

Abstract
Purpose of Review
Advances in the field of monocyte and macrophage biology have dramatically changed our understanding of their role during homeostasis and inflammation. Here we review the role of these important innate immune effectors in the lung during inflammatory challenges including lung transplantation.
Recent Findings
Neutrophil extravasation into lung tissue and the alveolar space have been shown to be pathogenic during acute lung injury as well as primary graft dysfunction following lung transplantation. Recent advances in lung immunology have demonstrated the remarkable plasticity of both monocytes and macrophages and demonstrated their importance as mediators of neutrophil recruitment and transendothelial migration during inflammation.
Summary
Monocytes and macrophages are emerging as key players in mediating both the pathogen response and sterile lung inflammation, including that arising from barotrauma and ischemia-reperfusion injury. Ongoing studies will establish the mechanisms by which these monocytes and macrophages initiate a variety of immune response that lay the fundamental basis of injury response in the lung.
Keywords: Monocytes, macrophages, lung transplantation, primary graft dysfunction
Introduction
Monocytes and macrophages are members of the mononuclear phagocyte system, a component of innate immunity. Monocytes are bone marrow derived leukocytes that circulate in the blood and spleen. They are characterized by their ability to recognize “danger signals” via pattern recognition receptors. Monocytes can phagocytose and present antigens, secrete chemokines, and proliferate in response to infection and injury. Once recruited to tissues, monocytes are capable of differentiating into macrophages and dendritic cells. Macrophages, on the other hand, are generally considered terminally differentiated cells that phagocytose pathogens or toxins, secrete chemokines to recruit other immune cells, and migrate to local lymph node beds via lymphatics where they present processed antigens. Here we will discuss the current understanding of monocyte and macrophage function in lung immunobiology based on animal and human studies. We will review their role in homeostasis and response to lung infection and tissue injury. Additionally, we will review their importance in lung transplantation with a focus on ischemia-reperfusion injury, primary graft dysfunction, and allograft rejection.
Monocytes and Macrophages in Lung Homeostasis
While once thought of as a lineage of cells with macrophages as the terminally differentiated cell in a progression from monocyte to macrophage, studies over the past twenty years have proven otherwise. Advances in our understanding of monocyte and macrophage biology have direct applications to the lung and maintenance of its homeostasis. Together, in a coordinated fashion, monocytes and macrophages survey the lung (Figure 1A).
Figure 1.
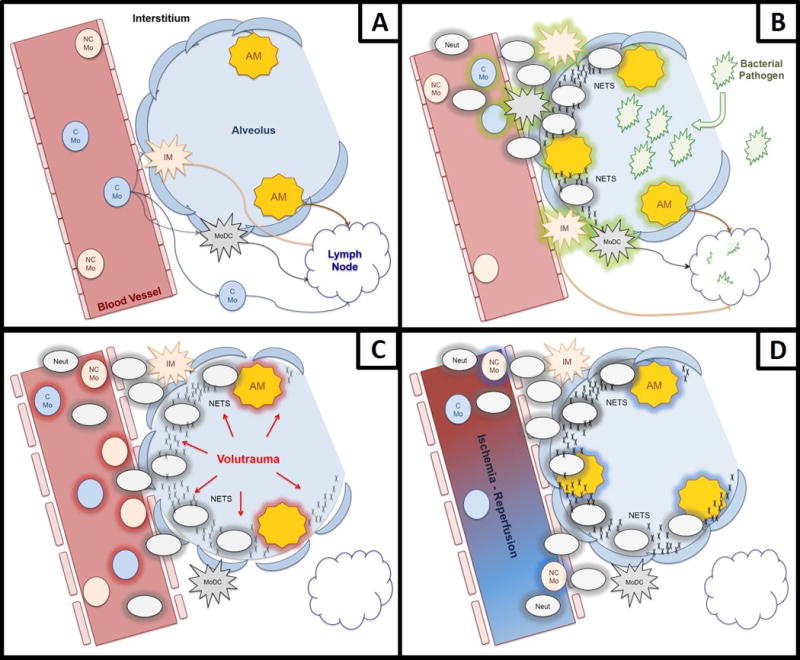
Role of Lung Monocytes and Macrophages in Homeostasis, Bacterial Infection, Barotrauma, and Ischemia-Reperfusion Injury/PGD. (A) In homeostasis, lung non-classical monocytes patrol the endothelium, while classical monocytes survey the parenchyma. Classical monocytes can differentiate into interstitial macrophages or monocyte-derived dendritic cells, or remain undifferentiated while monitoring for infection or injury signals. Alveolar macrophages are resident in the lung, independent from monocytes and remain in the alveolar space. (B) During pathogen challenge in the alveolar space, alveolar macrophages are the first to be activated, releasing chemokines and cytokines which then recruit monocytes and neutrophils. Neutrophils, on arrival to the site of infection, are capable of degranulating and generating neutrophil extracellular traps in an effort to combat infection. (C) During mechanical ventilation, stretch-induced injury activates endothelium, alveolar macrophages, and epithelium, causing recruitment and activation of both classical and non-classical monocytes and the recruitment of neutrophils. (D) During ischemia-reperfusion, alveolar macrophages and monocytes become activated, recruiting neutrophils, and if the injury is severe enough, they can lead to primary graft dysfunction.
