

Abstract
Patient-derived tumor xenografts (PDTX) generally represent a kind of more reliable model of human disease, by which a potential drugs’ preclinical efficacy could be evaluated. To date, no stable gastrointestinal stromal tumor (GIST) PDTX models have been reported. In this study, we aimed to establish stable GIST PDTX models and to evaluate whether these models accurately reflected the histological feature of the corresponding patient tumors and create a reliable GIST PDTX models for our future experiment. By engrafting fresh patient GIST tissues into immune-compromised mice (BALB/c athymic mice), 4 PDTX models were established. Histological features were assessed by a qualified pathologist based on H&E staining, CD117 and DOG-1. We also conduct whole exome sequencing(WES) for the 4 established GIST PDTX models to test if the model still harbored the same mutation detected in corresponding patient tumors and get a more intensive vision for the genetic profile of the models we have established, which will help a lot for our future experiment. To explore the tumorigenesis mechanism for GIST, we also have a statistical analysis for the genes detected as nonsynchronous-mutated simultaneously in 4 samples. All 4 GIST PDTX models retained the histological features of the corresponding human tumors, with original morphology type and positive stains for CD117 and DOG-1. Between the GIST PDTX models and their parental tumors, a same mutation site was detected, which confirmed the genetic consistency. The stability of molecular profiles observed within the GIST PDTX models provides confidence in the utility and translational significance of these models for in vivo testing of personalized therapies. To date, we conducted the first study to successfully establish a GIST PDTX model whose genetic profiles were revealed by whole exome sequencing. Our experience could be of great use.
Keywords: Patient-derived tumor xenografts, gastrointestinal stromal tumor, whole exome sequencing
Introduction
Gastrointestinal stromal tumor (GIST) is the most common mesenchymal tumor of the alimentary canal with a morbidity of 12-20 patients per million annually [1]. Compared with other solid tumors, etiology of GIST is well characterized. More than 80% cases were caused by mutation of c-KIT and PDGFRA. Though recognized as the central event of development of GIST, mutations of c-KIT and PDGFRA were still not enough to explain the whole group because less than 20% cases are detected no mutation in either c-KIT or PDGFRA. This subgroup is called wild type GISTs (WT GISTs) [2].
Imatinib, a small molecule inhibitor of tyrosine kinases for c-KIT and BCR-ABL, have been recognized as the first-line drug for unresectable and resectable high-risk GIST patients [3]. The introduction of Imatinib in the treatment of GIST has revolutionized the result of GIST patients. More than 80% patients with metastatic or unresectable GIST treated with imatinib achieved a partial response or stable disease [4]. GIST has been the paradigm for the treatment of solid tumors. Despite the great success it achieved, Imatinib is rarely curative. GIST patients who were initially sensitive to the treatment of Imatinib could develop drug resistance in 2 years [5,6]. Among the secondary resistant group, nearly 50% cases could be attributed to the development of another c-KIT mutation. It has also been reported that some patients develop Imatinib resistance for c-KIT overexpression and amplification [7]. GIST patients who harbor PDGFRA D842 or BRAF mutation are primary resistant to Imatinib [8]. Most of the mechanism had not been completely elucidated. Sunitinib is the second-line drug for GIST. For GIST patients who develop secondary resistance, Sunitinib could be the best choice. However, its efficacy is limited, leaving GIST patients without an alternative approved treatment option [8]. It is urgent need to develop new more effective drug for GIST.
In the drug testing procedure, the most commonly cited reasons for high failure rate of new antineoplastic agents is the lack of representative preclinical models which include the whole gene expression profile in patients [9]. Nowadays, standard cell line derived xenografts and patient-derived tumor xenograft (PDTX) were most frequently used in the study of GIST [10-13]. Standard cell line derived xenografts models are constructed by injecting the tumor cells to immune deficiency mice subcutaneously. The tumor cells are easily obtained and tumor cells could undergo genetic modification during the cell line’s in-vitro culture [14]. However, the model is not enough when it comes to the test of new drug for GIST because various sites of mutations in GIST could not be represented by a single cell line. PDTX models are established by directly engrafting fresh human tumor tissues into immune deficient mice. PDTX models are preferred in the study of anticancer efficacy of antineoplastic agents, which inherit the complexity and biological characteristic of the original human tumors [15]. Previous study has firmly confirmed that PDTX model can faithfully represent the biology characteristics under the genome level.
Though the feasibility has been verified in the study of GIST [10-13], the expression profile of the model has not been elucidated in any of the studies before. Whole exons sequencing (WES) is a newly emerging technology developed to detect exon mutations in cells, tissues, organs [16]. Not only could the technology be applied to produce molecular fingerprints of disease processes and predict new potential targets for drug discovery, but also to quantify the gene expression profiles for comparison of original tumors and PDTX tumors [16]. WES has been applied in many studies before. Ioannis concocts WES in a spindle cell sarcoma and discover several fusion genes characterized by rearrangements of chromosome arm 12q and MDM2 amplification [17]. Kimbung identified liver metastasis-selective genes of breast cancer and revealed the adverse outcome was associated with the Extracellular matrix (stromal) genes by WES [18]. WES was also applied by Ho to reveal significantly dysregulated genes and signaling pathways in hepatocellular carcinoma [19].
In this study, we create four groups of GIST PDTX models which represent different clinical response to imatinib treatment. For the first time, WES was conducted to explore the expression profile of GIST PDTX models. By doing so, we could not only have a more intensive understanding of the model which would enable our future experiment, but primarily explore the associated pathway in the development of GISTs by conducting a statistical analysis for the data collected from the WES for the four GIST PDTX models.
