

Abstract
Purpose: It is a challenge to find a better microorganisms DNA extraction method for samples taken from the lower airways for metagenomic sequencing, as the concentrations of bacteria in the alveoli and small airways are likely considerably less than that of the mouth or lower digestive tract. Background DNA from the host, and extraction biases can significantly interfere with microbiota assessment and increase the cost of sequencing. This study aimed to develop an optimized DNA extraction method, which would enable a higher concentration of microbial DNA to be extracted from the samples. Methods: We compared the microbiota profiles of the lower airway communities in twelve individuals with IIP. DNA was extracted using three different extraction methods: QIAamp UCP PurePathogen Blood Kit named kit3 in this study, QIAamp UCP Pathogen Mini Kit named kit2, and QIAamp DNA Microbiome Kit named kit1. DNA libraries were constructed according to the manufacturer’s instructions (Illumina). The same workflows from Illumina were used to perform cluster generation, template hybridization, isothermal amplification, linearization, blocking, denaturing, and hybridization of the sequencing primers. Raw data was uploaded to MG-RAST v3 and analyzed. Results: A great number of bacterium inhabits the lower airways of patients with IIP, though there is no airway infection. More bacterium was found in mouth or upper airway. DNA concentrations of DNA samples isolated with kit1 with Benzonase were significantly lower than those isolated with the other two kits for BALF and mouthwash samples. Moreover, the ratio of human genome in clean reads of samples isolated with kit1 with Benzonase was remarkably smaller than those isolated with kit2 and kit3. The relative abundance of total bacteria, the total number of taxa, and the relative abundance of taxa in BALF samples as opposed to mouthwash samples with kit1 were significantly higher than for those extracted the other kits. Conclusion: A microbial DNA extraction method with pretreatment of depletion of host nucleic acid by Benzonase can enable a higher yield of microbial DNA from samples with a higher fraction of host cells to be obtained. The lower airways of patients with IIP without airway infection were inhabited by a great number of bacterium.
Keywords: DNA, BALF, metagenomic data
Introduction
Metagenomics is to consider the microbial population as a whole ‘metagenome’ by applying high-throughput shot-gun sequencing to the entire population to identify the community members present and their genetically encoded functional capacity. Mao et al. determined that, particularly in comparisons restricted to a specific type of sample (e.g., only from human fecal samples), technical differences in experimental protocols between laboratories, including DNA extraction methods, the instruments used to determine the nucleotide sequences, and the manner in which samples are obtained and stored, might all produce variability that could outweigh biological differences [1].
There are a lot of studies published about the effects of DNA extraction methods on deep sequencing analyses of microbial communities from bacterial samples of the human sites [2-6]. However, these studies mostly used bacterial 16S rDNA gene PCR amplification targeted to region of the 16S rRNA gene and pyrosequencing, and did not test for methods to reduce background DNA. The development of effective and efficient decontamination methods also suitable for high-throughput use or development of ultrapure reagents could potentially further reduce background DNA [7,8]. The difference is that the reports of metagenomic high throughput sequencing include host or background DNA sequence reads while supplying DNA sequence reads of microorganisms. Host DNA sequence reads are not included in the following analysis. Therefore, if a situation whereby less data is discarded can be arrived at, then the cost of sequencing will be lowered and the results derived from the subsequent analysis will be more accurate. To date there is a dearth of research on metagenomic explorations of lower airway microorganisms from bronchoalveolar lavage fluid (BALF) or lung tissues. As observed by Hilty et al. the concentrations of bacteria in the alveoli and small airways are likely considerably less than that of the mouth or lower digestive tract-at most comparable to that of the stomach or small intestine-thus the majority of research has tended to focus on those sites [9]. Accordingly, it is a ch-allenge to find a better microorganism DNA extraction method of BALF.
Idiopathic interstitial pneumonias (IIPs) are the most prevalent diseases of the group of interstitial lung diseases (ILDs) including a heterogeneous group of disorders, mostly with unknown causes. Progressive pulmonary fibrosis appears to be the coalescence of a complex mix of environmental and genetic factors, but the mechanism by which connective tissue proliferation occurs is unknown [10]. Much interest has been focused on the potential role of viruses as cofactors in accelerating the progression of IIPs, and several studies have implicated viral infection as a cause of ongoing epithelial injury in idiopathic pulmonary fibrosis, and therefore it is an important factor in pathogenesis [11]. However, the possible role of other infectious agents has been largely neglected, and other uncultivated microorganisms could colonize IIP patients and play a role in the physiopathology of these diseases [12].
We therefore have studied the effect of DNA extraction methods on metagenomic sequence characteristics, total bacteria community diversity, and bacterial community structure of airway DNA samples from patients with IIP. This report aimed to develop an optimized DNA extraction method, which would enable a higher concentration of microbial DNA to be extracted from the samples, thereby reducing the cost of sequencing and increasing the accuracy of the analysis of the sequencing data.
Materials and methods
Patient information
IIP was diagnosed according to the international guidelines by ATS/ERS [13]. Enrolled individuals with an indication for bronchoscopy at Beijing Chao-Yang Hospital had to be ≥18 years of age and able to provide informed consent. A complete clinical, functional and radiological evaluation of all patients was made. Exclusion criteria for this study were: recent bacterial/viral respiratory tract infection within 1 month prior to bronchoalveolar lavage (BAL), present active lung disease other than IIP, HIV-positivity and subjects that had received antibiotic therapy within 1 month prior to BAL as this was previously shown to affect the airway microbiota [14,15].
All participants, or their legally authorized representatives, provided a written informed consent upon enrollment. The study conformed to the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the Institutional Review Board of Beijing Hospital.
Human mouthwash and bronchoalveolar lavage samples collection
Two types of respiratory samples (mouthwash and bronchoalveolar lavage) were taken from each individual.
Patients gargled with a 10 ml volume of sterile saline (0.9%) for 30 seconds before local anaesthesia for bronchoscopy, and wash fluid was collected in a sputum pot. To obtain the BALF samples three 50 ml-aliquots of sterile saline (0.9% w/v) were instilled, under local anaesthetic, in the third generation bronchus of the middle lobe (lingual) or in the area containing most lung infiltrates, using a fibrotic bronchoscope (Type 40; Olympus, Tokyo, Japan). Each aliquot was recovered immediately by suction.
All specimens collected were delivered immediately from our hospital to the laboratory in an ice bag using insulating polystyrene foam containers. In the laboratory each specimen was divided into 1.5 ml aliquots, and stored at -80°C until processing for DNA extraction.