Monocytes
Much of our knowledge about monocytes comes from studies using mouse tissues and human blood. In both humans and mice, monocytes can be subdivided into: (1) classical inflammatory monocytes that are Ly6ChighCCR2+ in mice and CD14+CD16−CCR2+ in humans; and (2) non-classical endothelial patrolling monocytes that are Ly6ClowCX3CR1high in mice and CD14dimCD16+CX3CR1high in humans (1). A third group of intermediate monocytes that are Ly6CintCX3CR1high in mice and CD14+CD16+CX3CR1high in humans have also been identified, but their specific role is incompletely characterized. It is likely that they have functions distinct from classical and non-classical monocytes (2).
Classical Monocytes
Classical monocytes are well-characterized. In response to infection or injury, they proliferate in the bone marrow, are released into circulation in a CCR2-dependent manner and home to the site of interest via a chemokine gradient (3). During bacterial infection, for example, these monocytes home to the site of infection and phagocytose pathogens, secrete a distinct set of chemokines that lead to recruitment of other immune cells, and present antigen via class II MHC (4). Murine studies have also revealed that these monocytes can exit the vasculature, and without further differentiating, survey the tissue microenvironment, before departing via the lymphatics (5). The spleen functions as a reservoir for monocytes. In response to signals emanating from a distant tissue injury, for instance release of angiotensin II during myocardial infarction, these cells can be mobilized to the site of injury from the spleen (6).
In the lung, these cells are capable of differentiating into pulmonary dendritic cells and macrophages. Utilizing a combination of fate-mapping techniques, parabionts, and fluorescent reporter murine studies, investigators have shown that Ly6Chigh classical monocytes differentiated into both the CD11b+CD103− and CD11b−CD103+ dendritic cell subsets (7). In addition, these cells differentiate into macrophages when recruited to the lung and help replenish the tissue resident macrophage pool when depleted either by injury or through experimental manipulation (8).
Non-classical and Intermediate Monocytes
Non-classical endothelial patrolling monocytes were first reported in 2007 (9) and studies of their biology and function remain ongoing. These cells are likely the descendants of classical monocytes that have returned to the bone marrow and under the control of Nur77 (Nr4a1) mature into non-classical monocytes (10). They display a distinct motility and crawling pattern. In vivo imaging studies have shown non-classical monocytes crawling along the luminal side of the endothelium. This integrin-dependent crawling is independent of the direction of blood flow. In fact, it is frequently against the direction of flow (11, 12). In the lung, non-classical monocytes are known to be capable of differentiating into CD11b+CD103− dendritic cells (7). Recent studies have further attempted to explore the role of these monocytes. Investigators have shown that non-classical monocytes are involved in intraluminal surveillance of the endothelium and phagocytosis of injured endothelium along with recruitment of neutrophils to the site of injury (13). In addition, these cells have also been shown to limit tumor metastases to the lung via the CX3CR1-CX3CL1 axis (14). Studies of human non-classical monocytes have shown that these monocytes sense nucleic acids and viruses via TLR7 signaling and can initiate the innate immune response by secreting cytokines (15). These monocytes can be mobilized from the marginalized vascular compartment during vigorous exercise, sepsis, and after cardiac surgery (16–18) and are depleted by high-dose glucocorticoid treatment (19, 20).
However, there are conflicting reports regarding the function of non-classical monocytes. Some studies suggest this population secretes pro-inflammatory cytokines, while others suggest they are anti-inflammatory and promote the resolution of inflammation and the initiation of healing and fibrosis (14). Difficulty differentiating non-classical monocytes from intermediate monocytes by cell surface marker expression may account for the conflicting reports regarding the function of these cells. In fact, as non-classical monocytes have become the subject of recent more intense investigation, it has become clear that there is a population of “intermediate” monocytes. Murine Ly6C and human CD14 are expressed in monocytes at high, intermediate, and low levels as a continuum, and intermediate levels of these surface markers along with expression of the cell surface marker 6-sulfo LacNac (slan) identify the intermediate monocyte subset. When studied as distinct populations, intermediate monocytes show higher expression of MHCII and are more closely related to classical than non-classical monocytes based on transcriptome analyses (21, 22).