Materials and methods
This study protocol was approved by the Ethical Committee of Zhongshan Hospital. After informed consent, tissue samples were obtained from the patient and PDTX models.
PDX establishment
In compliance with the protocol approved by the Institutional Review Board and with the subject’s informed consent, a fragment of surgically resected tumor tissue was used for xenotransplantation to establish PDTX models as described earlier [20]. Briefly, patient samples (designated as PA) were implanted subcutaneously in 6- to 8-week old female BALB/c athymic mice (Shanghai SLAC Laboratory Animal Co., Ltd., Shanghai). Once the first generation of xenografts (designed as P0) was established serial implantations were performed to expand the xenograft tumors (i.e. P1 and beyond). Tumor was measured using a digital caliper (Cal Pro, Sylvac, Switzerland). Tumor volume was calculated as 0.5 × length × width2. Tumor fragments (~200 mm3) at each passage were viably frozen in a freezing solution (10% DMSO, 20% FBS, and 70% RPMI 1640 medium) and stored in liquid nitrogen for future re-implantation. Additional fragments were either snap-frozen in liquid nitrogen, or fixed for histology. All procedures and protocols were approved by the Institutional Animal Care and Use Committee of Zhongshan Hospital.
Immunohistochemistry (IHC)
Five μm thick formalin-fixed paraffin-embedded sections were immunostained for CD-117 and DOG-1. Briefly, after de-paraffinization and antigen retrieval, the slides were incubated with CD117 antibody (1:200, ab5506, Abcam), DOG-1 antibody (1:200, ab111160, Abcam) overnight at 4°C. The slides were then washed and detected with REAL™ EnVision™ Detection System, Peroxidase/DAB+, Rabbit/Mouse (DAKO, #K5007).
Tissue processing for genomic studies
Genomic DNA was isolated using a QIAamp DNA mini kit (Qiagen). The expression was quantified using NanoDrop ND-1000 spectrophotometer (NanoDrop, Wilmington, DE). DNA samples with A260/280 ratios between 1.8 and 2.0 and A260/230 ratios above 2.0 and proven to be high quality by gel electrophoresis were used for WES and SNP 6.0 array analyses.
Whole exome sequencing
One microgram of each DNA sample isolated from P3 xenograft tumors was used for library construction using a TruSeq DNA sample preparation kit (Illumina, San Diego, CA). Libraries were pooled (500 ng each) for exome capture using the TruSeq Exome Enrichment kit (Illumina). Sequencing was then performed with paired-end 2 × 100 base reads on the Illumina HiSeq 2000 platform (Illumina). Raw FASTQ files were first processed by a proprietary algorithm to filter out mouse sequence contaminations. We have shown that this filter step does not affect the human SNP detection [21,22]. After mouse sequence removed, data was aligned to human reference genome hg19/GRCh37 by BWA 0.6.1 and processed to variants calling by GATK 1.6.
Statistical analysis
Mutated genes associated with cancer behaviors, no matter synchronous or non-synchronous, were selected from the data derived from the WES (Supplemental 1). To make a full use of the data, we select the 30 nonsynchronous-mutated genes existed simultaneously in 4 samples to conduct GO and Pathway analysis The details were listed below.
GO analysis
GO analysis was applied to analyze the main function of the differential expression genes according to the Gene Ontology which is the Wey functional classification of NCBI [23]. Generally, Fisher’s exact test and c2 test were used to classify the GO category, and the false discovery rate (FDR) was calculated to correct the P-value, the smaller the FDR, the small the error in judging the P-value [24]. The FDR was defined as FDR = 1-Nκ/T, where Nκ refers to the number of Fisher’s test P-values less than X2 test P-values. We computed P-values for the GOs of all the differential genes. Enrichment provides a measure of the significance of the function: as the enrichment increases, the corresponding function is more specific, which helps us to find those GOs with more concrete function description in the experiment. Within the significant category, the enrichment Re was given by: Re = (nf/n)/(Nf/N), where nf is the number of differential genes within the particular category, n is the total number of genes within the same category, Nf is the number of differential genes in the entire experimental data, and N is the total number of genes in the experimental data. The positive criteria is Log2FC > 1 or Log2FC < -1, FDR < 0.05 and P value < 0.05.
Pathway analysis
Similarly, pathway analysis was used to find out the significant pathway of the differential genes according to KEGG, Biocarta and Reactome [25-27]. Still, we turn to the Fisher’s exact test and X2 test to select the significant pathway, and the threshold of significance was defined by P-value and FDR. The significant pathway was identified by P-value < 0.05 and FDR < 0.05. The enrichment was calculated like the equation above. [26,28,29] The positive criteria is Log2FC > 1 or Log2FC < -1, FDR < 0.05 and P value < 0.05.
Results
PDTX model establishment
Original tumors were obtained from the GIST patients who underwent surgeries in our department in 2014. To make sure the success rate we get are repeatable, we choose a skillful researcher to conduct all the procedure, including the coverage and implantation. Among the 4 human GIST tissue engrafted mouse models, patient tumors grew up in 4 mouse models (P0) within three months post implantation. Furthermore, P1 tumor tissues continued growth after implantation into BALB/c athymic mice and were able to grow for more than three passages (P2, P3). The time required for the tumor becoming acquirable in P1, P2 and P3 was highly heterogeneous, ranging from 77 to 211 days (Figure 1). The final success rate for the procedure is 24 percent (4/17).
Figure 1.