DNA extraction
All DNA extractions were performed with 3.0 ml of the original samples. Three DNA extraction methods were tested: QIAamp UCP PurePathogen Blood Kit (Catalogue 50112, Qiagen, Hilden, Germany) named kit3 in this study, QIAamp UCP Pathogen Mini Kit (Catalogue 50214, Qiagen, Hilden, Germany) named kit2, and QIAamp DNA Microbiome Kit (Catalogue 51704, Qiagen, Hilden, Germany) named kit1. To increase DNA yields, DNA extracted with all methods was eluted with relatively small volume (30 μl) of recommended elution buffer. DNA was isolated exactly as the manufacturer’s instructions. DNA concentration was measured by Qubit® 2.0 Fluorometer (Life Technologies, Invitrogen, USA).
As a negative control, the same procedure was used with sterile water; no PCR products were detected in any experiment, indicating lack of contamination of any of the reagents used.
DNA library construction and sequencing
DNA libraries were constructed according to the manufacturer’s instructions (Illumina). The same workflows from Illumina were used to perform cluster generation, template hybridization, isothermal amplification, linearization, blocking, denaturing and hybridization of the sequencing primers. We performed pairedend sequencing on 2 × 100 base pairs (bp) for all libraries. The base-calling pipeline (Casava 1.8.2 with parameters ‘-use-bases-mask y100n, I6n, Y100n, -mismatches 1, -adaptor-sequence’) was used to process the raw fluorescent images and call sequences. The same insert size inferred by Agilent 2100 was used for all libraries.
Sequence processing and statistical analysis
After upload to MG-RAST v3 [16], data is preprocessed by using SolexaQA [17] to trim low-quality regions from FASTQ data. MG-RAST v3 uses DRISEE (Duplicate Read Inferred Sequencing Error Estimation) [18] to analyze the sets of Artificial Duplicate Reads (ADRs) [19] and determine the degree of variation among prefix-identical sequences derived from the same template. The data was compared to M5NR using the following parameters: a maximum e-value of 1e-5, a minimum identity of 90%, and a minimum alignment length of 15 aa for protein and bp for RNA databases. The displayed data has been normalized to values between 0 and 1 to allow for comparison of differently sized samples based on abundance. The taxonomic profiles use the NCBI taxonomy. We used the best hit classification to report the functional and taxonomic annotation of the best hit in the M5nr for each feature. Raw sequences were analyzed using Mothur v1.21 [17] to remove sequences containing homopolymers greater than 8 bp, mismatches in the barcode or primer, one or more ambiguous bases, or an average quality score below 35 over a moving window of 50 bp. Remaining sequences that were at least 200 bp but less than 590 bp in length were further curated to remove chimeric sequences using UCHIME [19] and to reduce sequencing noise by a preclustering methodology [20] before being assigned to operational taxonomic units (OTUs) using an average neighbor algorithm with a 0.03 dissimilarity cutoff. The consensus taxonomy of each OTU was identified at the genus level using the Bayesian method [21]. The total number of reads for each community was normalized to 498, the smallest number of reads among the samples included in the study, to control for differences in sequencing depth before alpha and beta diversity measures were calculated. Community diversity was measured using non-parametric Shannon indices [22]. The number of observed OTUs was used as a measure of community richness. Community evenness was measured with Shannon indices-based measure of evenness. Beta diversity was measured using Bray-Curtis dissimilarity coefficients.
The alpha diversity estimate is a single number that summarizes the distribution of species level annotations in a dataset. The Shannon diversity index is an abundance-weighted average of the logarithm of the relative abundances of annotated species. Mean and Standard Error were calculated by IBM SPSS 22.0 software package. One-way ANOVA by IBM SPSS 22.0 software package was used to compare the relative abundance of total bacteria, and community diversity, richness, and evenness between sets of samples. A P<0.05 was considered statistically significant.
Results
Study patients, DNA concentration, and sequence characteristics
A total of 12 patients were enrolled in this study. The study subjects had an overall mean age of (60.58 ± 12.82) years. Seven (58.33%) were women. None of the 12 study subjects had evidence of infections of respiratory tract or had received antibiotic treatment within 1 month prior to BALF. The 12 study subjects were randomly divided into three extraction methods groups: group kit1 (QIAamp DNA Microbiome Kit, Catalogue: 51704), group kit2 (QIAamp UCP Pathogen Mini Kit, Catalogue: 50214), group kit3 (QIAamp UCP PurePathogen Blood Kit, catalogue: 50112). We collected two types of respiratory samples including mouthwash and bronchoalveolar lavage from each individual and categorised those samples into the following groups according to type of samples and DNA extraction method: group kit1A (patient 1-4, BALF samples), group kit2A (patient 5-8, BALF samples), group kit3A (patient 9-12, BALF samples), group kit1B (patient 1-4, mouthwash samples), group kit2B (patient 5-8, mouthwash samples), group kit3B (patient 9-12, mouthwash samples).
Prior to DNA library construction we measured the DNA concentration of all DNA samples (Table 1). For DNA samples from BALF, we found that the DNA concentrations of DNA samples in group kit1A were much lower than those in group kit2A and kit3A; that is to say DNA concentrations of DNA samples isolated with QIAamp DNA Microbiome Kit were significantly lower than those isolated with other two kits. This result was mirrored in the mouthwash DNA samples.
Table 1.