Macrophages
Macrophages exist in tissues as sentinels, sensing pathogens and injury. While once thought of as being constantly replenished by circulating monocytes, murine studies over the past decade have revealed tissue-resident macrophages to be capable of self-renewal. These include the Kupffer cells in the liver, red pulp macrophages in the spleen, Langerhans cells in the skin, cardiac-resident macrophages, and alveolar macrophages in the lung (23, 24).
Cardiac-resident and lung-resident macrophages have marked heterogeneity. A recent study identified distinct populations of cardiac macrophages, some of which were capable of self-renewal, and others which were replenished by circulating monocytes (25). Similarly, in the lung, alveolar macrophages and lung-resident interstitial macrophages appear to be distinct populations with independent lineages (26). These populations, while heterogenous, appear capable of carrying out the functions traditionally considered to be of macrophages, including phagocytosis of pathogen, efferocytosis of injured host cells, secretion of cytokines upon activation, and migration via lymphatics to present antigen, initiating the adaptive immune response.
Macrophage activation occurs in two distinct varieties. The first is “classical” or “M1” activation, which leads to a pro-inflammatory phenotype. In response to extra- or intra-cellular pathogens, M1 macrophages upregulate inducible nitric oxide synthase and secrete pro-inflammatory chemokines and cytokines via pattern recognition receptors. They also present antigen via MHC class II, initiating inflammation, recruitment of granulocytes, and a Type-1 helper T cell response. The second, “alternative” or “M2” activation is more varied in its phenotype. In response to IL-4 and IL-13 during allergy response or parasite infection, M2 macrophages secrete histamine and promote killing and encapsulation of parasites. Outside of the realm of pathogen response, M2-activated macrophages are capable of down-regulating the initial inflammatory response and promoting the resolution of inflammation and initiation of tissue healing and fibrosis (27). Importantly, the M1/M2 classification of activation is likely too dichotomous in its characterization and the actual activation states of macrophages likely is better represented along a continuum in response to a variety of stimuli with responses ranging from pro-inflammatory to anti-inflammatory, demonstrating the remarkable plasticity of macrophages (28, 29).
Monocyte and Macrophage Responses to Mechanisms of Host Injury after Lung Transplant
Patients undergoing lung transplant are subject to a multitude of infectious and inflammatory insults. These include ischemia-reperfusion injury (IRI), mechanical ventilation, and infection by commensal and lung pathogens. Any of these insults can result in primary graft dysfunction (PGD), acute lung injury, and acute respiratory distress syndrome, depending on the etiology (Figure 1B through 1D). In the worst lung injury phenotypes, regardless of etiology, neutrophils infiltrate the alveolar space and neutrophil extracellular traps (NETS) are formed (30–33). When released, NETS are meant to help capture and facilitate removal of pathogens (34). However, when the inflammatory response becomes overwhelming, NETS can become pathogenic and contribute to the cycle of atelectrauma, ineffective gas exchange across the alveolar-capillary barrier, and respiratory failure. A review of neutrophils is beyond the scope of this review. However, it is important to understand their role in inflammation, as they are frequently the effector cells recruited by monocytes and macrophages. Infiltration of the lung by neutrophils and the presence of NETS are also frequently used as signs of severe lung injury.
Pathogen Response
Lung transplant recipients are particularly susceptible to pathogens. Long-standing lung disease is frequently complicated by prior infections, some frequent and with repeated hospitalizations. Long-standing or incompletely treated infections, in addition to donor-acquired pathogens, immunosuppression, mechanical ventilation, and mandatory ICU stay, all place recipients at high risk of lung injury from pathogens.
Both monocytes and macrophages utilize pattern recognition-receptors to recognize pathogens and initiate their response. Classical monocytes are mobilized in response to bacterial, fungal, protozoal, and viral pathogens (35). On arrival to inflamed or infected tissue, they differentiate into either dendritic cells or macrophages. This distinction is not clear-cut, but rather based on cytokine profile and immunophenotyping. For example, in models of mouse colitis, Ly6Chigh monocytes are recruited to the intestine and give rise to CX3CR1+ dendritic cells which then phagocytose bacteria and transport them to mesenteric lymph nodes to induce T-cell response (36). The same circulating Ly6Chigh monocytes are capable of differentiating into TNF-α and inducible nitric oxide producing DCs or differentiate into macrophages which, depending on environmental cues, can either contribute to a pro-inflammatory milieu or resolution of inflammation (37, 38). Current guidelines recommend referring to these cells as simply “monocyte-derived cells” or MoDCs to avoid confusion, as the functions can overlap (39).
In another layer of complexity, non-classical monocytes are also recruited, albeit in a less robust fashion, in the initial response to inflammation. In the initial comprehensive report of their biology, they participated in the initial inflammatory response through the secretion of TNF and chemokines (9). Their response has been particularly well studied in response to circulating nucleic acids or viral infection via TLR7/8, endothelial injury via TLR7, or tumor metastasis control via CX3CR1 (14, 15). Altogether, the studies on non-classical monocytes portray a population that appears to be resident vascular endothelial macrophages.