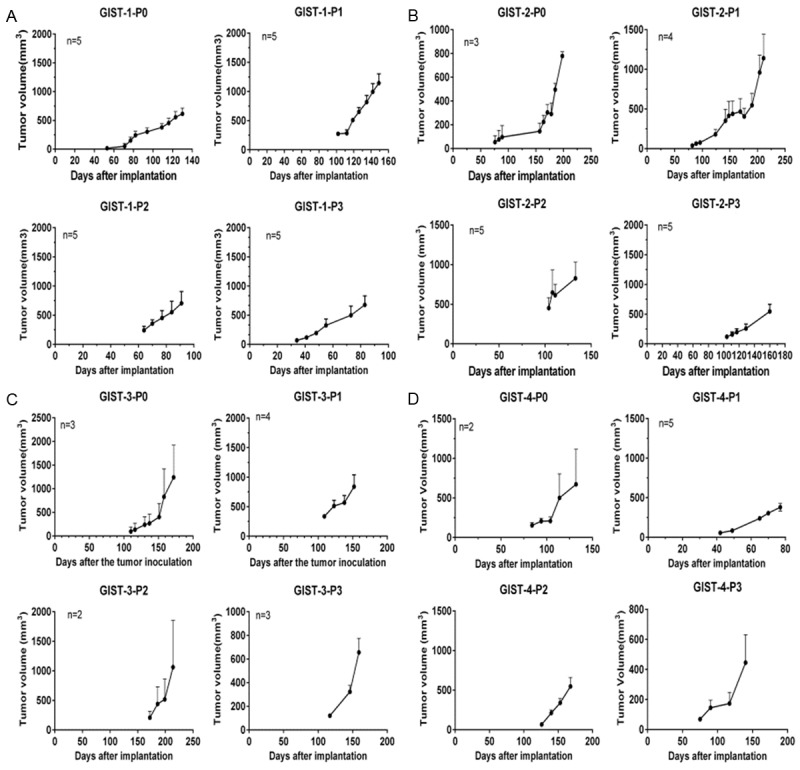
Growth curve for 4 GIST PDTX samples. GIST tumor tissues continued growth after implantation into BALB/c athymic mice and were passageable for more than three passages (P2, P3). The time required for the tumor becoming acquirable in P1, P2 and P3 was highly heterogeneous, ranging from 77 to 211 days. A-D. Indicate the period of cultivation process for GIST1, GIST2, GIST3 and GIST4, respectively.
To evaluate the potential impact of clinicopathological parameters of GIST patient on PDTX model establishment success rate, we compared patients’ clinical parameters by dividing the GIST patient tumors into ‘Established model’ and ‘Failed model’ groups as listed in Table 1. Statistical analysis revealed that the success rate of model establishment was independent of most patients’ pathological parameters such as gender, age, tumor diameter, mitotic index, occurring position or mutation site. However, models were more likely to be successfully established if derived from recurrent patients.
Table 1.
Clinical characteristics of 17 primary GISTs patients
| Parameter | Established model | Failed Model | P-value* | |
|---|---|---|---|---|
| Gender | Male | 3 | 6 | 0.59 |
| Female | 1 | 7 | ||
| Age | > 59 | 0 | 5 | 0.26 |
| ≤ 59 | 4 | 8 | ||
| Diameter (cm) | ≤ 10 | 2 | 7 | 1.00 |
| > 10 | 2 | 6 | ||
| Mitotic index (HPF) | ≤ 5 | 1 | 8 | 0.29 |
| > 5 | 3 | 5 | ||
| Position | Gastrointestinal tract | 1 | 8 | 0.29 |
| Outside the gastrointestinal tract | 3 | 5 | ||
| Mutation | c-KIT 11 exon | 1 | 8 | 0.29 |
| Non-c-KIT 11 exon | 3 | 5 | ||
| Sample | Primary | 1 | 12 | 0.02 |
| Recurrence | 3 | 1 |
p-value: Fisher exact test.
PDTX model stability
Histological assessment for H&E staining, CD117 and DOG-1 was performed by pathologist for all 4 GIST patient tumors and corresponding established PDTX models (P3). The original tumors and corresponding established PDTX models (P3) were highly consistent, with 4 GIST patient tumors and corresponding established PDTX models were all diagnosed as epithelial cell and spindle cells mix type. Though different expression level of CD117 and DOG-1 exist between original tumors and corresponding PDTX models, CD117 and DOG-1 were all detected positive in established PDTX models, confirming the diagnosis of GIST in PDTX models (Figure 2). The mutation in c-KIT or PDGFRA for PDTX models, which had been revealed by WES, was completely consistent with the mutation detected in the patients (Supplemental 1). All the results indicate that our PDTX models perfectly maintained the same histological and genetic features as their parental human tumors.
Figure 2.
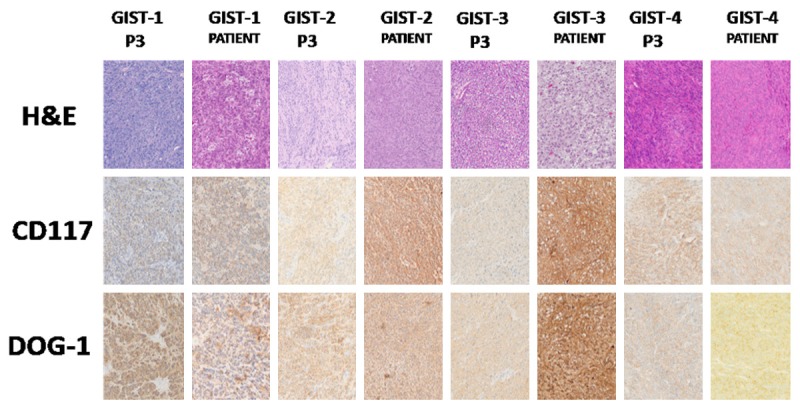
H&E and IHC for GIST PDTX models and corresponding patient tumors. The corresponding established PDTX models (P3) and original tumors were highly consistent, with 4 GIST patient tumors and corresponding established PDTX models all diagnosed as epithelial cell and spindle cells mix type. CD117 and DOG-1 were all detected positive in established PDTX models. The expression level of PDTX models (P3) and original tumors were not all consistent.
