

Abstract
Background:
Mitochondria are sensitive to environmental toxicants due to their lack of repair capacity. Changes in mitochondrial DNA (mtDNA) content may represent a biologically relevant intermediate outcome in mechanisms linking air pollution and fetal growth restriction.
Objective:
We investigated whether placental mtDNA content is a possible mediator of the association between prenatal nitrogen dioxide (NO2) exposure and birth weight.
Methods:
We used data from two independent European cohorts: INMA (n = 376; Spain) and ENVIRONAGE (n = 550; Belgium). Relative placental mtDNA content was determined as the ratio of two mitochondrial genes (MT-ND1 and MTF3212/R3319) to two control genes (RPLP0 and ACTB). Effect estimates for individual cohorts and the pooled data set were calculated using multiple linear regression and mixed models. We also performed a mediation analysis.
Results:
Pooled estimates indicated that a 10-μg/m3 increment in average NO2 exposure during pregnancy was associated with a 4.9% decrease in placental mtDNA content (95% CI: –9.3, –0.3%) and a 48-g decrease (95% CI: –87, –9 g) in birth weight. However, the association with birth weight was significant for INMA (–66 g; 95% CI: –111, –23 g) but not for ENVIRONAGE (–20 g; 95% CI: –101, 62 g). Placental mtDNA content was associated with significantly higher mean birth weight (pooled analysis, interquartile range increase: 140 g; 95% CI: 43, 237 g). Mediation analysis estimates, which were derived for the INMA cohort only, suggested that 10% (95% CI: 6.6, 13.0 g) of the association between prenatal NO2 and birth weight was mediated by changes in placental mtDNA content.
Conclusion:
Our results suggest that mtDNA content can be one of the potential mediators of the association between prenatal air pollution exposure and birth weight.
Citation:
Clemente DB, Casas M, Vilahur N, Begiristain H, Bustamante M, Carsin AE, Fernández MF, Fierens F, Gyselaers W, Iñiguez C, Janssen BG, Lefebvre W, Llop S, Olea N, Pedersen M, Pieters N, Santa Marina L, Souto A, Tardón A, Vanpoucke C, Vrijheid M, Sunyer J, Nawrot TS. 2016. Prenatal ambient air pollution, placental mitochondrial DNA content, and birth weight in the INMA (Spain) and ENVIRONAGE (Belgium) birth cohorts. Environ Health Perspect 124:659–665; http://dx.doi.org/10.1289/ehp.1408981
Introduction
In recent years, traffic-related air pollution has been considered an important risk factor for adverse reproductive health effects. Prenatal exposure to nitrogen dioxide (NO2) has been associated with low birth weight, intrauterine growth restriction, and preterm birth (Pedersen et al. 2013; Stieb et al. 2012). Infants with low birth weight are at higher risk of mortality and morbidity and impaired cognitive development compared with infants with higher birth weight (Gluckman et al. 2008; Risnes et al. 2011).
Mitochondria are intracellular organelles that are essential for the aerobic production of adenosine triphosphate (ATP) by oxidative phosphorylation (OXPHOS). These “power plants of the cell” also play a critical role in signaling transduction for cell proliferation, apoptosis, calcium storage, and metabolism (Clay Montier et al. 2009; Lamson and Plaza 2002; Lee and Wei 2005). Several studies have identified the generation of oxidative stress, by producing reactive oxygen species (ROS), as one of the major mechanisms by which air pollution exerts adverse biological effects (Chahine et al. 2007; Li et al. 2008). Mitochondria are the major intracellular sources of ROS, which are generated under normal conditions as by-product of OXPHOS (Hou et al. 2010). On the other hand, mitochondria are also the primary targets of oxidative stress because, in comparison with nuclear DNA (nDNA), mitochondrial DNA (mtDNA) lacks the protective strategies associated with nDNA, such as protective histones, chromatine structure, and sufficient DNA repair capacity (Lee and Wei 2000). Consequently, mtDNA is particularly vulnerable to ROS-induced damage and has a high mutation rate (Lamson and Plaza 2002). Mitochondria compensate for these mutations by increasing their number and their replication rate, resulting in a change in mtDNA content, which therefore reflects mitochondrial damage and dysfunction (Clay Montier et al. 2009; Hou et al. 2010; Lamson and Plaza 2002; Sahin et al. 2011).
The placenta plays a unique role in the transfer of gases, nutrients, and waste between the mother and developing child. It is both a metabolic and an endocrine organ. However, the placenta has a limited capability to metabolize a large number of foreign compounds (Storvik et al. 2014). The placenta requires energy to maintain its function, and this energy provision is regulated by mitochondrial function of placenta cells (Myllynen et al. 2005). Air pollution exposure is hypothesized to affect the fetus directly through transplacental exposure or indirectly by affecting maternal health and body functions (Morello-Frosch et al. 2010). This could impair the placental exchange of nutrients and gases. Under poor nutritional conditions the fetus can adapt its mitochondrial structure and metabolism. Therefore, this “metabolic reprogramming” could be at the origin of adverse birth outcomes (Gemma et al. 2006).
Recently, it has been shown that exposure to particulate air pollution during pregnancy was associated with placental mtDNA content (Janssen et al. 2012). We hypothesized that changes in mtDNA content may represent a biological causal effect along the path linking air pollution exposure with the potential adverse health effects of the offspring. Given two independent European birth cohorts (INMA and ENVIRONAGE), we aimed to assess the role of mediating effects of placental mtDNA content on the association of prenatal NO2 exposure with birth weight.
Materials and Methods
Study design and population. The Spanish population-based birth cohort study INMA (INfancia y Medio Ambiente; Childhood and Environment) recruited pregnant women in four centers (Valencia, Sabadell, Gipuzkoa, and Asturias), following a common protocol (Guxens et al. 2012). A total of 2,616 pregnant women were enrolled between 2004 and 2008 during the first trimester of pregnancy at public primary health care centers or public hospitals if they fulfilled the inclusion criteria: ≥ 16 years of age, a singleton pregnancy, intention to deliver at the reference hospital, no problems of communication, and no assisted conception. Of all eligible pregnant women, 57% agreed to participate. The present analysis included 390 mother–newborn pairs from the INMA cohort with placental mtDNA content data. A comparison of this INMA subcohort with the whole INMA cohort (n = 2,616) did not show differences in maternal age, pregestational body mass index (BMI), parity, and ethnicity.