Distinct from the intravascular monocyte are the resident alveolar macrophage. In the lung, alveolar macrophages serve as the “first responders” to pathogens, particulate matter, and tissue injury. Along with alveolar epithelial cells, they comprise the mucosal barrier of the lung. As a result, alveolar macrophages have been implicated in both inflammatory and fibrotic lung states. While depletion of alveolar macrophages has been shown to decrease lung injury, it is also associated with decreased pathogen clearance, demonstrating the crucial role for macrophages in host defense (40).
Ventilator-Induced Lung Injury
As all lung transplant recipients are mechanically ventilated during the post-operative period, and some for prolonged periods of time, we review here current knowledge of the contribution of monocytes and macrophages to mechanical ventilator-induced lung injury (VILI). VILI has been well studied in animal models. The prevailing hypothesis is that the mechanical stress induced on the alveolar unit causes release of damage-associated molecular patterns (DAMPs), which can ligate toll-like receptors (TLRs), nucleotide-binding oligomerization domain-like receptors (NLRs) and the receptor for advanced glycosylation end products (RAGE). DAMPs can be recognized by both alveolar macrophages and monocytes and act downstream through several effector mechanisms.
Animal studies have shown that depletion of alveolar macrophages attenuates the severity of VILI (41). Mechanistic studies have revealed the response to be inflammasome-mediated, where the stretch injury causes NLR-containing pyrin domain 3 (NLRP3) activation and induces the release of IL-1β from alveolar macrophages (42–44). Additional studies showed alveolar macrophage high-mobility group box-1 (HMGB1) and possibly transient receptor potential vanilloid 4 (TRPV4) to be involved in neutrophil recruitment to the alveolar space and lung vascular permeability and lung edema (45–47). Human studies have correlated BAL levels of NLRP3 mRNA with short periods of high tidal volume ventilation (44).
An additional line of investigation has implicated lung-marginated monocytes in the pathophysiology of VILI. In these studies, classical monocytes were recruited to the lung during high tidal volume ventilation and depletion of these monocytes was protective against lung injury (48, 49). In a recent study, the mechanism of transduction of stretch injury to the lung was revealed to first involve pulmonary endothelium, then alveolar epithelium, and lastly alveolar macrophages (50). Together, the studies suggest that, in animal models, both pulmonary monocytes and macrophages mediate VILI.
Ischemia-Reperfusion Injury and Primary Graft Dysfunction
Ischemia-reperfusion injury is the sterile inflammation that results from the resumption of blood flow after the mandatory ischemic period during clinical lung transplantation. This total ischemia time period consists of cold ischemia, during which the organ is transported in cold storage; and warm ischemia, during which the organ is taken out of cold storage for implantation. The total ischemia for double lung transplantation is generally six hours or less in which each lung undergoes about 1–2 hours of warm ischemia. Warm ischemia has been studied using hilar clamping, during which either an isolated pulmonary artery or the entire pulmonary hilum is temporarily clamped for a time period of 30 minutes to two hours (51). This model of pulmonary IRI is associated with severe lung injury mediated by TLR and NLR inflammasome signaling. Cold ischemia and primary graft dysfunction have been studied using a murine single lung transplant model, with up to 18 hours of allograft cold ischemia time prior to implantation (52).
The majority of work to-date has focused on the role of the alveolar macrophage in IRI and PGD. Both IRI and PGD are characterized by early neutrophil infiltration of the alveolar spaces and recent studies have shown that this leads to the formation of neutrophil extracellular traps (NETs) and lung injury (33). Indeed, macrophage-induced early reperfusion injury and neutrophil recruitment has been postulated and verified by several groups using animal models of both warm and cold ischemia (53–57), but there is some conflicting evidence as to whether depletion of alveolar macrophages is protective or injurious (58).
However, there is also evidence that implicates the involvement of monocytes in IRI. In one study utilizing intravital multiphoton microscopy of the lung and the murine lung transplant model, monocytes and neutrophils were found surveying the lung at steady-state. Four hours after allograft implantation, dynamic clusters of neutrophils associated with monocytes which were absent when blood monocytes were depleted, suggesting a role for monocyte-mediated extravasation of neutrophils during inflammation (59). Furthermore, our recent data shows that depletion of intravascular monocytes utilizing clodronate-loaded liposomes in mice abrogates neutrophil extravasation at 24 hours after single-lung transplant (manuscript under preparation). We have found that there are pulmonary intravascular monocytes that mediate transendothelial migration of neutrophils in lung allografts, leading to PGD (60). Additionally, in a recent study of the leukocyte filter trap in an ex vivo lung perfusion (EVLP) circuit, human monocytes capable of differentiating into MoDCs were identified (61). Given the protective effect of EVLP against PGD, this finding may implicate monocytes as a critical player in the pathogenesis of PGD.