Statistical analysis
To characterize the gene profiles of GIST and further explore the tumorgenesis mechanism, we conducted GO and Pathway analysis based on the 30 nonsynchronous-mutated genes existed simultaneously in 4 samples. The GO terms considered as most important for GIST include lymphocyte differentiation, embryonic organ development and histone-serine phosphorylation in biological process (BP), histone serine kinase activity, 1-phosphatidylinositol-3-kinase activity and protein kinase activity in molecular function (MF), chromosome passenger complex, apindle and protein complex in cellular component (CC). Pathway terms include transcriptional misregulation, non-small cell lung cancer and colorectal cancer (Figure 3, Supplemental 2).
Figure 3.
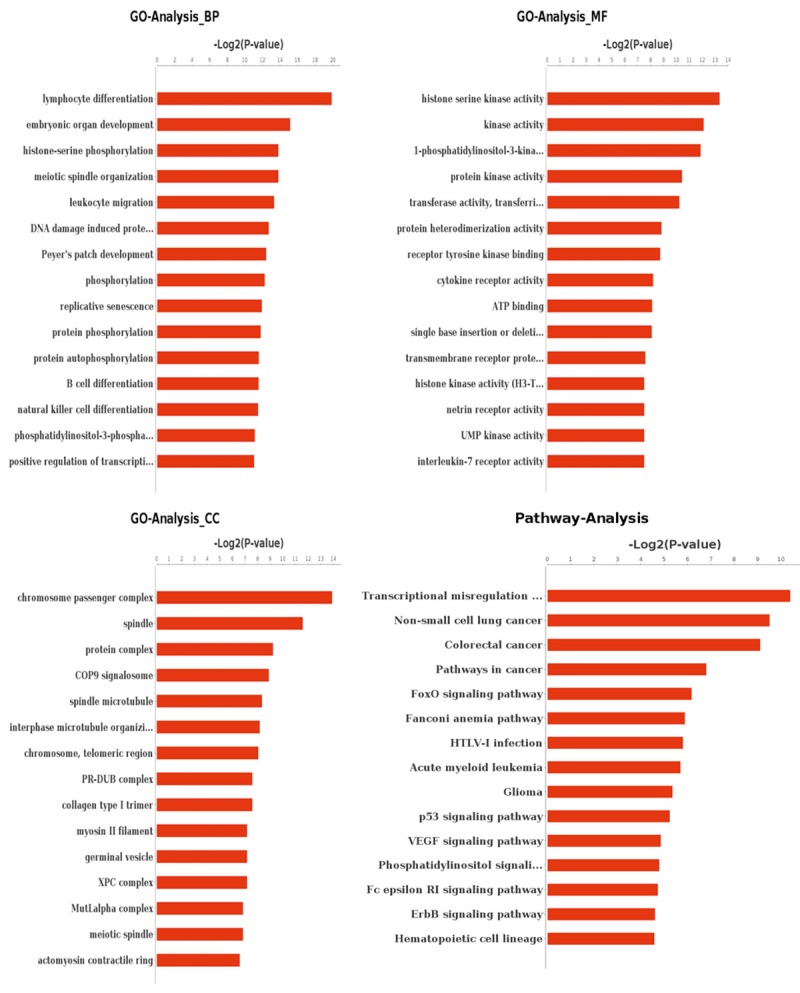
GO and Pathway analysis based on the 30 nonsynchronous-mutated genes existed simultaneously in 4 samples.
Discussion
The application of PDTX models has accelerated the development of new anti-cancer drugs. The establishment of PDTX models enables new drugs to be tested in different cancer simultaneously. Also, the concept of using PDTX models could protect patients who undergo new anti-cancer drug test from the potential toxicity [30]. Despite all the advantage PDTX models provide, questions still exists, especially for the stability of the models.
Regarding the stability of PDTX models, the most important question is whether the process of engraftment and expansion alters the genetic features of the tumors. Genetic studies have revealed that PDTX models maintain the key genes and global pathway activity in original tumors [15,31,32]. For example, in gastric cancer PDTX models, H&E staining and Genomic comparison was performed for several biomarkers including ERBB1, ERBB2, ERBB3, FGFR2, MET and PTEN. The results were proved highly consistent between the PDTX models and original tumors [15]. In non-small-cell lung cancer (NSCLC) PDTX models, unsupervised hierarchical clustering of genome-wide gene-expression profiles demonstrated that nine of the 17 original tumors clustered directly with the derived PDTX models, with correlation coefficients ranging from 0.78 to 0.95. Besides, 10 of the 17 primary-PDTX tumor pairs exhibited correlation coefficients > 0.90 indicating a high degree of similarity between the primary cancer and the corresponding PDTX model [32]. In pancreatic cancer PDTX models, KRAS mutation and SMAD4 expression status were found to be concordant for PDTX models and original tumors [31]. Previous studies inspire that PDTX model can fully represent the biological characteristics of the original tumors.
In our study, results of IHC were highly consistent in 4 GIST PDTX model samples, with CD117 and DOG-1 all positive. However, expression level were not all consistent between original tumors and PDTX models. Considering the quantity of our samples was small, it is difficult to predict whether the variation of expression level in that 4 sample was associated with the different mutation site. The possibility need further study to elaborate.
In our experiences, the establishment process of GIST PDTX models was relatively difficult for the low success rate of tumor implantation and long period of cultivation. Until now, seldom has a study been reported for successfully building a reliable PDTX model. The only study of the GIST PDTX models was reported by Jason [10]. It is the also the first orthotopic GIST PDTX models reported. Our models were different because the sites we choose for implantation were all subcutaneous. Because of the more complex procedure in building an orthotopic model. Our experiences could be of greater value for the majority.