Sequence characteristics of bronchoalveolar lavage fluid and mouthwash, isolated with different extraction methods
| Sample | DNA Concentration (ng/ml) | Raw reads | Raw bases | Clean reads | Clean bases | Ratio1 | Align_hg192 | Ratio3 |
|---|---|---|---|---|---|---|---|---|
| Group kit1A | ||||||||
| A1 | 0.72 | 6,037,378 | 911,644,078 | 5,275,216 | 796,557,616 | 87.38% | 1,562,208 | 29.61% |
| A2 | <0.05 | 310,942 | 46,952,242 | 263,056 | 39,721,456 | 84.60% | 33,535 | 12.75% |
| A3 | <0.05 | 465,010 | 70,216,510 | 356,812 | 53,878,612 | 76.73% | 11,056 | 3.10% |
| A4 | <0.05 | 4,484,096 | 564,996,096 | 4,125,850 | 519,857,100 | 92.01% | 198,685 | 4.82% |
| Group kit2A | ||||||||
| A5 | >600 | 3,477,108 | 438,115,608 | 3,449,730 | 434,665,980 | 99.21% | 3,157,491 | 91.53% |
| A6 | >600 | 3,227,836 | 406,707,336 | 3,206,374 | 404,003,124 | 99.34% | 2,927,491 | 91.30% |
| A7 | 92.4 | 5,108,128 | 643,624,128 | 5,076,382 | 639,624,132 | 99.38% | 4,667,233 | 91.94% |
| A8 | 2.11 | 4,395,052 | 553,776,552 | 4,348,518 | 547,913,268 | 98.94% | 3,936,642 | 90.53% |
| Group kit3A | ||||||||
| A9 | 1.76 | 4,830,772 | 608,677,272 | 4,789,136 | 603,431,136 | 99.14% | 4,332,321 | 90.46% |
| A10 | 3.36 | 5,150,432 | 648,954,432 | 5,110,978 | 643,983,228 | 99.23% | 4,668,790 | 91.35% |
| A11 | <0.05 | 886,084 | 111,646,584 | 758,522 | 95,573,772 | 85.60% | 662,632 | 87.36% |
| A12 | 7.58 | 53,281,532 | 6,713,473,032 | 52,783,514 | 6,650,722,764 | 99.07% | 48,142,856 | 91.21% |
| Group kit1B | ||||||||
| B1 | 16 | 6,685,644 | 842,391,144 | 6,465,234 | 814,619,484 | 96.70% | 175,539 | 2.72% |
| B2 | 3.17 | 6,766,884 | 852,627,384 | 6,532,778 | 823,130,028 | 96.54% | 197,003 | 3.02% |
| B3 | 70.3 | 1,406,798 | 177,256,548 | 1,145,564 | 144,341,064 | 81.43% | 144,264 | 12.59% |
| B4 | 1.25 | 3,619,584 | 456,067,584 | 3,294,628 | 415,123,128 | 91.02% | 1,090,245 | 33.09% |
| Group kit2B | ||||||||
| B5 | 36.2 | 68,589,090 | 8,642,225,340 | 67,541,700 | 8,510,254,200 | 98.47% | 61,038,323 | 90.37% |
| B6 | 3.59 | 42,519,012 | 5,357,395,512 | 42,198,100 | 5,316,960,600 | 99.25% | 35,839,622 | 84.93% |
| B7 | 67.3 | 46,783,892 | 5,894,770,392 | 46,138,672 | 5,813,472,672 | 98.62% | 40,468,246 | 87.71% |
| B8 | 70.6 | 6,380,916 | 803,995,416 | 6,336,480 | 798,396,480 | 99.30% | 5,658,884 | 89.31% |
| Group kit3B | ||||||||
| B9 | 0.254 | 2,083,784 | 262,556,784 | 1,957,590 | 246,656,340 | 93.94% | 1,276,697 | 65.22% |
| B10 | 98.7 | 5,896,044 | 742,901,544 | 5,818,674 | 733,152,924 | 98.69% | 4,831,836 | 83.04% |
| B11 | 20.3 | 5,887,346 | 741,805,596 | 5,818,388 | 733,116,888 | 98.83% | 2,546,041 | 43.76% |
| B12 | 2.29 | 3,889,076 | 490,023,576 | 3,829,382 | 482,502,132 | 98.47% | 3,221,984 | 84.14% |
values of column “Clean reads”/values of column “Raw reads”;
Reads of alignments to hg19 (human genome);
values of column “Align_hg19”/values of column “Clean reads”, i.e. ratio of human genome in clean reads of each sample.
A: samples from BALF; B: samples from mouthwash; code on the right of “A” or “B” is patient’s code. kit1= QIAamp DNA Microbiome Kit (Catalogue 51704), kit2= QIAamp UCP Pathogen Mini Kit (Catalogue 50214), kit3= QIAamp UCP PurePathogen Blood Kit (Catalogue 50112).
Through preliminary analysis of sequence characteristics, for DNA samples from BALF, we found that the ratio of human genome in clean reads of each sample in group kit1A was remarkably lower than those from group kit2A and kit3A (Table 1, Figure 1). Again, this result also happened in the mouthwash DNA samples.
Figure 1.
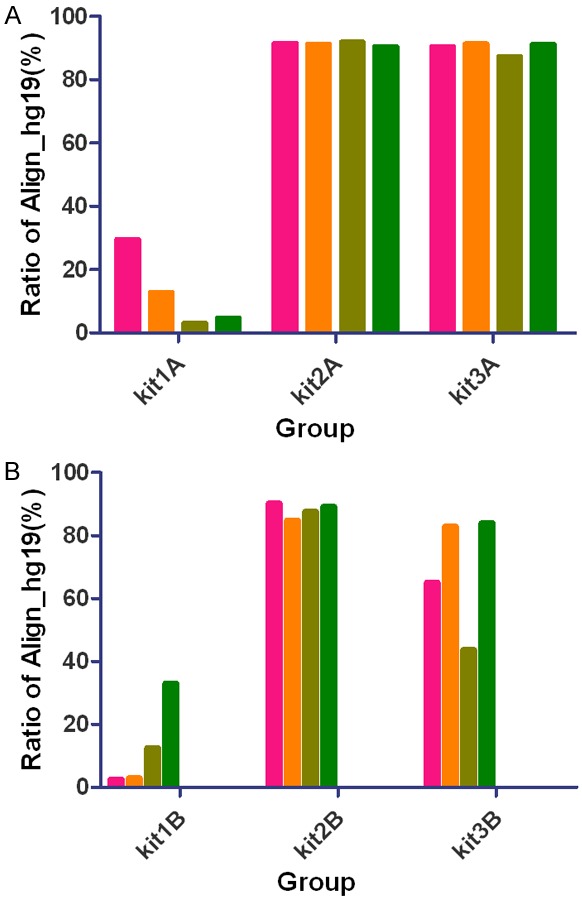
Impact of DNA extraction methods on sequence characteristic. A. Ratio of human genome in clean reads of each sample from BALF in group kit1A, group kit2A and group kit3A, showing all ratios in group kit1A were remarkably lower than those in group kit2A and kit3A. B. Ratio of human genome in clean reads of each sample from mouthwash in group kit1B, group kit2B and group kit3B, showing all ratios in group kit1B were remarkably lower than those in group kit2B and kit3B. Each column stands for one sample. Kit1= QIAamp DNA Microbiome Kit (Catalogue 51704), kit2= QIAamp UCP Pathogen Mini Kit (Catalogue 50214), kit3= QIAamp UCP PurePathogen Blood Kit (Catalogue 50112).
Impact of DNA extraction methods on total bacteria relative abundance, community diversity, richness and evenness
In order to analyse the impact of DNA extraction methods on total microorganisms, firstly we explored community composition with clean reads without human genome. This data was calculated for metagenomes for each individual. The data was compared to M5NR using a maximum e-value of 1e-5, a minimum identity of 90%, and a minimum alignment length of 15 measured in aa for protein and bp for RNA databases. The displayed data has been normalized to values between 0 and 1 to allow for comparison of differently sized samples based on abundance. We used normalized values to calculate P-values between groups. For samples from BALF, there were significant differences found on bacteria (P=0.0015), archea (P=0.0023), unassigned sequences (P=0.0029), unclassified sequences (P=0.0294), and other sequences (P=0.0306), between the three groups (group kit1A, group kit2A, group kit3A). The parameters for group kit1A were wider than those of group kit2A and group kit3A (Figure 2A, Table S1). However, there was significant difference only on eukaryota (P=0.0638) between the three groups for mouthwash samples (Figure 2B, Table S1).