In the population-based birth cohort study ENVIRONAGE (ENVIRonmental influence ON AGEing), 556 pregnant women were enrolled between 2010 and 2013 at the South-East-Limburg Hospital in Genk (Belgium) when they arrived for delivery. The inclusion criteria were ≥ 18 years of age, singleton pregnancy, and ability to fill out questionnaires in Dutch. The overall participation rate of eligible mothers was 56%. A comparison of the ENVIRONAGE birth cohort with all births in Flanders (Cox et al. 2013) did not show differences in maternal age, pregestational BMI, parity, and ethnicity.
Study approval was obtained from the ethics committees of each participating center and written informed consent was obtained from the mothers. In both cohorts information on maternal age, ethnicity, maternal smoking status, place of residence, prepregnancy BMI, and parity was obtained. In INMA they were obtained by questionnaires and interviews, in ENVIRONAGE by questionnaires. Perinatal parameters such as newborn’s sex, birth date, birth weight, gestational age, and delivery by cesarean section were collected by birth records. Details about maternal and child characteristics were standardized to perform a harmonized, pooled analysis.
Sample collection. In the INMA cohort, placentas were randomly collected in approximately one of four deliveries (n = 502). The entire placentas were frozen after delivery at –20°C until they were transferred on dry ice to the Hospital Universitario San Cecilio (HUSC) Biobank in Granada (Spain) and stored at –86°C. No information was available about the time between delivery and storage at the different Spanish hospitals involved in the study. MtDNA content was measured in 390 out of the 502 randomly selected placentas in INMA. In the ENVIRONAGE cohort, all placentas (n = 556) were collected after delivery and were frozen within 10 min at –20°C. In both cohorts, placentas were thawed minimally to obtain tissue biopsies for DNA extraction. To minimize the impact of within-placenta variability, biopsies were all taken 1–1.5 cm below the chorioamniotic membrane at a fixed location and preserved at –80°C (Janssen et al. 2012).
DNA extraction and measurement of mtDNA content. In the INMA cohort, DNA was extracted from placental tissue cells using the DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA, USA) following the manufacturer’s instructions. In the ENVIRONAGE cohort, DNA was extracted from placental tissue cells using the QIAamp DNA minikit (Qiagen Inc., Venlo, Netherlands) following the manufacturer’s instructions. In both cohorts, DNA samples were quantified using the Nanodrop spectrophotometer (ND-1000; Isogen Life Science, De Meern, Netherlands) and the Quant-iT™ PicoGreen® dsDNA Assay Kit (Life Technologies, Foster City, CA, USA) using the Omega Fluostar plate reader (BMG LABTECH, Ortenberg, Germany).
MtDNA content was measured in placental tissue cells by determining the ratio of two mitochondrial genes [mitochondrial encoded NADH dehydrogenase subunit 1 (MT-ND1) and mitochondrial forward primer for nucleotide 3212 and reverse primer from nucleotide 3319 (MTF3212/R3319)] to two nuclear control genes [acidic ribosomal phosphoprotein P0 (RPLP0), and beta-actin (ACTB)] using a quantitative real-time polymerase chain reaction (qPCR) assay (Janssen et al. 2012). qPCR was performed using 2.5 μL of extracted DNA (5 ng/μL) and 7.5 μL of master mix consisting of Fast SYBR® Green I dye 2× (5 μL/reaction; Life Technologies), forward (0.3 μL/reaction–300 nM) and reverse (0.3 μL/reaction–300 nM) primer (Biolegio, Nijmegen, Netherlands) and RNase free water (1.9 μL/reaction). Samples were run in triplicate in a 384-well format. qPCR was performed using the 7900HT Fast Real-Time PCR System (Life Technologies) with the following thermal cycling profile: 20 sec at 95°C (activation), followed by 40 cycles of 1 sec at 95°C (denaturation) and 20 sec at 60°C (annealing/extension). At the end of each run, a melting curve analysis was performed to confirm amplification specificity and absence of primer dimers (15 sec at 19°C, 15 sec at 60°C, 15 sec at 95°C). qBase software (Biogazelle, Zwijnaarde, Belgium) was used to normalize data and correct for run-to-run differences (Hellemans et al. 2007).
Ambient air pollution assessment. In INMA, ambient concentrations of NO2 were measured with the aid of passive samplers (Radiello, Fundazione Salvatore Maugeri, Padua, Italy) distributed outside across the study areas according to geographic criteria, taking into account the expected pollution gradients and the distribution of the residences of the participating women. The samplers remained exposed during various 7-day sampling periods through pregnancy. The methodology has been described in detail elsewhere (Aguilera et al. 2008; Iñiguez et al. 2009), and further sampling information is given in Supplemental Material, Table S1. Temporally adjusted land use regression (LUR) models were used to predict NO2 levels at women’s residential addresses and taking into account residential changes if women lived at least 2 months of pregnancy in the new residence. To calculate individual exposure during pregnancy, annual average NO2 estimates from the LUR were temporally adjusted using serial records from the network of monitoring stations covering each of the four INMA study areas. The validation statistics gave a spatial explained variance (R 2) for annual mean NO2 from 0.52 to 0.75 in the four INMA subcohorts (see Supplemental Material, Table S1).
In ENVIRONAGE, regional background levels of NO2 for each woman’s home address were calculated using a kriging interpolation method (Jacobs et al. 2010; Janssen et al. 2008) that uses land cover data obtained from satellite images combined with a dispersion model (Lefebvre et al. 2011). This model chain provided NO2 values, combining data from the Belgian telemetric air quality network, point sources and line sources, which were then interpolated to a high-resolution receptor grid. This method was used to obtain NO2 levels at women’s residential addresses, taking into account any residential change during pregnancy. The validation statistics gave a temporal explained variance (R 2) for hourly averages > 0.80 and a spatial explained variance (R 2) for annual mean NO2 of 0.82.
To explore potentially critical exposures during pregnancy, we calculated individual NO2 concentrations for the three trimesters of pregnancy using the same procedure used for the entire pregnancy: 1–13 weeks starting from date of conception (trimester 1), 14–28 weeks (trimester 2), and 29 weeks to delivery (trimester 3).
More details on exposure measurements for INMA and ENVIRONAGE can be found in Supplemental Material, Table S1.
