

Abstract
Coenzyme Q10, (CoQ10) an electron transporter and an antioxidant, protects a variety of cell types against oxidative stress and apoptosis. However, protective effect of CoQ10 on oxidative stress-induced apoptosis in lymphocytes has not been studied in detail. In this study, we investigated the effect of CoQ10 on oxidative stress-induced apoptosis in lymphocytes. An exposure of peripheral blood lymphocytes to oxidative stressors, rotenone or hydrogen peroxide, lead to apoptosis. Pre-treatment of lymphocytes with CoQ10 resulted in a significantly reduced level of oxidative stress-induced apoptosis, which was associated with decreased reactive oxygen species production, an inhibition of mitochondrial membrane depolarization, and inhibition of activation of caspase-9 and caspase-3. Furthermore, CoQ10 inhibited oxidative stress induced apoptosis in both CD4+ T, and CD8+ T, and CD19+ B cells. Our findings suggest that CoQ10 may provide new therapeutic strategies for preventing oxidative stress-induced cell death and dysfunction in lymphocytes and lymphocyte subsets.
Keywords: Coenzyme Q10, oxidative stress, apoptosis, lymphocytes
Introduction
Oxidative stress is characterized by an imbalance between the cellular production of oxidants and the capacity of cellular antioxidant defenses to scavenge these oxidants. Oxidative stress is produced in cells by reactive oxygen species (ROS), which include free radicals and peroxides. They are produced at a low level by normal aerobic metabolism. Increasing evidence suggests that oxidative stress is a major inducer of cell death by apoptosis via mitochondrial pathway [1,2].
Coenzyme Q10 (CoQ10), also known as ubiquinone 10, is an electron transporter, that transports electrons from electron transport complex (ETC) I and complex II to complex III. CoQ10, has been shown to be an important antioxidant, and acts as a modulator of mitochondrial permeability transition pore, mitochondrial membrane potential and an inhibitor of ROS generation [3-5]. CoQ10 is synthesized endogenously and small quantities are consumed through diet.
Previous studies have shown that CoQ10 protects human keratocytes, keratinocytes and leukemia cells from oxidative stress-induced apoptosis [5-7]. Furthermore, CoQ10 has been shown ameliorates H2O2-induced DNA damage in human lymphocytes [8]. In this study, we investigated the effects of CoQ10 on oxidative stress-induced apoptosis of lymphocyte and lymphocyte subsets.
Materials and methods
Chemicals and reagents
Coenyme Q10 (CoQ10), Lutrol F127, Rotenone and H2O2 were purchased from Sigma (St Louis, MO). Annexin V-FITC, TMRE, DHR 123, and In Situ Death Detection Kit, were obtained from Life technologies (Grand island NY). Caspase -9 (FAM-LEHD-FMK) and caspases 3 (FAM-DEVD-FMK) Colorimetric assay kits were purchased from Biovision Research Products, (Palo Alto, CA).
CoQ10 solution was prepared by dissolving it in absolute alcohol and then added to AIM V medium containing 0.04% Lutrol 127. Leutrol was used as a vehicle to ensure cellular uptake of hydrophobic CoQ10.
Subjects
Peripheral blood was obtained from healthy volunteers. The study was approved by Institutional Review Board (Human) of the University of California, Irvine.
Isolation of lymphocytes and treatments
Peripheral blood mononuclear cells (MNCs) were separated from peripheral blood by density gradient centrifugation. MNCs were treated with various concentration of CoQ10 for 24 hrs prior to exposure to apoptotic stimuli. Cells treated with Lutrol F127 alone served as controls. Rotenone and hydrogen peroxide (H2O2) were used as apoptotic stimuli. They were used at concentrations previously established to induce maximun apoptosis: rotenone 6.25 μM and H2O2 25 μM.
Detection of apoptosis
Apoptosis was measured by TUNEL and Annexin V-FITC binding assays, according to manufacturer’s instructions.
TUNEL assay (terminal deoxyribonucleotidyl transferase (TDT)-mediated dUTP-nick end labeling
Briefly, cells were fixed with 2% paraformaldehyde, and permeabilized with sodium citrate buffer containing 0.1% Triton X-100 Following washing, cells were incubated for 1 hour with FITC-conjugated dUTP in the presence of TdT enzyme solution containing 1 M potassium cacodylate and 125 mM Tris-Hcl, pH 6.6 (In Situ Cell Death Detection Kit), Cells were washed with phosphate buffered saline (PBS) and 10,000 cells were acquired with FACSCalibur and analyzed by using Cell Quest software.
Annexin-V-FITC assay
In few experiments, MNCs (0.5×106) were stained with 10 µL of PerCP-conjugated anti-CD19, anti-CD4 or anti-CD8 monoclonal antibodies, washed twice with PBS, and resuspended in 100 µL of binding buffer containing 5 µL of FITC-conjugated Annexin V. The cells were incubated in dark at room temperature for 15 minutes, after which 400 µL of binding buffer added, and 5000 cells were acquired and anlyzed by FACSCalibur. FL3 channels were used to gate CD19+ B cells, CD4+ T cells and CD8+ T and the proportions of annexin V positive cells were determined.
Determination of mitochondrial potential (ΔΨm)
Mitochondrial membrane potential was analyzed with TMRE, using FACSCalibur. Briefly, Cells (1×106/ml) that were exposed to apoptotic stimuli were incubated in medium containing 50 nM TMRE and incubated for 10 minutes at 37°C. Cells were transferred on ice, Stained with 10 µL of PerCP-conjugated anti-CD19, anti-CD4 or anti-CD8 monoclonal antibodies. TMRE staining in lymphocyte subsets was analyzed by dual color flowcytometry. FL3 channels were used to gate CD19+ B cells, CD4+ T cells and CD8+ T cells and FL2 channel was used to collect the red fluorescence of TMRE. A reduction in red fluorescence indicates loss of ΔΨm.