Figure 2.
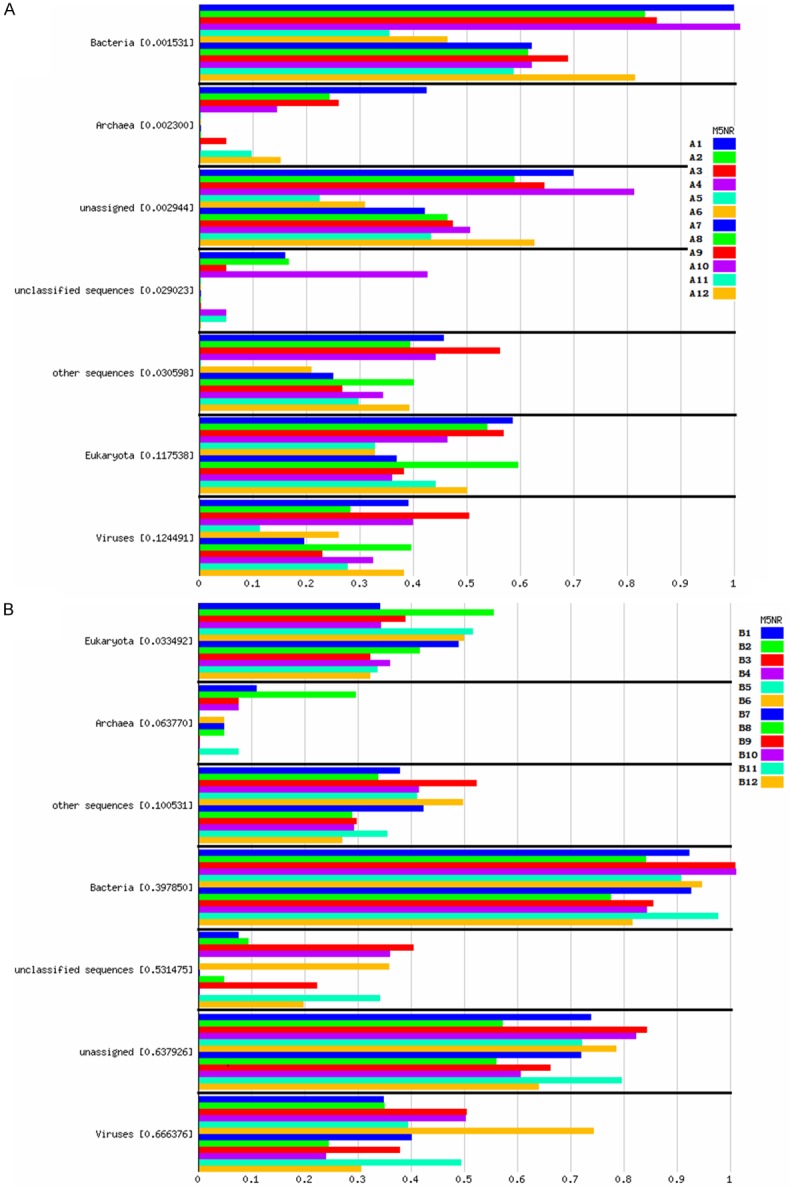
Impact of DNA extraction methods on total microorganisms’ community composition with clean reads without human genome of samples. A. Samples from BALF; B. Samples from mouthwash. Each horizontal bar stands for one sample, values in brackets are P-values between groups by ANOVA.
While observing the impact of DNA extraction methods on total bacteria, we compared total bacteria relative abundance and community diversity, richness, and evenness between groups (Figure 3, Table S2). We found that the relative abundance of total bacteria in group kit1A (90%; range 79%-95%) was significantly greater than that of group kit2A (12%; range 2%-22%) and group kit3A (35%; range 20%-45%; P<0.001, one-way ANOVA). Among these three groups, the relative abundance of total bacteria in group kit2A was the lowest. There was also a significant difference in the relative abundance of total bacteria between group kit1B (92%; range 90%-94%) and group kit2B (72%; range 66%-81%). Significant differences were also found between group kit3B (89%; range 86%-93%) and group kit2B (72%; range 66%-81%). Group kit1B had greater relative abundance of total bacteria than both kit2B and group kit3B.
Figure 3.
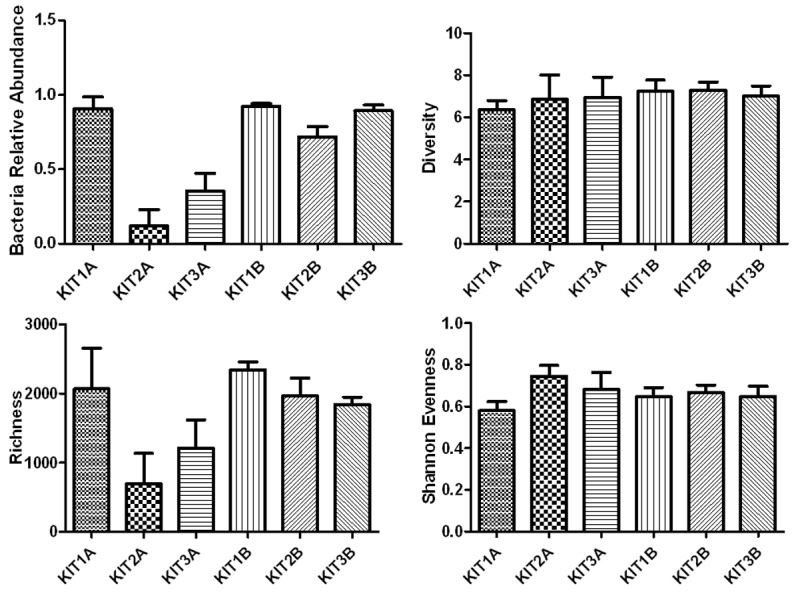
Impact of DNA extraction methods on total bacteria relative abundance, community diversity, richness and evenness. The relative abundance of total bacteria in group kit1A (90%; range 79%-95%) was significantly greater than that of group kit2A (12%; range 2%-22%) and group kit3A (35%; range 20%-45%; P<0.001, oneway ANOVA). Among these three groups, the relative abundance of total bacteria in group kit2A was the lowest. There was also a significant difference in the relative abundance of total bacterias between group kit1B (92%; range 90%-94%) and group kit2B (72%; range 66%-81%). Significant differences were also found between group kit3B (89%; range 86%-93% ) and group kit2B (72%; range 66%-81%). Group kit1B had greater relative abundance of total bacteria than both kit2B and group kit3B. For BALF samples, the measure of bacterial community diversity was lower in group kit1A compared with group kit2A and group kit3A, although without significant differences. However, the richness (total number of taxa) and evenness (relative abundance of taxa) were significantly higher in group kit1A compared with group kit2A and group kit3A (richness: P=0.009, evenness: P=0.014, respectively, oneway ANOVA). For the mouthwash samples there were significant differences in richness between the three groups (group kit1B, group kit2B and group kit3B, P=0.007, oneway ANOVA). Kit1= QIAamp DNA Microbiome Kit (Catalogue 51704), kit2= QIAamp UCP Pathogen Mini Kit (Catalogue 50214), kit3= QIAamp UCP PurePathogen Blood Kit (Catalogue 50112).