Statistical analysis. Continuous data were checked for normality using the Shapiro–Wilk test statistic. Placental mtDNA content data were right skewed and therefore logarithmically transformed (log10). Generalized additive models (GAMs) were used to assess the linearity of the associations between a) prenatal NO2 exposure and mtDNA content, b) mtDNA content and birth weight, and c) prenatal NO2 exposure and birth weight (see Supplemental Material, Figure S1). Multiple linear regression models were used in ENVIRONAGE. Multiple linear mixed models with a random cohort effect were used in INMA and in the pooled data set (four INMA cohorts and ENVIRONAGE). Covariates used in the model were gestational age (linear and quadratic term), newborn’s sex, maternal age, ethnicity, parity, smoking status, education, season of birth (January–March, April–June, July–September, October–December), and prepregnancy maternal BMI. For the present analysis, we excluded 14 mother–newborn pairs from INMA and 6 mother–newborn pairs from ENVIRONAGE with missing values in the outcome, exposure, and confounders. After these exclusions, the final study population consisted of 376 subjects for INMA and 550 subjects for ENVIRONAGE.
To determine whether placental mtDNA content is a potential mediator of the association between prenatal NO2 exposure and birth weight, we performed a mediation analysis. The direct effect (DE), indirect effect (IE), and total effect (TE) were estimated using the SAS macro developed by Valeri and VanderWeele (2013). When assumptions of the mediation analysis hold, the DE represents the effect of prenatal NO2 exposure on birth weight after controlling for mtDNA content, and the IE is the estimated effect of NO2 exposure during pregnancy operating through mtDNA content (Valeri and VanderWeele 2013). The proportion of mediation by placental mtDNA content was calculated as the ratio of IE to TE.
A sensitivity analysis was performed, in which all nonvaginal deliveries were excluded, because it has been suggested that the fetus could be exposed to different levels of oxidative stress depending on the type of delivery (Inanc et al. 2005). In another sensitivity analysis we used cohort as a fixed effect instead of a random effect. Further, we tested the interaction between mtDNA content and sex on birth weight by including its interaction term in the full model. In INMA we also performed an additional sensitivity analysis taking into account the time–activity patterns of the women during pregnancy. Because it has been indicated that time–activity patterns during pregnancy should be considered to improve the accuracy of exposure measurement and reduce exposure misclassification (Estarlich et al. 2011), we calculated the time spent at home from self-reported information (questionnaire at week 32) and restricted in INMA our analysis to women who spent ≥ 15 hr/day at home (Estarlich et al. 2011). This information was not available for the ENVIRONAGE cohort.
All statistical analysis were conducted using SAS software (version 9.3; SAS Institute Inc., Cary, NC, USA).
Results
Characteristics of the study population. Characteristics of the 376 and 550 mother–newborn pairs in, respectively, the INMA and the ENVIRONAGE cohort are shown in Table 1. INMA mothers were more likely to be primiparous, older, lower educated, of European origin, and had lower BMI compared with mothers from ENVIRONAGE. The mean birth weight was lower and placental mtDNA content higher in INMA newborns compared with newborns from ENVIRONAGE.
Table 1.
Characteristics of INMA and ENVIRONAGE participants.
| Characteristics | INMA (n = 376) | ENVIRONAGE (n = 550) |
|---|---|---|
| Maternal | ||
| Age (years) | 32.2 ± 3.9* | 29.0 ± 4.6* |
| Smoking | ||
| Never | 170 (45.2)* | 354 (64.4)* |
| Quit smoking before week 12 | 143 (38.0)* | 119 (21.6)* |
| During entire pregnancy | 63 (16.8)* | 77 (14.0)* |
| Education | ||
| Primary school or none | 75 (20.0)* | 67 (12.2)* |
| Secondary school | 167 (44.4)* | 203 (36.9)* |
| University | 134 (35.6)* | 280 (50.9)* |
| Parity | ||
| 1 | 212 (56.4) | 299 (52.4) |
| 2 | 138 (36.7) | 195 (35.5) |
| ≥ 3 | 26 (6.9) | 56 (10.2) |
| Prepregnancy BMI (kg/m2) | 23.5 ± 4.4 | 24.1 ± 4.5 |
| Ethnicity | ||
| European | 343 (91.2) | 485 (88.2) |
| Non-European | 33 (8.8) | 65 (11.8) |
| Cohort | ||
| Valencia | 63 (16.8) | NA |
| Asturias | 37 (9.8) | NA |
| Sabadell | 120 (31.9) | NA |
| Gipuzkoa | 156 (41.5) | NA |
| ENVIRONAGE | NA | 550 (100.0) |
| Time spent at home | ||
| > 15 hr/day | 214 (56.9) | NA |
| ≤ 15 hr/day | 162 (43.1) | NA |
| Newborn | ||
| Gestational age (weeks) | 39.9 ± 1.3 | 39.3 ± 1.2 |
| Sex | ||
| Male | 194 (51.6) | 277 (50.4) |
| Female | 182 (48.4) | 273 (49.4) |
| Season at birth | ||
| January–March | 99 (26.3) | 156 (28.4) |
| April–June | 102 (27.1) | 131 (23.8) |
| July–September | 92 (24.5) | 143 (26.0) |
| October–December | 83 (22.1) | 120 (21.8) |
| Preterm delivery (< 37 weeks) | ||
| Yes | 7 (1.9) | 14 (2.6) |
| No | 369 (98.1) | 536 (97.5) |
| Vaginal delivery | ||
| No | 322 (85.6) | 521 (94.7) |
| Yes | 54 (14.4) | 29 (5.3) |
| Birth weight (g) | 3,290 ± 423* | 3,429.6 ± 432* |
| Placental mtDNA content | 1.3 (1.1–1.5)* | 1.0 (0.7–1.4)* |
| NA, not applicable. Continuous covariates expressed by mean ± SD or geometric mean and 25–75th percentile; categorical covariates are described by frequencies (%). Differences between cohorts were assessed using independent t-tests. Subjects without available information have been excluded before performing the independent t-tests.*p < 0.05. | ||
The mean (± SD) pregnancy average exposure to NO2 was 25.5 ± 11.4 μg/m3 and 21.1 ± 4.2 μg/m3 in INMA and ENVIRONAGE, respectively. Similar differences in exposure levels between cohorts were observed in the mean trimester concentrations (Table 2).
Table 2.