While alveolar macrophage and epithelial cell injury and dysfunction can cause lung injury, there is a component of endothelial dysfunction that has yet to be incorporated into current understanding of primary graft dysfunction pathogenesis. The endothelial dysfunction that occurs during IRI and PGD increases expression of cell adhesion molecules necessary for arrest of monocytes and neutrophils, implicating a possible role for classical monocytes. The injured endothelium also generates injurious reactive oxygen species and nitric oxide, and as non-classical monocytes are responsible for phagocytosis and disposal of injured or necrotic endothelial cells, their role in PGD and IRI is worth investigating.
Allograft Rejection
While primary graft dysfunction is a phenomenon of innate immunity, allograft rejection has traditionally been thought to be exclusively within the domain of adaptive immunity. However, there is a strong association between primary graft dysfunction and chronic allograft rejection (62) and few studies have been done to link the two entities. In one line of investigation, experimental depletion of monocytes in murine models of heart and kidney transplantation was shown to blunt T cell-mediated rejection (63). In another study of murine lung transplant, CD4 T cells isolated from allogeneic lung recipients lacking circulating classical monocytes were protected against allorecognition, but allografts still experienced acute rejection (64). Thus, evidence exists that monocytes and macrophages may link the innate and adaptive immune responses after transplantation, but further studies are necessary to fully elucidate these processes.
Conclusion
Monocytes and macrophages in the lung are crucial players in host innate immune defense against pathogens and processes of sterile inflammation involved in lung transplantation, including mechanical ventilation, ischemia-reperfusion, and primary graft dysfunction. A link between monocytes, macrophages, and allograft rejection likely exists and should be further characterized. Further studies are needed to elucidate the role of monocytes and macrophages in the pathogenesis of primary graft dysfunction and lung allograft rejection.
Key Points.
Monocytes and macrophages in the lung are crucial players in host innate immune defense against pathogens.
Processes of sterile inflammation involved in lung transplantation, including mechanical ventilation, ischemia-reperfusion, and primary graft dysfunction have been shown to be monocyte and macrophage dependent.
Further studies are needed to elucidate the role of monocytes and macrophages in the pathogenesis of primary graft dysfunction and lung allograft rejection.
Acknowledgments
We thank Ms. Elena Susan for administrative assistance in submission of this manuscript.
Financial support and sponsorship: S.C. is supported by NIH Grant #T32 DK077662. A.B. is supported by NIH Grant #K08 HL125940, an AATS John H. Gibbon Jr. Research Award, and grants from the American Lung Association, Gift of Hope, and the LUNGevity Foundation.
Footnotes
Conflicts of interest: None.
References and Recommended Reading
- 1 *.Mitchell AJ, Roediger B, Weninger W. Monocyte homeostasis and the plasticity of inflammatory monocytes. Cellular immunology. 2014;291(1–2):22–31. doi: 10.1016/j.cellimm.2014.05.010. In this comprehensive review the authors discuss the monocyte development and homeostatic control of monocyte populations. [DOI] [PubMed] [Google Scholar]
- 2 *.Ziegler-Heitbrock L. Blood Monocytes and Their Subsets: Established Features and Open Questions. Frontiers in immunology. 2015;6:423. doi: 10.3389/fimmu.2015.00423. This article discusses the cell surface markers to differentiate monocyte populations from other immune cells in humans. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nature immunology. 2006;7(3):311–7. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 4.Landsman L, Varol C, Jung S. Distinct differentiation potential of blood monocyte subsets in the lung. Journal of immunology. 2007;178(4):2000–7. doi: 10.4049/jimmunol.178.4.2000. [DOI] [PubMed] [Google Scholar]
- 5.Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. 2013;39(3):599–610. doi: 10.1016/j.immuni.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325(5940):612–6. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jakubzick C, Tacke F, Ginhoux F, Wagers AJ, van Rooijen N, Mack M, et al. Blood monocyte subsets differentially give rise to CD103+ and CD103- pulmonary dendritic cell populations. Journal of immunology. 2008;180(5):3019–27. doi: 10.4049/jimmunol.180.5.3019. [DOI] [PubMed] [Google Scholar]
- 8.Landsman L, Jung S. Lung macrophages serve as obligatory intermediate between blood monocytes and alveolar macrophages. Journal of immunology. 2007;179(6):3488–94. doi: 10.4049/jimmunol.179.6.3488. [DOI] [PubMed] [Google Scholar]