For BALF samples, the measure of bacterial community diversity was lower in group kit1A compared with group kit2A and group kit3A, although without significant differences. However, the richness (total number of taxa) and evenness (relative abundance of taxa) were significantly higher in group kit1A compared with group kit2A and group kit3A (richness: P=0.009, evenness: P=0.014, respectively, one-way ANOVA). For the mouthwash samples there were significant differences in richness between the three groups (group kit1B, group kit2B and group kit3B, P=0.007, one-way ANOVA).
Effect of relative abundance of total bacteria on bacterial community structure
The tree diagrams in Figure 4 show comparisons of datasets against a hierarchy (e.g., Subsystems or the NCBI taxonomy). The hierarchy is displayed as a rooted tree, and the abundance (normalized for dataset size) for each dataset in the various categories is displayed as a bar chart (Figure 2) for each category. Here, we elected to display only domain “bacteria” in order that we could see how different DNA extraction methods impact bacterial community structure. In Figure 4 (Table S3), colour shading of the family names indicates class membership. This figure displays leaf weights as stacked bar, maximum level as order, and colour by phylum. We found that for samples from BALF, the proteobacteria accounted for majority of phylum, the top 6 types were proteobacteria, firmicutes, bacteroidetes, actinobacteria, tenericutes, and cyanobacteria. From the mouthwash samples the top 6 phyla were proteobacteria, firmicutes, bacteroidetes, actinobacteria, fusobacteria, and fibrobacteres (Figure 5, Table S4).
Figure 4.
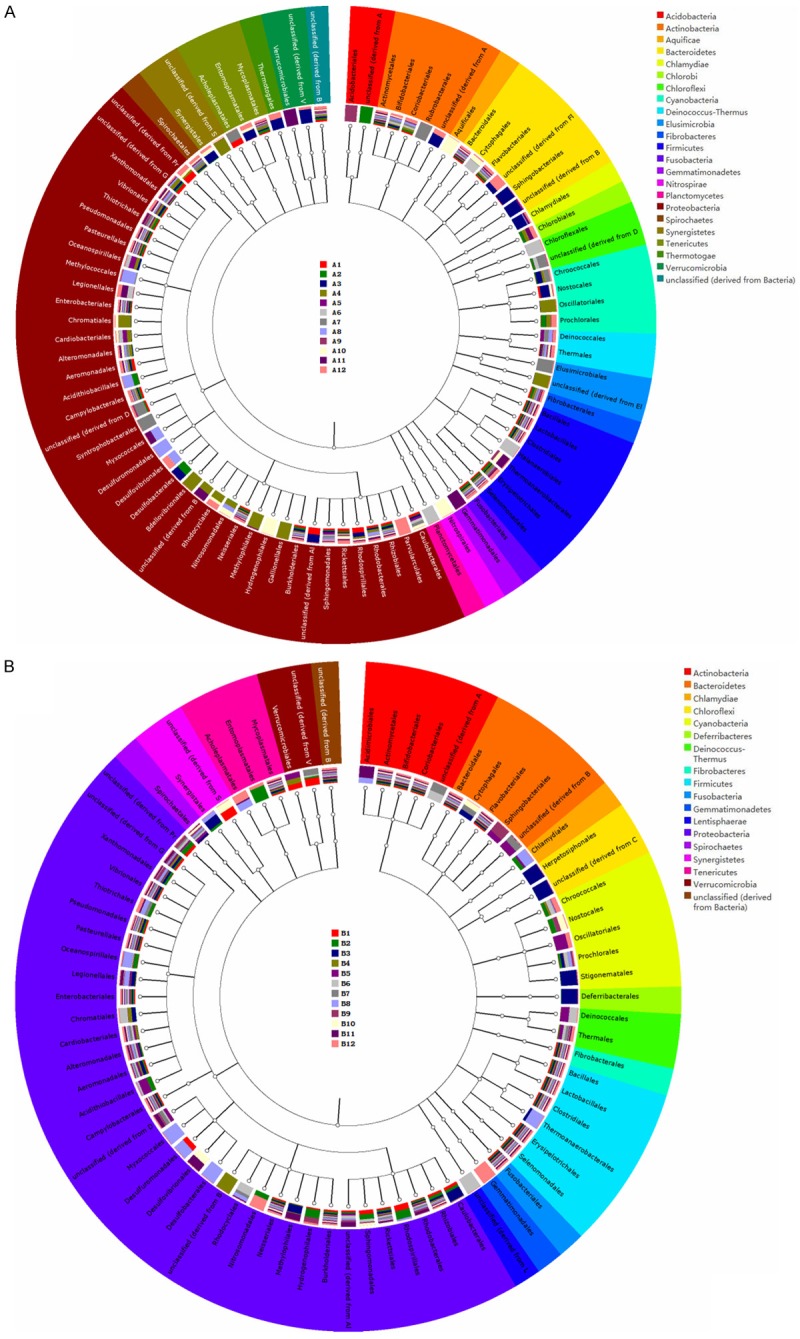
Tree diagrams which allow comparison of datasets against a hierarchy. The hierarchy is displayed as a rooted tree, and the abundance (normalized for dataset size) for each dataset in the various categories is displayed as a bar chart for each category. We elected to restrict view to domain “bacteria”. Colour shading of the family names indicates class membership. This figure displays leaf weights as stacked bar, maximum level as order, colour by phylum. A. Samples from BALF; B. Mouthwash samples.
Figure 5.
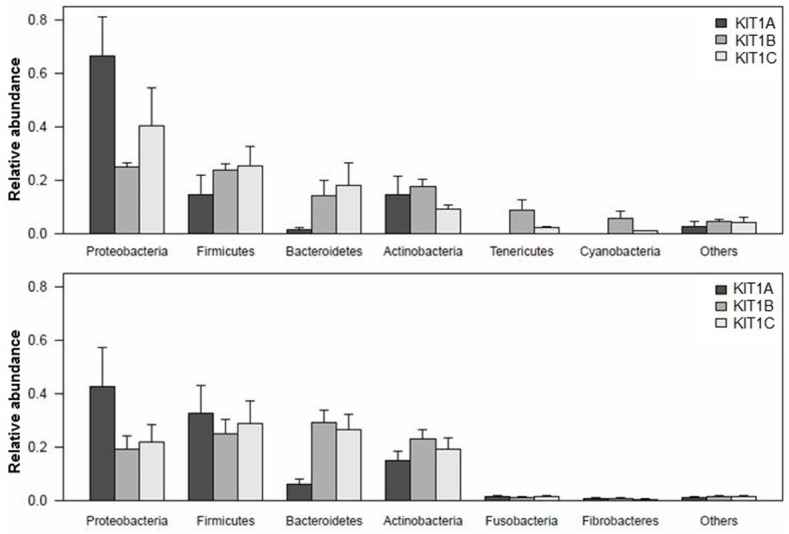
Average relative abundance of the 6 main phyla in each sample. Shown in error bars is the standard deviation per group of the variation between DNA extraction methods. Samples from BLAF are in the upper plot, the lower plot includes samples from mouthwash. Kit1= QIAamp DNA Microbiome Kit (Catalogue 51704), kit2= QIAamp UCP Pathogen Mini Kit (Catalogue 50214), kit3= QIAamp UCP PurePathogen Blood Kit (Catalogue 50112).