Descriptive statistics of prenatal NO2 exposure (μg/m3).
| NO2 exposure (μg/m3) | Mean ± SD | P5 | P25 | P50 | P75 | P95 | ra |
|---|---|---|---|---|---|---|---|
| INMA (n = 376) | |||||||
| Trimester 1 | 26.1 ± 12.9 | 5.6 | 16.4 | 23.1 | 33.7 | 74.2 | 0.91* |
| Trimester 2 | 25.6 ± 11.6 | 5.7 | 16.4 | 24.8 | 31.2 | 74.7 | 0.93* |
| Trimester 3 | 25.7 ± 12.1 | 5.7 | 16.9 | 23.8 | 32.3 | 74.4 | 0.92* |
| Entire pregnancy | 25.5 ± 11.4 | 5.7 | 17.2 | 24.0 | 32.3 | 66.7 | — |
| ENVIRONAGE (n = 550) | |||||||
| Trimester 1 | 20.7 ± 6.1 | 7.3 | 16.3 | 20.2 | 24.9 | 39.2 | 0.61* |
| Trimester 2 | 20.8 ± 6.0 | 8.6 | 16.2 | 20.5 | 25.1 | 46.0 | 0.86* |
| Trimester 3 | 21.4 ± 6.1 | 9.2 | 16.9 | 20.8 | 25.6 | 40.3 | 0.66* |
| Entire pregnancy | 21.1 ± 4.2 | 12.6 | 18.2 | 20.8 | 23.7 | 40.3 | — |
| INMA + ENVIRONAGE (n = 926) | |||||||
| Trimester 1 | 22.7 ± 9.8 | 5.6 | 16.1 | 21.2 | 26.8 | 74.2 | 0.86* |
| Trimester 2 | 22.6 ± 9.1 | 5.7 | 15.9 | 21.3 | 27.3 | 74.7 | 0.91* |
| Trimester 3 | 23.0 ± 9.3 | 5.7 | 16.7 | 21.5 | 27.3 | 74.4 | 0.88* |
| Entire pregnancy | 22.7 ± 8.3 | 5.7 | 17.6 | 21.2 | 25.6 | 66.7 | — |
| P, percentile. Continuous covariates expressed by mean ± SD.aPearson correlation between the pregnancy average and trimester-specific exposures. *p < 0.001. | |||||||
Association between placental mtDNA content and NO2 exposure. In the INMA cohort, NO2 exposure during each trimester and the entire pregnancy was negatively and significantly associated with placental mtDNA content (Table 3). These results were consistent in the direction of the associations in the four different INMA subcohorts (see Supplemental Material, Table S2). Each 10-μg/m3 increment in average pregnancy exposure was associated with a lower placental mtDNA content of 5.5% [95% confidence interval (CI): –8.8, –2.1%]. In the ENVIRONAGE cohort, NO2 exposure was also negatively associated with placental mtDNA content in each trimester and in the entire pregnancy, but it was statistically significant only during the second (–11.1%; 95% CI: –19.9, –1.2%) and third trimesters of pregnancy (–13.5%; 95% CI: –20.1, –6.4%) (Table 3). The pooled analysis showed a statistically significant association with exposure during the second and third trimesters as well as in the entire pregnancy (Table 3).
Table 3.
Percent change in placental mtDNA content in association with prenatal NO2 exposure in INMA, ENVIRONAGE, and in the pooled sample.
| Pregnancy period | Differences in placental mtDNA content (%) (95% CI) | p-Value |
|---|---|---|
| INMA (n = 376)a,b | ||
| Trimester 1 | –4.1 (–7.1, –1.1) | 0.007 |
| Trimester 2 | –5.0 (–8.0, –2.0) | 0.002 |
| Trimester 3 | –4.9 (–7.9, –1.8) | 0.003 |
| Entire pregnancy | –5.5 (–8.8, –2.1) | 0.002 |
| ENVIRONAGE (n = 550) | ||
| Trimester 1 | –5.1 (–15.5, 6.6) | 0.38 |
| Trimester 2 | –11.1 (–19.9, –1.24) | 0.03 |
| Trimester 3 | –13.5 (–20.1, –6.4) | 0.003 |
| Entire pregnancy | –10.1 (–20.1, 1.24) | 0.08 |
| INMA + ENVIRONAGE (n = 926)c | ||
| Trimester 1 | –2.5 (–6.4, 1.6) | 0.22 |
| Trimester 2 | –4.4 (–8.4, –0.3) | 0.04 |
| Trimester 3 | –5.2 (–9.1, –1.2) | 0.01 |
| Entire pregnancy | –4.9 (–9.3, –0.3) | 0.04 |
| Effect size was estimated for each 10-μg/m3 increment in exposure to NO2 at each mother’s residence during the corresponding period. Models were adjusted for newborn’s sex, maternal age, maternal smoking status, gestational age (linear and quadratic), prepregnancy BMI, parity, ethnicity, season of birth, and education.aResults followed the same direction in all four INMA subcohorts (see Supplemental Material, Table S2). bFour INMA subcohorts were included as random effect. cCohorts were included as random effect. | ||
Association between birth weight and NO2 exposure. The association between birth weight and prenatal NO2 exposure was significant in the INMA cohort for all three trimesters of pregnancy (Table 4). Each 10-μg/m3 increment in average pregnancy NO2 exposure was associated with a 66.4 g (95% CI: –111.0, –22.7) decrease in birth weight (Table 4). These results were consistent in the direction of the associations in the four different INMA subcohorts (see Supplemental Material, Table S3). In the ENVIRONAGE cohort, estimates were in the same direction although the estimated effects were smaller than in INMA and did not reach significance (–19.8 g; 95% CI: –101.1, 61.7). After both cohorts were pooled, the association between birth weight and NO2 was significant in all pregnancy trimesters and in the entire pregnancy (–47.5 g; 95% CI: –86.6, –8.5) (Table 4).
Table 4.
Association between prenatal NO2 exposure and birth weight in INMA, ENVIRONAGE, and in the pooled sample.