- 9.Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317(5838):666–70. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- 10.Hanna RN, Carlin LM, Hubbeling HG, Nackiewicz D, Green AM, Punt JA, et al. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C- monocytes. Nature immunology. 2011;12(8):778–85. doi: 10.1038/ni.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11 *.Collison JL, Carlin LM, Eichmann M, Geissmann F, Peakman M. Heterogeneity in the Locomotory Behavior of Human Monocyte Subsets over Human Vascular Endothelium In Vitro. Journal of immunology. 2015;195(3):1162–70. doi: 10.4049/jimmunol.1401806. This paper characterizes the locomotory behaviour of endothelial bound monocytes. [DOI] [PubMed] [Google Scholar]
- 12.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19(1):71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 13.Carlin LM, Stamatiades EG, Auffray C, Hanna RN, Glover L, Vizcay-Barrena G, et al. Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell. 2013;153(2):362–75. doi: 10.1016/j.cell.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomas G, Tacke R, Hedrick CC, Hanna RN. Nonclassical patrolling monocyte function in the vasculature. Arteriosclerosis, thrombosis, and vascular biology. 2015;35(6):1306–16. doi: 10.1161/ATVBAHA.114.304650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cros J, Cagnard N, Woollard K, Patey N, Zhang SY, Senechal B, et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity. 2010;33(3):375–86. doi: 10.1016/j.immuni.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steppich B, Dayyani F, Gruber R, Lorenz R, Mack M, Ziegler-Heitbrock HW. Selective mobilization of CD14(+)CD16(+) monocytes by exercise. American journal of physiology Cell physiology. 2000;279(3):C578–86. doi: 10.1152/ajpcell.2000.279.3.C578. [DOI] [PubMed] [Google Scholar]
- 17.Fingerle G, Pforte A, Passlick B, Blumenstein M, Strobel M, Ziegler-Heitbrock HW. The novel subset of CD14+/CD16+ blood monocytes is expanded in sepsis patients. Blood. 1993;82(10):3170–6. [PubMed] [Google Scholar]
- 18.Fingerle-Rowson G, Auers J, Kreuzer E, Fraunberger P, Blumenstein M, Ziegler-Heitbrock LH. Expansion of CD14+CD16+ monocytes in critically ill cardiac surgery patients. Inflammation. 1998;22(4):367–79. doi: 10.1023/a:1022316815196. [DOI] [PubMed] [Google Scholar]
- 19.Fingerle-Rowson G, Angstwurm M, Andreesen R, Ziegler-Heitbrock HW. Selective depletion of CD14+ CD16+ monocytes by glucocorticoid therapy. Clinical and experimental immunology. 1998;112(3):501–6. doi: 10.1046/j.1365-2249.1998.00617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dayyani F, Belge KU, Frankenberger M, Mack M, Berki T, Ziegler-Heitbrock L. Mechanism of glucocorticoid-induced depletion of human CD14+CD16+ monocytes. Journal of leukocyte biology. 2003;74(1):33–9. doi: 10.1189/jlb.1202612. [DOI] [PubMed] [Google Scholar]
- 21.Frankenberger M, Hofer TP, Marei A, Dayyani F, Schewe S, Strasser C, et al. Transcript profiling of CD16-positive monocytes reveals a unique molecular fingerprint. European journal of immunology. 2012;42(4):957–74. doi: 10.1002/eji.201141907. [DOI] [PubMed] [Google Scholar]
- 22.Hofer TP, Zawada AM, Frankenberger M, Skokann K, Satzl AA, Gesierich W, et al. slan-defined subsets of CD16-positive monocytes: impact of granulomatous inflammation and M-CSF receptor mutation. Blood. 2015;126(24):2601–10. doi: 10.1182/blood-2015-06-651331. [DOI] [PubMed] [Google Scholar]
- 23.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38(1):79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38(4):792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 2014;40(1):91–104. doi: 10.1016/j.immuni.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26 *.Bharat A, Bhorade SM, Morales-Nebreda L, McQuattie-Pimentel AC, Soberanes S, Ridge K, et al. Flow Cytometry Reveals Similarities Between Lung Macrophages in Humans and Mice. Am J Respir Cell Mol Biol. 2016;54(1):147–9. doi: 10.1165/rcmb.2015-0147LE. This paper characterizes the monocyte and macrophage populations in murine and human lungs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445–55. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hussell T, Bell TJ. Alveolar macrophages: plasticity in a tissue-specific context. Nature reviews Immunology. 2014;14(2):81–93. doi: 10.1038/nri3600. [DOI] [PubMed] [Google Scholar]
- 29.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, et al. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. The American journal of pathology. 2011;179(1):199–210. doi: 10.1016/j.ajpath.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo L, Zhang S, Wang Y, Rahman M, Syk I, Zhang E, et al. Proinflammatory role of neutrophil extracellular traps in abdominal sepsis. American journal of physiology Lung cellular and molecular physiology. 2014;307(7):L586–96. doi: 10.1152/ajplung.00365.2013. [DOI] [PubMed] [Google Scholar]