Most of the previous studies have only reported the establishment process of the PDTX models and the test of a new drug. To have a further insight of the PDTX model, we believe that the gene profile of the model should be fully tested. The results of WES could not only inspire us to apply or develop anti-cancer drug for GISTs, but conduct a statistical analysis for find in different subgroups of GISTs. Though only 4 sample were included in the statistical analysis, we still believed the process was of potential use for wild type, IM-sensitive and IM-resistant GIST were included in our study. The result of GO and Pathway analysis for the common aberration genes indicated that proteins associated with lymphocyte differentiation, histone serine kinase activity and chromosome passenger complex and transcriptional misregulation pathway should be shed more light on, which lead new directions for our future experiment. After reviewing the previous studies, we find us to be the first to apply WES in the GIST PDTX models, and also the first to study the tumorgenesis mechanism of GIST by conducting GO and PATHWAY analysis for common aberrant genes in different GISTs subgroups.
The function of the aberrant genes detected in our study has been confirmed to be associated with the biological behaviors of GISTs by previous studies. Some of the proteins expressed by corresponding aberrant genes are proved to be the central event of tumor survival and some could be less important. Of all the aberrant genes we detect, ZNF has been a promising target because several studies has revealed the crucial role ZNF-overexpression played in the development of imatinib resistance for GIST. The first study was conducted by Rink from Fox Chase Cancer Center, Philadelphia, Pennsylvania, United States of America [33]. The studies indicated the genes associated with ZNF were highly specific to IM and another tyrosine kinase inhibitor (TKI), sunitinib, in GIST cells. They further conducted an RNAi approaches targeting the associated genes in GIST cells and find it becoming more sensitive to imatinib treatment. Also, overexpression of ZNF have been proved as predictors of poor response to imatinib treatment [34]. In our results, ZNF mutations are detected mutated in all 4 specimens. ETV1 was also a promising target for the treatment of GISTs. Several studies have confirmed the central role it plays in the development of GISTs [35-38]. Chi utilized three expression datasets to search for GIST specific genes that might provide new molecular insights, finally choosing ETV1 as the most promising one. They further studied the protein under the level of mouse models, GIST tissues and GIST cell lines which come to an impressed conclusion that abnormal expression of EVT1 could be the central event of GISTs [38]. Following the previous studies, the researchers published their new studies discovering that destabilizing ETV1 by inhibition of MAP kinase and KIT signaling could suppress GIST tumor growth [35]. Our results yet indicated that ETV1 mutations only exist in 3 specimens harboring c-KIT and PDGFRA mutation, not in WT specimen. It may indicate that the tumorigenesis mechanism of WT GISTs is different from GISTs with c-KIT and PDGFRA mutation. TET1 has been the hotspot in the study of WT GISTs because global epigenomic divergence has been discovered between GISTs with c-KIT and PDGFRA mutation and WT GISTs. TET1 has been considered to be the core of the theory [39,40]. TET1 mutations were also detected in 4 specimens.Despite the specimens volume of our study was small, the previous studies we mentioned proved the data we collected were valuable and worth further study.
Of all the aberrant genes detected, SDH mutation has attracted our supreme attention. Succinate dehydrogenase (SDH), also known as complex II, is an essential part of the Krebs cycle. Located on the inner membrane of mitochondria, SDH can not only catalyze conversion of succinate to fumarate, but also plays a role in the electron transport chain. Over the last 15 years, many tumor syndromes associated with SDH and accessory factor gene mutations have been identified, which include renal cell carcinomas (RCCs), wild type (WT) gastrointestinal stromal tumors (GISTs) and hereditary paragangliomas/pheochromocytomas [41]. SDH has been widely linked with the WT GISTs because lots of WT GISTs are detected with SDH gene mutation [42]. However, it is still not revealed whether the phenomenon is identical in GISTs harboring c-KIT and PDGFRA mutation. According to our results of the whole exons sequencing, we discover that 3 GISTs specimens with c-KIT mutation are harboring a SDHD mutation respectively. The results were inspiring because we give a primary impression that GISTs with c-KIT and PDGFRA mutation could also be SDH-deficient. Besides, existing data has always approved that SDHA mutation was most frequently seen in WT GISTs, which is consistent with the testing result of our WT GISTs specimen detected with a synchronous SDHA mutation. However, based on the SDHD mutation we detected in the 3 GISTs specimens with c-KIT mutation, we have to reconsider the belief we hold and expand the number of samples to further test the results.
The reason we shed so much light on SDH is that we believe it could be a new target for GIST treatment. By reviewing the previous studies, we find a chain of evidence that could support the hypotheses. As previous described, most of WT GISTs are SDH-deficient. In additions, they are not sensitive to treatment with a tyrosine kinase inhibitor, whether the first-line inhibitor imatinib or the second-line multikinase inhibitor sunitinib [43]. It inspired us to study the possible link between the two phenomenons. After review of the literature, we come up with our hypotheses. When SDH is inhibited in cells, succinate will accumulate, which will lead to a high concentration of succinate in the cytoplasm. A high concentration of succinate inhibits degradation of HIF-1a, which leads to HIF-1a overexpression and HIF translocation into nuclei [41]. The HIF-1 complex can bind to hypoxia response element regions and promote the expression of specific genes, such as NIX and Bnip3, leading to autophagy. It has also been proved that HIF-1a can induce endoplasmic reticulum stress, which will also cause autophagy [44]. Autophagy is involved in the development of imatinib resistance of gastrointestinal stromal tumors [45]. The chain of evidence has made us believe that SDH dysfunction can play an important role in the development of imatinib resistance for WT GISTs. Besides. we recently reported a germline SDHA-mutated WT GIST patient complicated with renal cell carcinoma, which need further study for the tumorgenesis mechanism [46]. After successfully establishing the WT GIST PDTX model, we have been able to test our theory under the animal level. Relevant studies are now underwent in our lab.