| Pregnancy period | Differences in birth weight (g) (95% CI) | p-Value |
|---|---|---|
| INMA (n = 376)a,b | ||
| Trimester 1 | –56.2 (–94.5, –17.8) | 0.004 |
| Trimester 2 | –56.3 (–96.2, –16.4) | 0.006 |
| Trimester 3 | –52.1 (–93.8, –12.5) | 0.01 |
| Entire pregnancy | –66.4 (–111.0, –22.7) | 0.004 |
| ENVIRONAGE (n = 550) | ||
| Trimester 1 | –20.0 (–91.3, 51.3) | 0.58 |
| Trimester 2 | –3.4 (–76.4, 69.5) | 0.93 |
| Trimester 3 | –29.9 (–98.2, 38.3) | 0.39 |
| Entire pregnancy | –19.8 (–101.1, 61.7) | 0.63 |
| INMA + ENVIRONAGE (n = 926)c | ||
| Trimester 1 | –44.1 (–77.4, –10.8) | 0.01 |
| Trimester 2 | –36.2 (–70.9, –1.6) | 0.04 |
| Trimester 3 | –37.5 (–71.4, –3.6) | 0.03 |
| Entire pregnancy | –47.5 (–86.6, –8.5) | 0.02 |
| Effect size was estimated for each 10-μg/m3 increment in exposure to NO2 at each mother’s residence during the corresponding period. Models were adjusted for newborn’s sex, season of birth, maternal age, maternal smoking status, parity, ethnicity, education, gestational age (linear and quadratic), and prepregnancy BMI.aResults followed the same direction in all four INMA subcohorts. (see Supplemental Material, Table S3). bFour INMA subcohorts were included as random effect. cCohorts were included as random effect. | ||
Association between placental mtDNA content and birth weight. Placental mtDNA content was positively and significantly associated with birth weight in both cohorts and in the pooled analysis (Table 5). Interaction tests showed that the interaction of mtDNA content with sex was significant for the individual cohorts and the pooled analysis. This suggests evidence of effect modification by sex. In the pooled analysis, an interquartile range (IQR) increase in mtDNA content was associated with a 66-g (95% CI: 18, 114) increase in mean birth weight in boys, compared with 26 g (95% CI: –67, 15) in girls (interaction p-value = 0.009) (Table 5).
Table 5.
Association between placental mtDNA content and birth weight (g) in INMA, EVIRONAGE, and in the pooled sample.
| INMAa,b | ENVIRONAGE | INMA + ENVIRONAGEc | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | Differences in birth weight (g) (95% CI) | p-Value | Interaction p-value | n | Differences in birth weight (g) (95% CI) | p-Value | Interaction p-value | n | Differences in birth weight (g) (95% CI) | p-Value | Interaction p-value | |
| All | 376 | 249.0 (83.6, 414.3) | 0.003 | 0.003 | 550 | 129.2 (7.8, 259.0) | 0.04 | 0.04 | 926 | 140.2 (43.2, 237.2) | 0.005 | 0.009 |
| Boys | 194 | 124.0 (45.6, 202.5) | 0.002 | NA | 277 | 34.0 (–34.4, 102.4) | 0.33 | NA | 471 | 65.9 (17.9, 114.0) | 0.007 | NA |
| Girls | 182 | –2.44 (–80.5, 75.6) | 0.95 | NA | 273 | –15.2 (–69.3, 39.0) | 0.58 | NA | 455 | 26.4 (–67.4, 14.6) | 0.21 | NA |
| NA, not applicable. Effect size was estimated for each IQR increment (INMA = 0.58; ENVIRONAGE = 0.77; pooled sample = 0.76) in mtDNA content. aModels were adjusted for gestational age (linear and quadratic), newborn’s sex, maternal age, maternal smoking status, prepregnancy BMI, parity, ethnicity, season of birth, education, and interaction term sex and mtDNA content. bFour INMA subcohorts were included as random effect. cCohorts were included as random effect. | ||||||||||||
Mediation analysis. In INMA, the mediation analysis suggested that 10% (95% CI: 6.6, 13.0) of the association between birth weight and pregnancy average NO2 exposure may be mediated by differences in placental mtDNA content (Figure 1A). The corresponding estimates for mediation of associations between birth weight and NO2 exposure during the first, second, and third trimesters, were 9.1% (95% CI: 5.3, 12.6), 11.4% (95% CI: 7.3, 15.2), and 12.2% (95% CI: 7.7, 16.3), respectively. When we limited the mediation analysis to boys in the INMA cohort, the analysis suggested a mediation effect of 16% (95% CI: 18.7, 13.2) (Figure 1B). Because in the ENVIRONAGE cohort prenatal NO2 exposure was not significantly associated with birth weight, we did not perform the subsequent mediation analysis. After pooling both cohorts, the estimated proportion of mediation by mtDNA content was not significant for the association between birth weight and pregnancy average NO2 exposure (2.2%; 95% CI: –2.0, 6.1), or for NO2 exposure during the different trimesters (see Supplemental Material, Table S4). Mediation analysis of the pooled data for boys suggested placental mtDNA content mediated 6.4% (95% CI: 2.4, 10.0) of the association between NO2 during trimester 3 and birth weight, but mediation was not statistically significant for average pregnancy NO2 or NO2 during trimester 1 or 2 (see Supplemental Material, Table S4).
Figure 1.
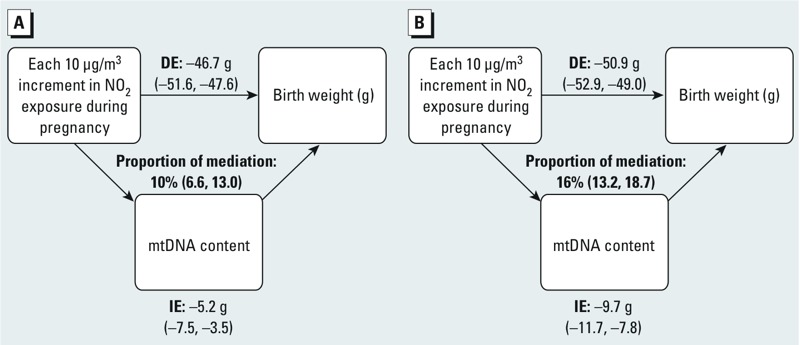
Mediation analysis of the estimated effect (95% CIs) of prenatal NO2 exposure (μg/m3) on birth weight through placental mtDNA content in the INMA cohort. (A) Whole INMA; (B) INMA boys. Results from mediation analysis with exposure to NO2 during the entire pregnancy were obtained using the SAS macro developed by Valeri and VanderWeele (2013). The figure shows placental mtDNA as a potential mediator, the estimates of indirect effect (IE), the estimates of the direct effect (DE), and proportion of mediation. Model was adjusted for gestational age (linear and quadratic term), newborn’s sex, maternal age, maternal smoking status, prepregnancy BMI, parity, ethnicity, season, education, and INMA subcohort.
Sensitivity analysis. The reported associations did not change after excluding mother–newborn pairs with nonvaginal deliveries (n = 83) (data not shown). Furthermore, when we used cohort as a fixed effect, our reported estimates did not change either (data not shown). Finally, associations were stronger when the analysis was limited to 214 INMA participants who spent ≥ 15 hr/day at home, including associations between NO2 and birth weight (e.g., for average pregnancy NO2: 84 g; 95% CI: –142, –26 compared with –66 g; 95% CI: –111, –23) (see Supplemental Material, Tables S5 and S6, respectively).