- 32.Yildiz C, Palaniyar N, Otulakowski G, Khan MA, Post M, Kuebler WM, et al. Mechanical ventilation induces neutrophil extracellular trap formation. Anesthesiology. 2015;122(4):864–75. doi: 10.1097/ALN.0000000000000605. [DOI] [PubMed] [Google Scholar]
- 33.Sayah DM, Mallavia B, Liu F, Ortiz-Munoz G, Caudrillier A, DerHovanessian A, et al. Neutrophil extracellular traps are pathogenic in primary graft dysfunction after lung transplantation. American journal of respiratory and critical care medicine. 2015;191(4):455–63. doi: 10.1164/rccm.201406-1086OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–5. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 35.Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nature reviews Immunology. 2011;11(11):762–74. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diehl GE, Longman RS, Zhang JX, Breart B, Galan C, Cuesta A, et al. Microbiota restricts trafficking of bacteria to mesenteric lymph nodes by CX(3)CR1(hi) cells. Nature. 2013;494(7435):116–20. doi: 10.1038/nature11809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 2003;19(1):59–70. doi: 10.1016/s1074-7613(03)00171-7. [DOI] [PubMed] [Google Scholar]
- 38.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327(5966):656–61. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guilliams M, van de Laar L. A Hitchhiker’s Guide to Myeloid Cell Subsets: Practical Implementation of a Novel Mononuclear Phagocyte Classification System. Frontiers in immunology. 2015;6:406. doi: 10.3389/fimmu.2015.00406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghoneim HE, Thomas PG, McCullers JA. Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. Journal of immunology. 2013;191(3):1250–9. doi: 10.4049/jimmunol.1300014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frank JA, Wray CM, McAuley DF, Schwendener R, Matthay MA. Alveolar macrophages contribute to alveolar barrier dysfunction in ventilator-induced lung injury. American journal of physiology Lung cellular and molecular physiology. 2006;291(6):L1191–8. doi: 10.1152/ajplung.00055.2006. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, Liu G, Dull RO, Schwartz DE, Hu G. Autophagy in pulmonary macrophages mediates lung inflammatory injury via NLRP3 inflammasome activation during mechanical ventilation. American journal of physiology Lung cellular and molecular physiology. 2014;307(2):L173–85. doi: 10.1152/ajplung.00083.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu J, Yan Z, Schwartz DE, Yu J, Malik AB, Hu G. Activation of NLRP3 inflammasome in alveolar macrophages contributes to mechanical stretch-induced lung inflammation and injury. Journal of immunology. 2013;190(7):3590–9. doi: 10.4049/jimmunol.1200860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuipers MT, Aslami H, Janczy JR, van der Sluijs KF, Vlaar AP, Wolthuis EK, et al. Ventilator-induced lung injury is mediated by the NLRP3 inflammasome. Anesthesiology. 2012;116(5):1104–15. doi: 10.1097/ALN.0b013e3182518bc0. [DOI] [PubMed] [Google Scholar]
- 45.Patel VS, Sitapara RA, Gore A, Phan B, Sharma L, Sampat V, et al. High Mobility Group Box-1 mediates hyperoxia-induced impairment of Pseudomonas aeruginosa clearance and inflammatory lung injury in mice. American journal of respiratory cell and molecular biology. 2013;48(3):280–7. doi: 10.1165/rcmb.2012-0279OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hamanaka K, Jian MY, Townsley MI, King JA, Liedtke W, Weber DS, et al. TRPV4 channels augment macrophage activation and ventilator-induced lung injury. American journal of physiology Lung cellular and molecular physiology. 2010;299(3):L353–62. doi: 10.1152/ajplung.00315.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ogawa EN, Ishizaka A, Tasaka S, Koh H, Ueno H, Amaya F, et al. Contribution of high-mobility group box-1 to the development of ventilator-induced lung injury. American journal of respiratory and critical care medicine. 2006;174(4):400–7. doi: 10.1164/rccm.200605-699OC. [DOI] [PubMed] [Google Scholar]
- 48.Wilson MR, O’Dea KP, Zhang D, Shearman AD, van Rooijen N, Takata M. Role of lung-marginated monocytes in an in vivo mouse model of ventilator-induced lung injury. American journal of respiratory and critical care medicine. 2009;179(10):914–22. doi: 10.1164/rccm.200806-877OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wakabayashi K, Wilson MR, Tatham KC, O’Dea KP, Takata M. Volutrauma, but not atelectrauma, induces systemic cytokine production by lung-marginated monocytes. Critical care medicine. 2014;42(1):e49–57. doi: 10.1097/CCM.0b013e31829a822a. [DOI] [PubMed] [Google Scholar]