Conclusion
In summary, we have successfully established 4 GIST PDTX models and demonstrated that these models faithfully recapitulate the histological and genetic characteristics of the primary human tumors. Furthermore, we are the first study to conduct WES for the GIST PDTX models and analyzed the data for our future experiment. Thus, this panel of GIST PDTX models represents a valuable tool in understanding this lethal disease and serves as a powerful resource in enabling preclinical efficacy testing in oncology drug discovery.
Acknowledgements
Supported by Grants from the Ministry of Health of the China, No. W2012RQ02; Shanghai Science and Technology Committee, No. 12nm0501402; and Shanghai Education Committee, No. 120311. Thanks to the technical help provided by WuXi AppTec Co., Ltd. Waigaoqiao Free Trade Zone, Shanghai, China. This study is part of the program achievement of Shanghai Municipal Commission of Health and Family Planning, Key developing disciplines, 2015ZB0201.
Disclosure of conflict of interest
None.
Supplemental 1
Supplemental 2
References
- 1.Reichardt P, Hogendoorn PC, Tamborini E, Loda M, Gronchi A, Poveda A, Schoffski P. Gastrointestinal stromal tumors I: pathology, pathobiology, primary therapy, and surgical issues. Semin Oncol. 2009;36:290–301. doi: 10.1053/j.seminoncol.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 2.Patil DT, Rubin BP. Gastrointestinal stromal tumor: advances in diagnosis and management. Arch Pathol Lab Med. 2011;135:1298–1310. doi: 10.5858/arpa.2011-0022-RA. [DOI] [PubMed] [Google Scholar]
- 3.Kingham TP, DeMatteo RP. Multidisciplinary treatment of gastrointestinal stromal tumors. Surg Clin North Am. 2009;89:217–233. x. doi: 10.1016/j.suc.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Peng B, Dimitrijevic S, Druker BJ, Corless C, Fletcher CD, Joensuu H. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 5.Blanke CD, Demetri GD, von Mehren M, Heinrich MC, Eisenberg B, Fletcher JA, Corless CL, Fletcher CD, Roberts PJ, Heinz D, Wehre E, Nikolova Z, Joensuu H. Long-term results from a randomized phase II trial of standardversus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J. Clin. Oncol. 2008;26:620–625. doi: 10.1200/JCO.2007.13.4403. [DOI] [PubMed] [Google Scholar]
- 6.Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, Issels R, van Oosterom A, Hogendoorn PC, Van Glabbeke M, Bertulli R, Judson I. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364:1127–1134. doi: 10.1016/S0140-6736(04)17098-0. [DOI] [PubMed] [Google Scholar]
- 7.Debiec-Rychter M, Cools J, Dumez H, Sciot R, Stul M, Mentens N, Vranckx H, Wasag B, Prenen H, Roesel J, Hagemeijer A, Van Oosterom A, Marynen P. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology. 2005;128:270–279. doi: 10.1053/j.gastro.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 8.Comandone A, Boglione A. [The importance of mutational status in prognosis and therapy of GIST] . Recenti Prog Med. 2015;106:17–22. doi: 10.1701/1740.18950. [DOI] [PubMed] [Google Scholar]
- 9.Johnson JI, Decker S, Zaharevitz D, Rubinstein LV, Venditti JM, Schepartz S, Kalyandrug S, Christian M, Arbuck S, Hollingshead M, Sausville EA. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br J Cancer. 2001;84:1424–1431. doi: 10.1054/bjoc.2001.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sicklick JK, Leonard SY, Babicky ML, Tang CM, Mose ES, French RP, Jaquish DV, Hoh CK, Peterson M, Schwab R, Lowy AM. Generation of orthotopic patient-derived xenografts from gastrointestinal stromal tumor. J Transl Med. 2014;12:41. doi: 10.1186/1479-5876-12-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Floris G, Wozniak A, Sciot R, Li H, Friedman L, Van Looy T, Wellens J, Vermaelen P, Deroose CM, Fletcher JA, Debiec-Rychter M, Schoffski P. A potent combination of the novel PI3K Inhibitor, GDC-0941, with imatinib in gastrointestinal stromal tumor xenografts: long-lasting responses after treatment withdrawal. Clin Cancer Res. 2013;19:620–630. doi: 10.1158/1078-0432.CCR-12-2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Floris G, Sciot R, Wozniak A, Van Looy T, Wellens J, Faa G, Normant E, Debiec-Rychter M, Schoffski P. The Novel HSP90 inhibitor, IPI-493, is highly effective in human gastrostrointestinal stromal tumor xenografts carrying heterogeneous KIT mutations. Clin Cancer Res. 2011;17:5604–5614. doi: 10.1158/1078-0432.CCR-11-0562. [DOI] [PubMed] [Google Scholar]