Discussion
Mitochondria, the energy producers of the cells, are particularly sensitive to environmental toxicants because they lack repair capacity (Clay Montier et al. 2009). Fetuses adapt their mitochondrial structure and metabolism when the supply of nutrients is limited (Gemma et al. 2006). We hypothesized that mitochondrial damage may be a causal intermediate in biological mechanisms linking air pollution to birth outcomes. In the present study, we evaluated placental mtDNA content, a proxy of mitochondrial damage, as a potential mediator of the association between reduced birth weight and prenatal NO2 exposure. The key findings, based on two independent European cohorts were that a) prenatal NO2 exposure is inversely associated with placental mtDNA content; b) placental mtDNA content is positively associated with birth weight; c) prenatal NO2 exposure is inversely associated with birth weight; and d) placental mtDNA content can be a potential mediator of the association between birth weight and prenatal NO2 exposure.
Ambient air contains a mixture of pollutants. NO2 is frequently used as a surrogate for traffic-related air pollution because it is considered to be a good proxy of other pollutants originating from the same sources (World Health Organization 2006). An appreciable number of epidemiologic studies have shown an association between fetal growth restriction and prenatal exposure to air pollution with evidence of statistical significant heterogeneity in the estimated effects among different locations (Parker et al. 2011; Stieb et al. 2012). An earlier observation on the full INMA cohort (n = 2,337) reported an estimated decrease in birth weight of 11 g for every 10-μg/m3 increment in NO2 (Estarlich et al. 2011). Data from 14 European mother–child cohorts, including INMA, reported a very weak association between birth weight of 1 g (95% CI: –6, 4 g) and NO2 during pregnancy (Pedersen et al. 2013). In our present study we estimated a 48-g reduction in birth weight (95% CI: –87, –9) with a 10-μg/m3 increment in pregnancy average NO2 based on the pooled analysis, with a stronger association in the INMA cohort (–66 g; 95% CI: –111, –23) than in the ENVIRONAGE cohort (–20 g; 95% CI: –101, 62). This association also varied among the four INMA subcohorts, ranging from –12 g (95% CI: –20, 12; n = 63) for the Valencia subcohort to –156 g (95% CI: –378, 67; n = 37) for the Asturias subcohort (see Supplemental Material, Table S4). Differences in effect sizes across the studied cohorts might be attributable to different levels of exposure, population variability, and variation in meteorological conditions, but also noncausal mechanisms could explain these differences—for example, differences in study design and conduct, exposure assessment, and differences in residual confounding. Nevertheless, our GAM plots showed linear associations between NO2 and mtDNA content, NO2 and birth weight, and mtDNA content and birth weight in both cohorts (see Supplemental Material, Figure S1).
We need to consider that our exposure assessment was limited to the residential address of the mothers: Exposure to other air pollutants (e.g., particulate matter and carbon monoxide), environmental and dietary contaminants that have been associated with lower birth weight, as well as exposure to NO2 during a commute, at work, and elsewhere was not taken into account. Time spent at home can influence associations between ambient air pollution measured at the mother’s residence and birth weight (Estarlich et al. 2011); in this regard we also found stronger associations within INMA for mothers who spend > 15 hr/day at home.
In contrast to the epidemiological evidence, the mechanisms responsible for fetal growth restriction due to air pollution are largely unknown. Hypotheses are that air pollutants could cause oxidative stress or inflammation, alter placental growth, decrease placental exchange of nutrients and gases, foster endocrine disruption, or cause maternal health effects—all of which could possibly lead to altered fetal growth (Kannan et al. 2006). Oxidative stress–induced DNA damage appears to be a particularly important mechanism of action of urban air pollutants (Møller et al. 2014). MtDNA is particularly vulnerable to ROS-induced damage and has been described as a proxy of air pollution–induced damage (Byun and Baccarelli 2014; Hou et al. 2010). In this study, we estimated that 10% (95% CI: 6.6, 13.0%) of low birth weight caused by prenatal NO2 exposure could be explained by placental mtDNA content. This finding was demonstrated in a subsample of 376 mother–child pairs of the INMA cohort. We are aware that epidemiological studies can only show associations, but cannot prove causality; nevertheless, our formal mediation analysis is based on a predefined hypothesis and is in line with experimental evidence.
Mechanisms through which prenatal exposure to traffic-related air pollution might cause placental inflammation and oxidative stress are unclear. The maternal and fetal circulation are separated by the placental barrier that is formed by the syncytiotrophoblast layer, which faces the maternal environment (Wick et al. 2010). This barrier contains placental transporters that can block or facilitate foreign compounds (Myllynen et al. 2005; Wick et al. 2010). Further, it has been reported that air pollution was associated with increased white blood cells in chronic obstructive pulmonary disease (COPD) patients, suggesting that air pollutants can elicit systemic inflammation (Bose et al. 2015). In addition, it has been observed that human plasma collected from individuals exposed to diesel exhaust for only short periods of time (1 hr) is proinflammatory to endothelial cells in vitro (Channell et al. 2012), implying that soluble, proinflammatory mediators circulate in the blood after inhalation of diesel. From these studies it might be hypothesized that maternal circulating proinflammatory mediators are responsible for associations of prenatal NO2 with placental mtDNA content and birth weight in our study population. Other mechanisms might include transient receptor potential (TRP) channels which are highly expressed in placenta, and their activation has been suggested to play important roles in placental development and regulating the fetal–maternal interface in mice models (Weldy et al. 2014). If air pollution exposure can result in systemic activation of TRP channels, we might speculate that placental TRP channels are also activated and may mediate our observed effects. In this context, it has been shown recently that TRP channels interact with a large number of mitochondrial proteins (Feng et al. 2013).
Induction of ROS levels stimulates autophagy and mitophagy as exemplified by lower mtDNA content in placental tissue (Kubli and Gustafsson 2012). In the present study, stratified analyses indicated a stronger inverse association between placental mtDNA content and prenatal NO2 exposure in newborn boys than in girls. Indeed sex-dependent susceptibility to oxidative stress has been shown and the antioxidant defenses are apparently different in XX and XY cells (Malorni et al. 2008).
Our study has some limitations. Ambient exposure to air pollution does not account for indoor exposure, which has also been associated with reduced birth weight (Nieuwenhuijsen et al. 2013). Although our results were consistent after multiple adjustments, some residual confounding by some unknown factors that are associated with ambient air pollution, mitochondrial function and mitochondrial DNA content, and birth weight cannot be excluded.