We subsequently analyzed the top ten for each sample by family level and genus level, respectively. The results show that there are 40 possible families which could feature in the top 10 for each sample from both BALF and mouthwash (Figure 6). Some families mostly appear in samples from mouthwash, like Neisseriaceae, Micrococcaceae, Pasteurellaceae, and Actinomycetaceae; while some families mostly appear in samples from BALF, like Enterobacteriaceae, Acholeplasmataceae, Erysipelotrichaceae, Xanthomonadaceae, and Sphingomonadaceae. And moreover, some families frequently appear in samples from both BALF and mouthwash, such as Alcaligenaceae, Methylobacteriaceae, Streptococcaceae, Prevotellaceae, and Veillonellaceae.
Figure 6.

Proportions of top 10 bacterial families in each sample inferred from metagenomic sequence data. Each column corresponds to an individual respiratory tract sample. Each row corresponds to a specific bacterial family. Rows were subjected to hierarchical clustering to emphasize families that show similar abundance patterns. The proportional representation (relative abundance) of each family is represented by the color code (key to the right). Codes of DNA samples are shown along the bottom.
There are 20 possible genera of bacteria which could feature in top 10 for each sample from BALF, but less than mouthwash (24 genera of bacteria) (Figure 7A, 7B). The genera mostly appearing in samples from mouthwash are Achromobacter, Actinomyces, Arthrobacter, Bacteroides, Bordetella, Bradyrhizobium, Candidatus Phytoplasma, Capnocytophaga, Coprobacillus, and additional 14 genera; the genera mostly appearing in samples from BALF are Achromobacter, Actinomyces, Bacteroides, Bordetella, Bradyrhizobium, Candidatus Phytoplasma, Coprobacillus, Cyanothece, Escherichia, Fusobacterium, and ten more genera. And there are four genera only appearing in samples from mouthwash: Arthrobacter, Capnocytophaga, Mesorhizobium, and Porphyromonas, the rest ones appear in samples from both BALF and mouthwash (Figure 7C).
Figure 7.
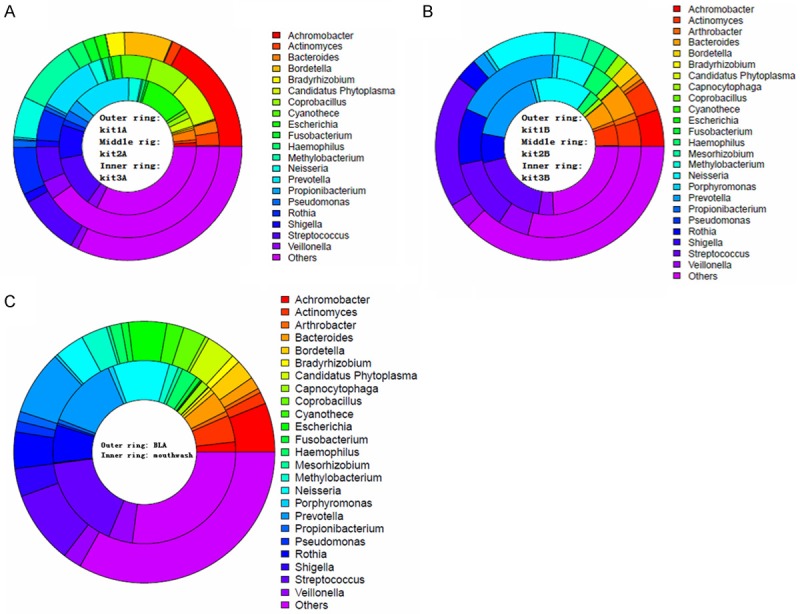
Proportions of top 10 bacterial genera in each sample inferred from metagenomic sequence data. A. Samples from BALF; B. Samples from mouthwash; C. Comparison of all samples from BALF with all mouthwash samples. Each sector represent proportion of relative abundance of a bacterial genus in each DNA sample, interpretations of colours are shown on the right of rings. Kit1= QIAamp DNA Microbiome Kit (Catalogue 51704), kit2= QIAamp UCP Pathogen Mini Kit (Catalogue 50214), kit3= QIAamp UCP PurePathogen Blood Kit (Catalogue 50112).
Discussion
In this study we explored the effects of DNA extraction methods on metagenomic sequence characteristics, total bacteria relative abundance, community diversity, richness and evenness and bacterial community structure.
Previous studies showed that DNA extraction utilizing a mechanical approach with bead beating would perform best in extracting representative DNA from faecal material and upper airway samples [2-6]. We chose three DNA extraction kits using mechanical lysis with bead beating made from the same company that have been used widely across the literatures [12,23,24]. Before DNA library construction, we measured the DNA concentration of all DNA samples and found that DNA concentrations of DNA samples in group kit1 were much lower than those in both group kit2 and kit3, for samples from both BALF and mouthwash. However, when analyzing preliminarily sequence characteristics, we found that the ratio of human genome in clean reads of each sample in groups using kit1 was remarkably lower than those in groups using kit2 and kit3, for DNA samples from both BALF and mouthwash. The main difference among the three protocols was the procedure of depletion of host cells or nucleic acid before the isolation of bacterial DNA. In the protocol of kit1, there are special steps of depletion of host cells or nucleic acid, which include adding Buffer RDD and Benzonase to samples, mixing well, and incubating at 37°C for 30 minutes at 600 rpm. In kit2, there are no special steps of depletion of host cells or nucleic acid before the isolation of bacterial DNA. In kit3, approximately 1 volume of Buffer APL1 is added to the remaining cell fraction. It is claimed that Buffer APL1 specifically lyses human blood cells while microbial cells stay intact. Our results show that adding Benzonase to samples before the isolation of bacterial DNA could enable DNA samples with a higher fraction of microbial DNA from samples with a higher fraction of host cells to be obtained. However, the total DNA concentrations of DNA samples isolated via the steps of depletion of host cells or nucleic acid by Benzonase are significantly lower than those isolated with other two kits.
