.1996.7.12-1405. [DOI] [PubMed] [Google Scholar]
- 25.Kouro T, Yokota T, Welner R, Kincade PW. In vitro differentiation and measurement of B cell progenitor activity in culture. Curr Protoc Immunol Chapter. 2005;22(Unit 22F):22. doi: 10.1002/0471142735.im22f02s66. [DOI] [PubMed] [Google Scholar]
- 26.Nishihara M, Wada Y, Ogami K, Ebihara Y, Ishii T, Tsuji K, Ueno H, Asano S, Nakahata T, Maekawa T. A combination of stem cell factor and granulocyte colony-stimulating factor enhances the growth of human progenitor B cells supported by murine stromal cell line MS-5. Eur J Immunol. 1998;28:855–864. doi: 10.1002/(SICI)1521-4141(199803)28:03<855::AID-IMMU855>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 27.An G, Miner CA, Nixon JC, Kincade PW, Bryant J, Tucker PW, Webb CF. Loss of Bright/ARID3a function promotes developmental plasticity. Stem Cells. 2010;28:1560–1567. doi: 10.1002/stem.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ichii M, Oritani K, Yokota T, Schultz DC, Holter JL, Kanakura Y, Kincade PW. Stromal cell-free conditions favorable for human B lymphopoiesis in culture. J Immunol Methods. 2010;359:47–55. doi: 10.1016/j.jim.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ichii M, Oritani K, Yokota T, Zhang Q, Garrett KP, Kanakura Y, Kincade PW. The density of CD10 corresponds to commitment and progression in the human B lymphoid lineage. PLoS One. 2010;5:e12954. doi: 10.1371/journal.pone.0012954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Igarashi H, Medina KL, Yokota T, Rossi MI, Sakaguchi N, Comp PC, Kincade PW. Early lymphoid progenitors in mouse and man are highly sensitive to glucocorticoids. Int Immunol. 2005;17:501–511. doi: 10.1093/intimm/dxh230. [DOI] [PubMed] [Google Scholar]
- 31.Chen X, Esplin BL, Garrett KP, Welner RS, Webb CF, Kincade PW. Retinoids accelerate B lineage lymphoid differentiation. J Immunol. 2008;180:138–145. doi: 10.4049/jimmunol.180.1.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: a human perspective. Cell Stem Cell. 2012;10:120–136. doi: 10.1016/j.stem.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 33.Notta F, Doulatov S, Laurenti E, Poeppl A, Jurisica I, Dick JE. Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science. 2011;333:218–221. doi: 10.1126/science.1201219. [DOI] [PubMed] [Google Scholar]
- 34.Jarvis JN, Petty HR, Tang Y, Frank MB, Tessier PA, Dozmorov I, Jiang K, Kindzelski A, Chen Y, Cadwell C, Turner M, Szodoray P, McGhee JL, Centola M. Evidence for chronic, peripheral activation of neutrophils in polyarticular juvenile rheumatoid arthritis. Arthritis Res Ther. 2006;8:R154. doi: 10.1186/ar2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dozmorov MG, Hurst RE, Culkin DJ, Kropp BP, Frank MB, Osban J, Penning TM, Lin HK. Unique patterns of molecular profiling between human prostate cancer LNCaP and PC-3 cells. Prostate. 2009;69:1077–1090. doi: 10.1002/pros.20960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller CL, Lai B. Human and mouse hematopoietic colony-forming cell assays. Methods Mol Biol. 2005;290:71–89. doi: 10.1385/1-59259-838-2:071. [DOI] [PubMed] [Google Scholar]
- 37.Pereira C, Clarke E, Damen J. Hematopoietic colony-forming cell assays. Methods Mol Biol. 2007;407:177–208. doi: 10.1007/978-1-59745-536-7_14. [DOI] [PubMed] [Google Scholar]
- 38.Blank U, Karlsson S. The role of Smad signaling in hematopoiesis and translational hematology. Leukemia. 2011;25:1379–1388. doi: 10.1038/leu.2011.95. [DOI] [PubMed] [Google Scholar]