The major strengths of this study are that we tested the different windows of exposure, and made use of two independent birth cohorts in Southern (Spain) and Western (Belgium) Europe. Also, we used new methods and their assumptions to study causal interference (Valeri and VanderWeele 2013). Nevertheless, this mediation analysis gives only estimates of the DE, IE, and TE and the method assumes no uncontrolled confounding. Furthermore, our results of the association between prenatal air pollution exposure and mtDNA content are in line with those of Janssen et al. (2012) in the same birth cohort (ENVIRONAGE) with a smaller sample size. We also added more information by performing a mediation analysis that supports our hypothesis that mitochondrial damage may be a causal intermediate in biological mechanisms linking air pollution exposure to birth outcomes, which may provide some mechanistic clues to the adverse effects of early exposure to air pollution observed in humans.
In conclusion, we have shown that prenatal NO2 exposure was inversely associated with both placental mtDNA content and birth weight. Considering the high levels of NO2 in urban areas, which are increasing worldwide, this study indicates the relevance of further exploring this biological pathway linking early air pollution exposure and complications at birth.
Supplemental Material
Acknowledgments
We thank all the participants and collaborators in the INMA and ENVIRONAGE cohorts.
Footnotes
The research leading to these results was funded by the Spanish Ministry of Health (FIS-PI11/00610, FIS-PI041436, FIS-PI081151, FIS-PI042018, and FIS-PI09/02311), the European Union (EU) (FP7-ENV-2011 cod 282957 and HEALTH.2010.2.4.5-1), the Instituto de Salud Carlos III (Red INMA G03/176, CB06/02/0041, FIS-FEDER 03/1615, 04/1509, 04/1112, 04/1931, 05/1079, 05/1052, 06/1213, 07/0314, 09/02647, 11/01007, 11/02591, CP11/00178, FIS-PI06/0867, and FIS-PS09/00090), the Conselleria de Sanitat Generalitat Valenciana, the Generalitat de Catalunya-CIRIT (1999SGR 00241), the Obre Social Cajastur, the Universidad de Oviedo, the Department of Health of the Basque Government (2005111093 and 2009111069), and the Provincial Government of Gipuzkoa (DFG06/004 and DFG08/001). The ENVIRONAGE cohort is supported by the EU Program “Ideas” (ERC-2012-StG 310898) and by the Flemish Fund for Scientific Research (FWO 1516112N and G073315N).
The authors declare they have no actual or potential competing financial interests.
References
- Aguilera I, Sunyer J, Fernández-Patier R, Hoek G, Aguirre-Alfaro A, Meliefste K, et al. Estimation of outdoor NOx, NO2, and BTEX exposure in a cohort of pregnant women using land use regression modeling. Environ Sci Technol. 2008;42:815–821. doi: 10.1021/es0715492. [DOI] [PubMed] [Google Scholar]
- Bose S, Hansel NN, Tonorezos ES, Williams DL, Bilderback A, Breysse PN, et al. Indoor particulate matter associated with systemic inflammation in COPD. J Environ Prot (Irvine, Calif) 2015;6:566–572. [Google Scholar]
- Byun HM, Baccarelli A. Environmental exposure and mitochondrial epigenetics: study design and analytical challenges. Hum Genet. 2014;133:247–257. doi: 10.1007/s00439-013-1417-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahine T, Baccarelli A, Litonjua A, Wright RO, Suh H, Gold DR, et al. 2007. Particulate air pollution, oxidative stress genes, and heart rate variability in an elderly cohort. Environ Health Perspect 115 1617 1622; doi: 10.1289/ehp.10318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Channell MM, Paffett ML, Devlin RB, Madden MC, Campen MJ. Circulating factors induce coronary endothelial cell activation following exposure to inhaled diesel exhaust and nitrogen dioxide in humans: evidence from a novel translational in vitro model. Toxicol Sci. 2012;127:179–186. doi: 10.1093/toxsci/kfs084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay Montier LL, Denk JJ, Bai Y. Number matters: control of mammalian mitochondrial DNA copy number. J Genet Genomics. 2009;36:125–131. doi: 10.1016/S1673-8527(08)60099-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox B, Martens E, Nemery B, Vangronsveld J, Nawrot TS. 2013. Impact of a stepwise introduction of smoke-free legislation on the rate of preterm births: analysis of routinely collected birth data. BMJ 346 f441; doi: 10.1136/bmj.f441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estarlich M, Ballester F, Aguilera I, Fernández-Somoano A, Lertxundi A, Llop S, et al. 2011. Residential exposure to outdoor air pollution during pregnancy and anthropometric measures at birth in a multicenter cohort in Spain. Environ Health Perspect 119 1333 1338; doi: 10.1289/ehp.1002918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Li H, Tai Y, Huang J, Su Y, Abramowitz J, et al. Canonical transient receptor potential 3 channels regulate mitochondrial calcium uptake. Proc Natl Acad Sci USA. 2013;110:11011–11016. doi: 10.1073/pnas.1309531110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemma C, Sookoian S, Alvariñas J, García SI, Quintana L, Kanevsky D, et al. Mitochondrial DNA depletion in small- and large-for-gestational-age newborns. Obesity (Silver Spring) 2006;14:2193–2199. doi: 10.1038/oby.2006.257. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 2008;359:61–73. doi: 10.1056/NEJMra0708473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guxens M, Ballester F, Espada M, Fernández MF, Grimalt JO, Ibarluzea J, et al. Cohort profile: the INMA—INfancia y Medio Ambiente—(Environment And Childhood) Project. Int J Epidemiol. 2012;41:930–940. doi: 10.1093/ije/dyr054. [DOI] [PubMed] [Google Scholar]
- Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J. 2007. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 8 R19; doi: 10.1186/gb-2007-8-2-r19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Zhu Z, Zhang X, Nordio F, Bonzini M, Schwartz J, et al. 2010. Airborne particulate matter and mitochondrial damage: a cross-sectional study. Environ Health 9 48; doi: 10.1186/1476-069X-9-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inanc F, Kilinc M, Kiran G, Guven A, Kurutas EB, Cikim IG, et al. Relationship between oxidative stress in cord blood and route of delivery. Fetal Diagn Ther. 2005;20:450–453. doi: 10.1159/000086830. [DOI] [PubMed] [Google Scholar]