Benzonase is a genetically engineered endonuclease from Serratia marcescens [25,26]. The enzyme is produced and purified from E. coli strain W3110, a mutant of strain K12, containing the proprietary pNUC1 production plasmid [27,28]. Structurally, the protein is a dimer of identical 245 amino acid, ~30 kDa subunits with two essential disulfide bonds [29-32]. This promiscuous endonuclease attacks and degrades all forms of DNA and RNA (single stranded, double stranded, linear and circular) and is effective over a wide range of operating conditions [33]. The enzyme completely digests nucleic acids to 5’-monophosphate terminated oligonucleotides 2-5 bases in length [26,34]. Although the nuclease is capable of cleavage at nearly all positions along a nucleic acid chain, sequence-dependent preferences have been demonstrated [35]. The enzyme prefers GC-rich regions in dsDNA while avoiding d(A)/d(T)-tracts. As we know, the main composition of cell walls for bacterial is peptidoglycan, and it is polysaccharide for fungal cells. Benzonase has no impact on them and the additional Benzonase will be degraded by Proteinase K, hence it will not hurt microbial DNA.
We also analyzed community composition with clean reads without human genome. For samples from BALF there were significant differences on bacteria, archea, unassigned sequences, unclassified sequences, other sequences between the three groups. Moreover, the parameters for group kit1A were wider than those for group kit2A and group kit3A. This result corresponds to the result for fraction of microbial DNA. However, for mouthwash samples the only significant difference between the three groups was eukaryota. As noted in MG-RAST Manual for version 3.6, said “The system supports the analysis of the prokaryotic content of samples, analysis of viruses and eukaryotic sequences is not currently supported”. Therefore, this difference is invalid. The differences we found between BALF samples did not appear in mouthwash samples. For the majority subjects, with the exception of patient 4 and patient 12, the clean sequence reads removed human genome from mouthwash samples, more so than those from BALF samples. These results suggest that the DNA extraction methods used do not significantly affect the results when comparing samples from different body sites. This is consistent with previous study [23]. However, the strong clustering by study in fecal samples from Western adults indicates that differences in experimental protocol, including DNA extraction protocol, and sequencing platform can be associated with significant differences in the observed diversity [23]. Experimental protocols must thus be carefully standardized for studies conducted within populations and age groups, especially when the effects of a biological parameter on the (gut) microbiota are expected to be subtle [23].
When observing exclusively bacteria, we found that the relative abundance of total bacteria, richness (total number of taxa) and evenness (relative abundance of taxa) in group kit1A were significantly higher than those in group kit2A and group kit3A. Richness here stands for number of observed taxa of each sample. Although we have selected patients sharing similar clinical manifestations to minimize bias by population difference, difference between individuals could exist, and the results of analysis of bacterial community structure actually would be affected by these differences between individuals. Age and geography/culture could drive major clustering patterns across studies of the gut (stool) microbiota [23]. However, in the present study, we have determined that there were bacteria in BALF of each individual chosen by our research, which meant the lower airways of patients with IIP without airway infection were not an absolute sterile environment, but were inhabited, by a great number of bacterium. Our findings are consistent with some of the results of previous studies based on 16S rRNA gene sequencing [12,24]. However, we obtained some interesting findings when performing clustering analysis and gene annotation with a larger volume of samples (data not shown).
Microbial DNA extraction method with pretreatment of depletion of host cells or nucleic acid by Benzonase enables a higher yield of microbial DNA from samples with a higher fraction of host cells, thereby reducing the cost of sequencing and increasing the accuracy of the sequencing data analysis. The lower airways of patients with IIP without airway infection were inhabited by a great number of bacterium.
Acknowledgements
We thank Dr. Zhen Wang, and the Genomics and Synthetic Biology Center of Tsinghua University team for their assistance with this effort. This work was supported by a grant from the National High Technology Research and Development Program of China (No. 2012AA02A511). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure of conflict of interest
None.
Authors’ contribution
Conceived and designed the experiments: YW FX CW. Performed the experiments: YW. Analyzed the data: YW. Wrote the paper: YW FX.
Supporting Information
References
- 1.Mao DP, Zhou Q, Chen CY, Quan ZX. Coverage evaluation of universal bacterial primers using the metagenomic datasets. BMC Microbiol. 2012;12:66. doi: 10.1186/1471-2180-12-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Salonen A, Nikkila J, Jalanka-Tuovinen J, Immonen O, Rajilic-Stojanovic M, Kekkonen RA, Palva A, de Vos WM. Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J Microbiol Methods. 2010;81:127–134. doi: 10.1016/j.mimet.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 3.Wu GD, Lewis JD, Hoffmann C, Chen YY, Knight R, Bittinger K, Hwang J, Chen J, Berkowsky R, Nessel L, Li H, Bushman FD. Sampling and pyrosequencing methods for characterizing bacterial communities in the human gut using 16S sequence tags. BMC Microbiol. 2010;10:206. doi: 10.1186/1471-2180-10-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zoetendal EG, Heilig HG, Klaassens ES, Booijink CC, Kleerebezem M, Smidt H, de Vos WM. Isolation of DNA from bacterial samples of the human gastrointestinal tract. Nat Protoc. 2006;1:870–873. doi: 10.1038/nprot.2006.142. [DOI] [PubMed] [Google Scholar]
- 5.Zoetendal EG, Ben-Amor K, Akkermans AD, Abee T, de Vos WM. DNA isolation protocols affect the detection limit of PCR approaches of bacteria in samples from the human gastrointestinal tract. Syst Appl Microbiol. 2001;24:405–410. doi: 10.1078/0723-2020-00060. [DOI] [PubMed] [Google Scholar]