- 39.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosmarin AG, Yang Z, Resendes KK. Transcriptional regulation in myelopoiesis: Hematopoietic fate choice, myeloid differentiation, and leukemogenesis. Exp Hematol. 2005;33:131–143. doi: 10.1016/j.exphem.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 41.Popowski M, Templeton TD, Lee B-K, Rhee C, Li H, Miner C, Dekker Joseph D, Orlanski S, Bergman Y, Iyer Vishwanath R, Webb Carol F, Tucker H. Bright/Arid3A Acts as a Barrier to Somatic Cell Reprogramming through Direct Regulation of Oct4, Sox2, and Nanog. Stem Cell Reports. 2014;2:26–35. doi: 10.1016/j.stemcr.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Panopoulos AD, Zhang L, Snow JW, Jones DM, Smith AM, El Kasmi KC, Liu F, Goldsmith MA, Link DC, Murray PJ, Watowich SS. STAT3 governs distinct pathways in emergency granulopoiesis and mature neutrophils. Blood. 2006;108:3682–3690. doi: 10.1182/blood-2006-02-003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanyal M, Tung JW, Karsunky H, Zeng H, Selleri L, Weissman IL, Herzenberg LA, Cleary ML. B-cell development fails in the absence of the Pbx1 proto-oncogene. Blood. 2007;109:4191–4199. doi: 10.1182/blood-2006-10-054213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramirez J, Lukin K, Hagman J. From hematopoietic progenitors to B cells: mechanisms of lineage restriction and commitment. Curr Opin Immunol. 2010;22:177–184. doi: 10.1016/j.coi.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Novershtern N, Subramanian A, Lawton LN, Mak RH, Haining WN, McConkey ME, Habib N, Yosef N, Chang CY, Shay T, Frampton GM, Drake AC, Leskov I, Nilsson B, Preffer F, Dombkowski D, Evans JW, Liefeld T, Smutko JS, Chen J, Friedman N, Young RA, Golub TR, Regev A, Ebert BL. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011;144:296–309. doi: 10.1016/j.cell.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iwasaki H, Akashi K. Hematopoietic developmental pathways: on cellular basis. Oncogene. 2007;26:6687–6696. doi: 10.1038/sj.onc.1210754. [DOI] [PubMed] [Google Scholar]
- 47.Weissman IL, Shizuru JA. The origins of the identification and isolation of hematopoietic stem cells, and their capability to induce donor-specific transplantation tolerance and treat autoimmune diseases. Blood. 2008;112:3543–3553. doi: 10.1182/blood-2008-08-078220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iwasaki H, Akashi K. Myeloid lineage commitment from the hematopoietic stem cell. Immunity. 2007;26:726–740. doi: 10.1016/j.immuni.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 49.Parekh C, Crooks GM. Critical differences in hematopoiesis and lymphoid development between humans and mice. J Clin Immunol. 2013;33:711–715. doi: 10.1007/s10875-012-9844-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kikushige Y, Yoshimoto G, Miyamoto T, Iino T, Mori Y, Iwasaki H, Niiro H, Takenaka K, Nagafuji K, Harada M, Ishikawa F, Akashi K. Human Flt3 is expressed at the hematopoietic stem cell and the granulocyte/macrophage progenitor stages to maintain cell survival. J Immunol. 2008;180:7358–7367. doi: 10.4049/jimmunol.180.11.7358. [DOI] [PubMed] [Google Scholar]
- 51.Broudy VC. Stem cell factor and hematopoiesis. Blood. 1997;90:1345–1364. [PubMed] [Google Scholar]
- 52.Dittel BN, LeBien TW. The growth response to IL-7 during normal human B cell ontogeny is restricted to B-lineage cells expressing CD34. J Immunol. 1995;154:58–67. [PubMed] [Google Scholar]
- 53.Johnson SE, Shah N, Panoskaltsis-Mortari A, LeBien TW. Murine and human IL-7 activate STAT5 and induce proliferation of normal human pro-B cells. J Immunol. 2005;175:7325–7331. doi: 10.4049/jimmunol.175.11.7325. [DOI] [PubMed] [Google Scholar]