- Iñiguez C, Ballester F, Estarlich M, Llop S, Fernandez-Patier R, Aguirre-Alfaro A, et al. Estimation of personal NO2 exposure in a cohort of pregnant women. Sci Total Environ. 2009;407:6093–6099. doi: 10.1016/j.scitotenv.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Jacobs L, Emmerechts J, Mathieu C, Hoylaerts MF, Fierens F, Hoet PH, et al. 2010. Air pollution-related prothrombotic changes in persons with diabetes. Environ Health Perspect 118 191 196; doi: 10.1289/ehp.0900942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen BG, Munters E, Pieters N, Smeets K, Cox B, Cuypers A, et al. 2012. Placental mitochondrial DNA content and particulate air pollution during in utero life. Environ Health Perspect 120 1346 1352; doi: 10.1289/ehp.1104458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen S, Dumont G, Fierens F, Mensink C. Spatial interpolation of air pollution measurements using CORINE land cover data. Atmos Environ. 2008;42:4884–4903. [Google Scholar]
- Kannan S, Misra DP, Dvonch JT, Krishnakumar A. 2006. Exposures to airborne particulate matter and adverse perinatal outcomes: a biologically plausible mechanistic framework for exploring potential effect modification by nutrition. Environ Health Perspect 114 1636 1642; doi: 10.1289/ehp.9081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubli DA, Gustafsson ÅB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res. 2012;111:1208–1221. doi: 10.1161/CIRCRESAHA.112.265819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamson DW, Plaza SM. Mitochondrial factors in the pathogenesis of diabetes: a hypothesis for treatment. Altern Med Rev. 2002;7:94–111. [PubMed] [Google Scholar]
- Lee HC, Wei YH. Mitochondrial role in life and death of the cell. J Biomed Sci. 2000;7:2–15. doi: 10.1007/BF02255913. [DOI] [PubMed] [Google Scholar]
- Lee HC, Wei YH. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int J Biochem Cell Biol. 2005;37:822–834. doi: 10.1016/j.biocel.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Lefebvre W, Vercauteren J, Schrooten L, Janssen S, Degraeuwe B, Maenhaut W, et al. Validation of the MIMOSA-AURORA-IFDM model chain for policy support: Modeling concentrations of elemental carbon in Flanders. Atmos Environ. 2011;45:6705–6713. [Google Scholar]
- Li N, Xia T, Nel AE. The role of oxidative stress in ambient particulate matter-induced lung diseases and its implications in the toxicity of engineered nanoparticles. Free Radic Biol Med. 2008;44:1689–1699. doi: 10.1016/j.freeradbiomed.2008.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malorni W, Straface E, Matarrese P, Ascione B, Coinu R, Canu S, et al. Redox state and gender differences in vascular smooth muscle cells. FEBS Lett. 2008;582:635–642. doi: 10.1016/j.febslet.2008.01.034. [DOI] [PubMed] [Google Scholar]
- Møller P, Danielsen PH, Karottki DG, Jantzen K, Roursgaard M, Klingberg H, et al. Oxidative stress and inflammation generated DNA damage by exposure to air pollution particles. Mutat Res Rev Mutat Res. 2014;762:133–166. doi: 10.1016/j.mrrev.2014.09.001. [DOI] [PubMed] [Google Scholar]
- Morello-Frosch R, Jesdale BM, Sadd JL, Pastor M. 2010. Ambient air pollution exposure and full-term birth weight in California. Environ Health 9 44; doi: 10.1186/1476-069X-9-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myllynen P, Pasanen M, Pelkonen O. Human placenta: a human organ for developmental toxicology research and biomonitoring. Placenta. 2005;26:361–371. doi: 10.1016/j.placenta.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Nieuwenhuijsen MJ, Dadvand P, Grellier J, Martinez D, Vrijheid M. 2013. Environmental risk factors of pregnancy outcomes: a summary of recent meta-analyses of epidemiological studies. Environ Health 12 6; doi: 10.1186/1476-069X-12-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JD, Rich DQ, Glinianaia SV, Leem JH, Wartenberg D, Bell ML, et al. 2011. The International Collaboration on Air Pollution and Pregnancy Outcomes: initial results. Environ Health Perspect 119 1023 1028; doi: 10.1289/ehp.1002725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen M, Giorgis-Allemand L, Bernard C, Aguilera I, Andersen AMN, Ballester F, et al. Ambient air pollution and low birthweight: a European cohort study (ESCAPE). Lancet Respir Med. 2013;1:695–704. doi: 10.1016/S2213-2600(13)70192-9. [DOI] [PubMed] [Google Scholar]
- Risnes KR, Vatten LJ, Baker JL, Jameson K, Sovio U, Kajantie E, et al. Birthweight and mortality in adulthood: a systematic review and meta-analysis. Int J Epidemiol. 2011;40:647–661. doi: 10.1093/ije/dyq267. [DOI] [PubMed] [Google Scholar]
- Sahin E, Colla S, Liesa M, Moslehi J, Müller FL, Guo M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–365. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stieb DM, Chen L, Eshoul M, Judek S. Ambient air pollution, birth weight and preterm birth: a systematic review and meta-analysis. Environ Res. 2012;117:100–111. doi: 10.1016/j.envres.2012.05.007. [DOI] [PubMed] [Google Scholar]
- Storvik M, Huuskonen P, Pehkonen P, Pasanen M. The unique characteristics of the placental transcriptome and the hormonal metabolism enzymes in placenta. Reprod Toxicol. 2014;47:9–14. doi: 10.1016/j.reprotox.2014.04.010. [DOI] [PubMed] [Google Scholar]
- Valeri L, VanderWeele TJ. Mediation analysis allowing for exposure–mediator interactions and causal interpretation: theoretical assumptions and implementation with SAS and SPSS macros. Psychol Methods. 2013;18:137–150. doi: 10.1037/a0031034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weldy CS, Liu Y, Liggitt HD, Chin MT. 2014. In utero exposure to diesel exhaust air pollution promotes adverse intrauterine conditions, resulting in weight gain, altered blood pressure, and increased susceptibility to heart failure in adult mice. PLoS One 9 e88582; doi: 10.1371/journal.pone.0088582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick P, Malek A, Manser P, Meili D, Maeder-Althaus X, Diener L, et al. 2010. Barrier capacity of human placenta for nanosized materials. Environ Health Perspect 118 432 436; doi: 10.1289/ehp.0901200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. Air Quality Guidelines: Global Update 2005: Particulate Matter, Ozone, Nitrogen Dioxide and Sulfur Dioxide. 2006 Available: http://www.euro.who.int/__data/assets/pdf_file/0005/78638/E90038.pdf?ua=1 [accessed 15 July 2014] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.