- 50.Woods SJ, Waite AA, O’Dea KP, Halford P, Takata M, Wilson MR. Kinetic profiling of in vivo lung cellular inflammatory responses to mechanical ventilation. American journal of physiology Lung cellular and molecular physiology. 2015;308(9):L912–21. doi: 10.1152/ajplung.00048.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gielis JF, Jungraithmayr W, Boulet GA, Bogers JP, Weder W, Cos P, et al. A murine model of lung ischemia and reperfusion injury: tricks of the trade. The Journal of surgical research. 2015;194(2):659–66. doi: 10.1016/j.jss.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 52.Krupnick AS, Lin X, Li W, Okazaki M, Lai J, Sugimoto S, et al. Orthotopic mouse lung transplantation as experimental methodology to study transplant and tumor biology. Nature protocols. 2009;4(1):86–93. doi: 10.1038/nprot.2008.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53 *.Spahn JH, Li W, Bribriesco AC, Liu J, Shen H, Ibricevic A, et al. DAP12 expression in lung macrophages mediates ischemia/reperfusion injury by promoting neutrophil extravasation. J Immunol. 2015;194(8):4039–48. doi: 10.4049/jimmunol.1401415. This recent study demonstrates the role of lung macrophages during ischemia reperfusion injury using a murine lung transplant model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsushima Y, Jang JH, Yamada Y, Schwendener R, Suzuki K, Weder W, et al. The depletion of donor macrophages reduces ischaemia-reperfusion injury after mouse lung transplantation. European journal of cardio-thoracic surgery: official journal of the European Association for Cardio-thoracic Surgery. 2014;45(4):703–9. doi: 10.1093/ejcts/ezt489. [DOI] [PubMed] [Google Scholar]
- 55.Prakash A, Mesa KR, Wilhelmsen K, Xu F, Dodd-o JM, Hellman J. Alveolar macrophages and Toll-like receptor 4 mediate ventilated lung ischemia reperfusion injury in mice. Anesthesiology. 2012;117(4):822–35. doi: 10.1097/ALN.0b013e31826a4ae3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao M, Fernandez LG, Doctor A, Sharma AK, Zarbock A, Tribble CG, et al. Alveolar macrophage activation is a key initiation signal for acute lung ischemia-reperfusion injury. American journal of physiology Lung cellular and molecular physiology. 2006;291(5):L1018–26. doi: 10.1152/ajplung.00086.2006. [DOI] [PubMed] [Google Scholar]
- 57.Naidu BV, Krishnadasan B, Farivar AS, Woolley SM, Thomas R, Van Rooijen N, et al. Early activation of the alveolar macrophage is critical to the development of lung ischemia-reperfusion injury. The Journal of thoracic and cardiovascular surgery. 2003;126(1):200–7. doi: 10.1016/s0022-5223(03)00390-8. [DOI] [PubMed] [Google Scholar]
- 58.Nakamura T, Abu-Dahab R, Menger MD, Schafer U, Vollmar B, Wada H, et al. Depletion of alveolar macrophages by clodronate-liposomes aggravates ischemia-reperfusion injury of the lung. The Journal of heart and lung transplantation: the official publication of the International Society for Heart Transplantation. 2005;24(1):38–45. doi: 10.1016/j.healun.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 59.Kreisel D, Nava RG, Li W, Zinselmeyer BH, Wang B, Lai J, et al. In vivo two-photon imaging reveals monocyte-dependent neutrophil extravasation during pulmonary inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(42):18073–8. doi: 10.1073/pnas.1008737107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chiu SZZ, Sun H, DeCamp MM, Kreisel D, Budinger GRS, Perlman H, Misharin A, Bharat A. A novel subset of donor-derived intravascular monocytes mediates ischemia-reperfusion injury following lung transplanation. Chicago Surgical Society Annual Resident Research Meeting. 2015 [Google Scholar]
- 61.Stone JP, Sevenoaks H, Sjoberg T, Steen S, Yonan N, Fildes JE. Mechanical removal of dendritic cell-generating non-classical monocytes via ex vivo lung perfusion. The Journal of heart and lung transplantation: the official publication of the International Society for Heart Transplantation. 2014;33(8):864–9. doi: 10.1016/j.healun.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 62.Bharat A, Kuo E, Steward N, Aloush A, Hachem R, Trulock EP, et al. Immunological link between primary graft dysfunction and chronic lung allograft rejection. The Annals of thoracic surgery. 2008;86(1):189–95. doi: 10.1016/j.athoracsur.2008.03.073. discussion 96–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oberbarnscheidt MH, Zeng Q, Li Q, Dai H, Williams AL, Shlomchik WD, et al. Non-self recognition by monocytes initiates allograft rejection. The Journal of clinical investigation. 2014;124(8):3579–89. doi: 10.1172/JCI74370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gelman AE, Okazaki M, Sugimoto S, Li W, Kornfeld CG, Lai J, et al. CCR2 regulates monocyte recruitment as well as CD4 T1 allorecognition after lung transplantation. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2010;10(5):1189–99. doi: 10.1111/j.1600-6143.2010.03101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]