- 54.Clark MR, Mandal M, Ochiai K, Singh H. Orchestrating B cell lymphopoiesis through interplay of IL-7 receptor and pre-B cell receptor signalling. Nat Rev Immunol. 2014;14:69–80. doi: 10.1038/nri3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gururajan M, Haga CL, Das S, Leu CM, Hodson D, Josson S, Turner M, Cooper MD. MicroRNA 125b inhibition of B cell differentiation in germinal centers. Int Immunol. 2010;22:583–592. doi: 10.1093/intimm/dxq042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Puissegur MP, Eichner R, Quelen C, Coyaud E, Mari B, Lebrigand K, Broccardo C, Nguyen-Khac F, Bousquet M, Brousset P. B-cell regulator of immunoglobulin heavy-chain transcription (Bright)/ARID3a is a direct target of the oncomir microRNA-125b in progenitor B-cells. Leukemia. 2012;26:2224–2232. doi: 10.1038/leu.2012.95. [DOI] [PubMed] [Google Scholar]
- 57.Andrade D, Redecha PB, Vukelic M, Qing X, Perino G, Salmon JE, Koo GC. Engraftment of peripheral blood mononuclear cells from systemic lupus erythematosus and antiphospholipid syndrome patient donors into BALB-RAG-2−/− IL-2Rgamma−/− mice: a promising model for studying human disease. Arthritis Rheum. 2011;63:2764–2773. doi: 10.1002/art.30424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Darce JR, Arendt BK, Wu X, Jelinek DF. Regulated expression of BAFF-binding receptors during human B cell differentiation. J Immunol. 2007;179:7276–7286. doi: 10.4049/jimmunol.179.11.7276. [DOI] [PubMed] [Google Scholar]
- 59.Boiers C, Carrelha J, Lutteropp M, Luc S, Green JC, Azzoni E, Woll PS, Mead AJ, Hultquist A, Swiers G, Perdiguero EG, Macaulay IC, Melchiori L, Luis TC, Kharazi S, Bouriez-Jones T, Deng Q, Ponten A, Atkinson D, Jensen CT, Sitnicka E, Geissmann F, Godin I, Sandberg R, de Bruijn MF, Jacobsen SE. Lymphomyeloid contribution of an immune-restricted progenitor emerging prior to definitive hematopoietic stem cells. Cell Stem Cell. 2013;13:535–548. doi: 10.1016/j.stem.2013.08.012. [DOI] [PubMed] [Google Scholar]
- 60.Rathinam C, Poueymirou WT, Rojas J, Murphy AJ, Valenzuela DM, Yancopoulos GD, Rongvaux A, Eynon EE, Manz MG, Flavell RA. Efficient differentiation and function of human macrophages in humanized CSF-1 mice. Blood. 2011;118:3119–3128. doi: 10.1182/blood-2010-12-326926. [DOI] [PubMed] [Google Scholar]
- 61.De Luca K, Frances-Duvert V, Asensio MJ, Ihsani R, Debien E, Taillardet M, Verhoeyen E, Bella C, Lantheaume S, Genestier L, Defrance T. The TLR1/2 agonist PAM(3)CSK(4) instructs commitment of human hematopoietic stem cells to a myeloid cell fate. Leukemia. 2009;23:2063–2074. doi: 10.1038/leu.2009.155. [DOI] [PubMed] [Google Scholar]
- 62.Sioud M, Floisand Y, Forfang L, Lund-Johansen F. Signaling through toll-like receptor 7/8 induces the differentiation of human bone marrow CD34+ progenitor cells along the myeloid lineage. J Mol Biol. 2006;364:945–954. doi: 10.1016/j.jmb.2006.09.054. [DOI] [PubMed] [Google Scholar]
- 63.Cain DW, O’Koren EG, Kan MJ, Womble M, Sempowski GD, Hopper K, Gunn MD, Kelsoe G. Identification of a tissue-specific, C/EBPbeta-dependent pathway of differentiation for murine peritoneal macrophages. J Immunol. 2013;191:4665–4675. doi: 10.4049/jimmunol.1300581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stoilova B, Kowenz-Leutz E, Scheller M, Leutz A. Lymphoid to myeloid cell trans-differentiation is determined by C/EBPbeta structure and post-translational modifications. PLoS One. 2013;8:e65169. doi: 10.1371/journal.pone.0065169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schmeier S, MacPherson CR, Essack M, Kaur M, Schaefer U, Suzuki H, Hayashizaki Y, Bajic VB. Deciphering the transcriptional circuitry of microRNA genes expressed during human monocytic differentiation. BMC Genomics. 2009;10:595. doi: 10.1186/1471-2164-10-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ichikawa M, Yoshimi A, Nakagawa M, Nishimoto N, Watanabe-Okochi N, Kurokawa M. A role for RUNX1 in hematopoiesis and myeloid leukemia. Int J Hematol. 2013;97:726–734. doi: 10.1007/s12185-013-1347-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.