- 13.Cohen NA, Zeng S, Seifert AM, Kim TS, Sorenson EC, Greer JB, Beckman MJ, Santamaria-Barria JA, Crawley MH, Green BL, Rossi F, Besmer P, Antonescu CR, DeMatteo RP, Zhang T, Zhang L, Fan S, Zhang M, Fu H, Liu Y, Yin X, Chen H, Xie L, Zhang J, Gavine PR, Gu Y, Ni X, Su X. Pharmacological Inhibition of KIT Activates MET Signaling in Gastrointestinal Stromal Tumors Patient-Derived Gastric Carcinoma Xenograft Mouse Models Faithfully Represent Human Tumor Molecular Diversity. Cancer Res. 2015;75:2061–2070. doi: 10.1158/0008-5472.CAN-14-2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daniel VC, Marchionni L, Hierman JS, Rhodes JT, Devereux WL, Rudin CM, Yung R, Parmigiani G, Dorsch M, Peacock CD, Watkins DN. A primary xenograft model of small-cell lung cancer reveals irreversible changes in gene expression imposed by culture in vitro. Cancer Res. 2009;69:3364–3373. doi: 10.1158/0008-5472.CAN-08-4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang T, Zhang L, Fan S, Zhang M, Fu H, Liu Y, Yin X, Chen H, Xie L, Zhang J, Gavine PR, Gu Y, Ni X, Su X. Patient-Derived Gastric Carcinoma Xenograft Mouse Models Faithfully Represent Human Tumor Molecular Diversity. PLoS One. 2015;10:e0134493. doi: 10.1371/journal.pone.0134493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang Z, Zhou X, Li R, Michal JJ, Zhang S, Dodson MV, Zhang Z, Harland RM. Whole transcriptome analysis with sequencing: methods, challenges and potential solutions. Cell Mol Life Sci. 2015;72:3425–3439. doi: 10.1007/s00018-015-1934-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Panagopoulos I, Bjerkehagen B, Gorunova L, Berner JM, Boye K, Heim S. Several fusion genes identified by whole transcriptome sequencing in a spindle cell sarcoma with rearrangements of chromosome arm 12q and MDM2 amplification. Int J Oncol. 2014;45:1829–1836. doi: 10.3892/ijo.2014.2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kimbung S, Johansson I, Danielsson A, Veerla S, Brage SE, Frostvik SM, Skoog L, Carlsson L, Einbeigi Z, Lidbrink E, Linderholm BK, Loman N, Malmstrom P, Soderberg M, Walz TM, Ferno M, Hatschek T, Hedenfalk IA. Transcriptional profiling of breast cancer metastases identifies liver metastasis-selective genes associated with adverse outcome in luminal A primary breast cancer. Clin Cancer Res. 2016;22:146–157. doi: 10.1158/1078-0432.CCR-15-0487. [DOI] [PubMed] [Google Scholar]
- 19.Ho DW, Kai AK, Ng IO. TCGA whole-transcriptome sequencing data reveals significantly dysregulated genes and signaling pathways in hepatocellular carcinoma. Front Med. 2015;9:322–330. doi: 10.1007/s11684-015-0408-9. [DOI] [PubMed] [Google Scholar]
- 20.Fang DD, Zhang B, Gu Q, Lira M, Xu Q, Sun H, Qian M, Sheng W, Ozeck M, Wang Z, Zhang C, Chen X, Chen KX, Li J, Chen SH, Christensen J, Mao M, Chan CC. HIP1-ALK, a novel ALK fusion variant that responds to crizotinib. J Thorac Oncol. 2014;9:285–294. doi: 10.1097/JTO.0000000000000087. [DOI] [PubMed] [Google Scholar]
- 21.Xu Q, Wang G, Fang DD, Gao YB, Chen YD, Sun LL, Yan XL, Li Q, Yang QY, Qian MX, Zhang B, Chen ZL, Chan CC, Sun HY, He J. Deep sequencing of Xenografts and case-matched blood and primary tumors reveals a 20 folds enrichment of loss of heterozygosity versus somatic mutations suggesting LOH plays an ever important role in tumorigenesis. [abstract] Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research. 2012 Mar 31-Apr 4; Chicago, IL. [Google Scholar]
- 22.Fang DD, Gao YB, Xu Q, Sun HY, Zhang B, Chen ZL, Chan CC, He J. Identification of somatic mutations in esophageal squamous cell carcinoma and corresponding xenograft by nextgeneration sequencing. [abstract] Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research. 2012 Mar 31-Apr 4; Chicago, IL. [Google Scholar]
- 23.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pawitan Y, Michiels S, Koscielny S, Gusnanto A, Ploner A. False discovery rate, sensitivity and sample size for microarray studies. Bioinformatics. 2005;21:3017–3024. doi: 10.1093/bioinformatics/bti448. [DOI] [PubMed] [Google Scholar]
- 25.Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He X, Mieczkowski P, Grimm SA, Perou CM, MacLeod JN, Chiang DY, Prins JF, Liu J. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38:e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joshi-Tope G, Gillespie M, Vastrik I, D’Eustachio P, Schmidt E, de Bono B, Jassal B, Gopinath GR, Wu GR, Matthews L, Lewis S, Birney E, Stein L. Reactome: a knowledgebase of biological pathways. Nucleic Acids Res. 2005;33:D428–432. doi: 10.1093/nar/gki072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yi M, Horton JD, Cohen JC, Hobbs HH, Stephens RM. WholePathwayScope: a comprehensive pathway-based analysis tool for highthroughput data. BMC Bioinformatics. 2006;7:30. doi: 10.1186/1471-2105-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Draghici S, Khatri P, Tarca AL, Amin K, Done A, Voichita C, Georgescu C, Romero R. A systems biology approach for pathway level analysis. Genome Res. 2007;17:1537–1545. doi: 10.1101/gr.6202607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, Arcaroli JJ, Messersmith WA, Eckhardt SG. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012;9:338–350. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rubio-Viqueira B, Jimeno A, Cusatis G, Zhang X, Iacobuzio-Donahue C, Karikari C, Shi C, Danenberg K, Danenberg PV, Kuramochi H, Tanaka K, Singh S, Salimi-Moosavi H, Bouraoud N, Amador ML, Altiok S, Kulesza P, Yeo C, Messersmith W, Eshleman J, Hruban RH, Maitra A, Hidalgo M. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res. 2006;12:4652–4661. doi: 10.1158/1078-0432.CCR-06-0113. [DOI] [PubMed] [Google Scholar]