- 6.Biesbroek G, Sanders EA, Roeselers G, Wang X, Caspers MP, Trzcinski K, Bogaert D, Keijser BJ. Deep sequencing analyses of low density microbial communities: working at the boundary of accurate microbiota detection. PLoS One. 2012;7:e32942. doi: 10.1371/journal.pone.0032942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corless CE, Guiver M, Borrow R, Edwards-Jones V, Kaczmarski EB, Fox AJ. Contamination and sensitivity issues with a real-time universal 16S rRNA PCR. J Clin Microbiol. 2000;38:1747–1752. doi: 10.1128/jcm.38.5.1747-1752.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klaschik S, Lehmann LE, Raadts A, Hoeft A, Stuber F. Comparison of different decontamination methods for reagents to detect low concentrations of bacterial 16S DNA by realtime-PCR. Mol Biotechnol. 2002;22:231–242. doi: 10.1385/MB:22:3:231. [DOI] [PubMed] [Google Scholar]
- 9.Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, Davies J, Ervine A, Poulter L, Pachter L, Moffatt MF, Cookson WO. Disordered microbial communities in asthmatic airways. PLoS One. 2010;5:e8578. doi: 10.1371/journal.pone.0008578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Behr J, Thannickal VJ. Update in diffuse parenchymal lung disease 2008. Am J Respir Crit Care Med. 2009;179:439–444. doi: 10.1164/rccm.200901-0011UP. [DOI] [PubMed] [Google Scholar]
- 11.Vannella KM, Moore BB. Viruses as co-factors for the initiation or exacerbation of lung fibrosis. Fibrogenesis Tissue Repair. 2008;1:2. doi: 10.1186/1755-1536-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friaza V, la Horra C, Rodriguez-Dominguez MJ, Martin-Juan J, Canton R, Calderon EJ, Del Campo R. Metagenomic analysis of bronchoalveolar lavage samples from patients with idiopathic interstitial pneumonia and its antagonic relation with Pneumocystis jirovecii colonization. J Microbiol Methods. 2010;82:98–101. doi: 10.1016/j.mimet.2010.03.026. [DOI] [PubMed] [Google Scholar]
- 13.Travis WD, Costabel U, Hansell DM, King TE Jr, Lynch DA, Nicholson AG, Ryerson CJ, Ryu JH, Selman M, Wells AU, Behr J, Bouros D, Brown KK, Colby TV, Collard HR, Cordeiro CR, Cottin V, Crestani B, Drent M, Dudden RF, Egan J, Flaherty K, Hogaboam C, Inoue Y, Johkoh T, Kim DS, Kitaichi M, Loyd J, Martinez FJ, Myers J, Protzko S, Raghu G, Richeldi L, Sverzellati N, Swigris J, Valeyre D ATS/ERS Committee on Idiopathic Interstitial Pneumonias. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:733–748. doi: 10.1164/rccm.201308-1483ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iwai S, Fei M, Huang D, Fong S, Subramanian A, Grieco K, Lynch SV, Huang L. Oral and airway microbiota in HIV-infected pneumonia patients. J Clin Microbiol. 2012;50:2995–3002. doi: 10.1128/JCM.00278-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jakobsson HE, Jernberg C, Andersson AF, Sjolund-Karlsson M, Jansson JK, Engstrand L. Short-term antibiotic treatment has differing long-term impacts on the human throat and gut microbiome. PLoS One. 2010;5:e9836. doi: 10.1371/journal.pone.0009836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyer F, Paarmann D, D’Souza M, Olson R, Glass EM, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A, Wilkening J, Edwards RA. The metagenomics RAST server-a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9:386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cox MP, Peterson DA, Biggs PJ. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics. 2010;11:485. doi: 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keegan KP, Trimble WL, Wilkening J, Wilke A, Harrison T, D’Souza M, Meyer F. A platformindependent method for detecting errors in metagenomic sequencing data: DRISEE. PLoS Comput Biol. 2012;8:e1002541. doi: 10.1371/journal.pcbi.1002541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gomez-Alvarez V, Teal TK, Schmidt TM. Systematic artifacts in metagenomes from complex microbial communities. ISME J. 2009;3:1314–1317. doi: 10.1038/ismej.2009.72. [DOI] [PubMed] [Google Scholar]
- 20.Huse SM, Huber JA, Morrison HG, Sogin ML, Welch DM. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 2007;8:R143. doi: 10.1186/gb-2007-8-7-r143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huson DH, Auch AF, Qi J, Schuster SC. MEGAN analysis of metagenomic data. Genome Res. 2007;17:377–386. doi: 10.1101/gr.5969107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Institute NHGR. Cost per raw megabase of dna sequence. 2012 [Google Scholar]
- 23.Lozupone CA, Stombaugh J, Gonzalez A, Ackermann G, Wendel D, Vazquez-Baeza Y, Jansson JK, Gordon JI, Knight R. Metaanalyses of studies of the human microbiota. Genome Res. 2013;23:1704–1714. doi: 10.1101/gr.151803.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garzoni C, Brugger SD, Qi W, Wasmer S, Cusini A, Dumont P, Gorgievski-Hrisoho M, Muhlemann K, von Garnier C, Hilty M. Microbial communities in the respiratory tract of patients with interstitial lung disease. Thorax. 2013;68:1150–1156. doi: 10.1136/thoraxjnl-2012-202917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eaves GN, Jeffries CD. Isolation And Properties Of an Exocellular Nuclease Of Serratia Marcescens. J Bacteriol. 1963;85:273–278. doi: 10.1128/jb.85.2.273-278.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nestle M, Roberts WK. An extracellular nuclease from Serratia marcescens. I. Purification and some properties of the enzyme. J Biol Chem. 1969;244:5213–5218. [PubMed] [Google Scholar]
- 27.Molin S, Givskov M, Riise E. Production in Escherichia coli of extracellular Serratia spp. hydrolases. Hvidovre, Denmark: Benzon Pharma, A/S; 1992. [Google Scholar]
- 28.Molin S, Givskov M, Riise E. Bacterial enzymes and method for their production. Hvidovre, Denmark: Benzon Pharma, A/S; 1992. [Google Scholar]
- 29.Miller MD, Tanner J, Alpaugh M, Benedik MJ, Krause KL. 2.1 A structure of Serratia endonuclease suggests a mechanism for binding to double-stranded DNA. Nat Struct Biol. 1994;1:461–468. doi: 10.1038/nsb0794-461. [DOI] [PubMed] [Google Scholar]
- 30.Friedhoff P, Gimadutdinow O, Pingoud A. Identification of catalytically relevant amino acids of the extracellular Serratia marcescens endonuclease by alignment-guided mutagenesis. Nucleic Acids Res. 1994;22:3280–3287. doi: 10.1093/nar/22.16.3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ball TK, Suh Y, Benedik MJ. Disulfide bonds are required for Serratia marcescens nuclease activity. Nucleic Acids Res. 1992;20:4971–4974. doi: 10.1093/nar/20.19.4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ball TK, Saurugger PN, Benedik MJ. The extracellular nuclease gene of Serratia marcescens and its secretion from Escherichia coli. Gene. 1987;57:183–192. doi: 10.1016/0378-1119(87)90121-1. [DOI] [PubMed] [Google Scholar]
- 33.Code No. W 220911. Darmstadt, Germany: Merck KGaA; 1999. Benzonase. [Google Scholar]
- 34.Janning P, Schrader W, Linscheid M. A new mass spectrometric approach to detect modifications in DNA. Rapid Commun Mass Spectrom. 1994;8:1035–1040. doi: 10.1002/rcm.1290081226. [DOI] [PubMed] [Google Scholar]
- 35.Meiss G, Friedhoff P, Hahn M, Gimadutdinow O, Pingoud A. Sequence preferences in cleavage of dsDNA and ssDNA by the extracellular Serratia marcescens endonuclease. Biochemistry. 1995;34:11979–11988. doi: 10.1021/bi00037a040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.