- 32.Fichtner I, Rolff J, Soong R, Hoffmann J, Hammer S, Sommer A, Becker M, Merk J. Establishment of patient-derived non-small cell lung cancer xenografts as models for the identification of predictive biomarkers. Clin Cancer Res. 2008;14:6456–6468. doi: 10.1158/1078-0432.CCR-08-0138. [DOI] [PubMed] [Google Scholar]
- 33.Rink L, Ochs MF, Zhou Y, von Mehren M, Godwin AK. ZNF-mediated resistance to imatinib mesylate in gastrointestinal stromal tumor. PLoS One. 2013;8:e54477. doi: 10.1371/journal.pone.0054477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee EJ, Kang G, Kang SW, Jang KT, Lee J, Park JO, Park CK, Sohn TS, Kim S, Kim KM. GSTT1 copy number gain and ZNF overexpression are predictors of poor response to imatinib in gastrointestinal stromal tumors. PLoS One. 2013;8:e77219. doi: 10.1371/journal.pone.0077219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ran L, Sirota I, Cao Z, Murphy D, Chen Y, Shukla S, Xie Y, Kaufmann MC, Gao D, Zhu S, Rossi F, Wongvipat J, Taguchi T, Tap WD, Mellinghoff IK, Besmer P, Antonescu CR, Chen Y, Chi P. Combined inhibition of MAP kinase and KIT signaling synergistically destabilizes ETV1 and suppresses GIST tumor growth. Cancer Discov. 2015;5:304–315. doi: 10.1158/2159-8290.CD-14-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jang BG, Lee HE, Kim WH. ETV1 mRNA is specifically expressed in gastrointestinal stromal tumors. Virchows Arch. 2015;467:393–403. doi: 10.1007/s00428-015-1813-9. [DOI] [PubMed] [Google Scholar]
- 37.Hayashi Y, Bardsley MR, Toyomasu Y, Milosavljevic S, Gajdos GB, Choi KM, Reid-Lombardo KM, Kendrick ML, Bingener-Casey J, Tang CM, Sicklick JK, Gibbons SJ, Farrugia G, Taguchi T, Gupta A, Rubin BP, Fletcher JA, Ramachandran A, Ordog T. Platelet-Derived Growth Factor Receptor-alpha Regulates Proliferation of Gastrointestinal Stromal Tumor Cells With Mutations in KIT by Stabilizing ETV1. Gastroenterology. 2015;149:420–432. e416. doi: 10.1053/j.gastro.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chi P, Chen Y, Zhang L, Guo X, Wongvipat J, Shamu T, Fletcher JA, Dewell S, Maki RG, Zheng D, Antonescu CR, Allis CD, Sawyers CL. ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature. 2010;467:849–853. doi: 10.1038/nature09409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mason EF, Hornick JL. Succinate dehydrogenase deficiency is associated with decreased 5-hydroxymethylcytosine production in gastrointestinal stromal tumors: implications for mechanisms of tumorigenesis. Mod Pathol. 2013;26:1492–1497. doi: 10.1038/modpathol.2013.86. [DOI] [PubMed] [Google Scholar]
- 40.Killian JK, Kim SY, Miettinen M, Smith C, Merino M, Tsokos M, Quezado M, Smith WI, Jahromi MS, Xekouki P, Szarek E, Walker RL, Lasota J, Raffeld M, Klotzle B, Wang Z, Jones L, Zhu Y, Wang Y, Waterfall JJ, O’Sullivan MJ, Bibikova M, Pacak K, Stratakis C, Janeway KA, Schiffman JD, Fan JB, Helman L, Meltzer PS. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013;3:648–657. doi: 10.1158/2159-8290.CD-13-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bardella C, Pollard PJ, Tomlinson I. SDH mutations in cancer. Biochim Biophys Acta. 2011;1807:1432–1443. doi: 10.1016/j.bbabio.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 42.Miettinen M, Lasota J. Succinate dehydrogenase deficient gastrointestinal stromal tumors (GISTs) - a review. Int J Biochem Cell Biol. 2014;53:514–519. doi: 10.1016/j.biocel.2014.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Janeway KA, Albritton KH, Van Den Abbeele AD, D’Amato GZ, Pedrazzoli P, Siena S, Picus J, Butrynski JE, Schlemmer M, Heinrich MC, Demetri GD. Sunitinib treatment in pediatric patients with advanced GIST following failure of imatinib. Pediatr Blood Cancer. 2009;52:767–771. doi: 10.1002/pbc.21909. [DOI] [PubMed] [Google Scholar]
- 44.Wu H, Chen Q. Hypoxia activation of mitophagy and its role in disease pathogenesis. Antioxid Redox Signal. 2015;22:1032–1046. doi: 10.1089/ars.2014.6204. [DOI] [PubMed] [Google Scholar]
- 45.Rubin BP, Debnath J. Therapeutic implications of autophagy-mediated cell survival in gastrointestinal stromal tumor after treatment with imatinib mesylate. Autophagy. 2010;6:1190–1191. doi: 10.4161/auto.6.8.13430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang Q, Zhang Y, Zhou YH, Hou YY, Wang JY, Li JL, Li M, Tong HX, Lu WQ. A novel germline mutation in SDHA identified in a rare case of gastrointestinal stromal tumor complicated with renal cell carcinoma. Int J Clin Exp Pathol. 2015;8:12188–12197. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.