Measurement of reactive oxygen species
Reactive oxygen species (ROS) was determined by utilizing oxidation-dependent fluorescence of dye dihydrorhodamine 123 (DHR123). DHR123 is a cell permeable nonfluorecent dye that becomes highly fluorescent when oxidized by ROS. Peripheral blood mononuclear cells (1×106/ml) pretreated with CoQ10 or vehicle control were loaded with 5 µM DHR123 and then exposed to rotenone for 15 minutes. Cells were stained with PerCP-conjugated anti-CD19, anti-CD4 or anti-CD8 antibodies and DHR fluorescence in lymphocyte subsets was analyzed by dual color flow cytometry. The data was analyzed using cell quest software and the mean fluorescence channel numbers (MFC#) were recorded.
Caspase activity
The activation of caspases-3 and caspase-9 was analyzed by flow cytometry with carboxyfluorescein-labeled cell permeable peptide substrates (Cell Technology, Fremont CA), that recognize cleaved caspase-9 (FAM-LEHD-FMK) and caspases-3 (FAM-DEVD-FMK) according to the protocol provided by the manufacturer. Briefly, MNCs treated with CoQ10 and subsequently exposed to apoptotic stimuli for 24 hours were incubated with caspase-9 or caspase-3 substrates for 1 hour at 37°C. The cells were stained with PerCP-conjugated anti-CD19, anti-CD4 or anti-CD8 antibodies, washed with wash buffer and analyzed by FACScalibur.
Statistics
All of the experiments were repeated with samples from 3 to 16 individual subjects. Statistical analysis was performed by student t test. The level of significance was set at P < 0.05.
Results
CoQ10 protects lymphocytes from oxidative stress induced apoptosis
Peripheral blood mononuclear cells (MNCs) from healthy subjects were treated with different concentrations of CoQ10 for 24 hours prior to exposure to oxidative stressors, rotenone and H2O2, and apoptosis was determined by annexin V binding and TUNEL assays. Treatment with CoQ10 resulted in, a concentration-dependent manner, inhibition of apoptosis (Figure 1). The maximal inhibition was observed at 10 μM and therefore was used in subsequent studies.
Figure 1.
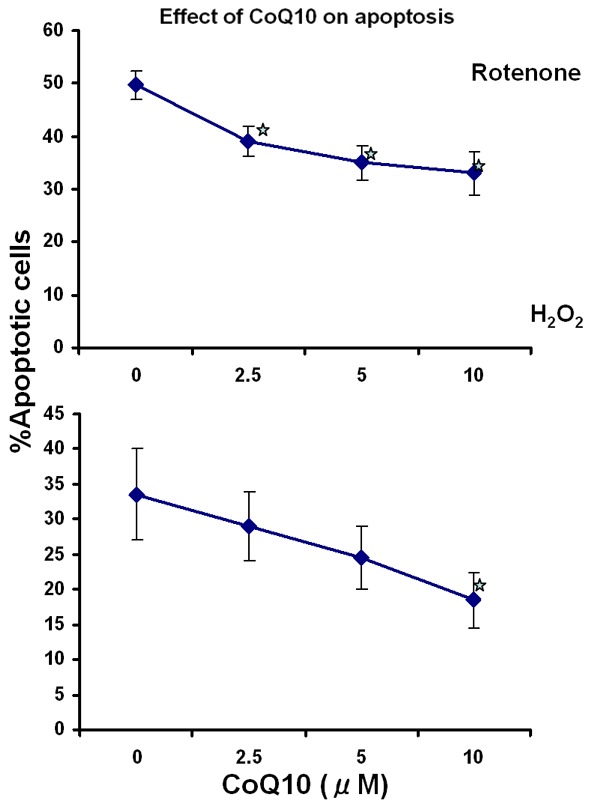
Dose response curve of CoQ10 protective effect on oxidative stress-induced apoptosis. MNCs were pretreated with the indicated concentrations of CoQ10 for 24 hours prior to addition of rotenone or H2O2 Apoptosis was determined by annexin V (upper panel) and tunnel (lower panel) assays. Results are the mean of 3 separate experiments each done on lymphocytes from a different donor *p ≤ 0.05.
Figure 2 shows protective effect of 10 μM CoQ10 on rotenone-induced and H2O2-induced apoptosis on lymphocytes of 16 and 12 subjects, respectively.
Figure 2.
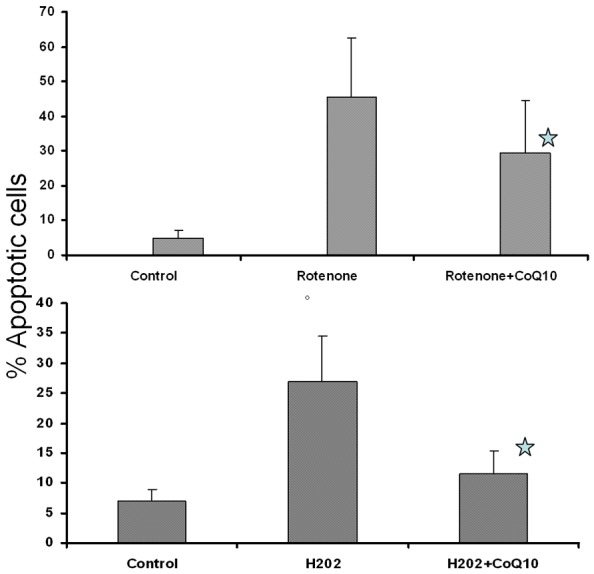
Protective effect of CoQ10 on oxidative stress-induced apoptosis. MNCs were pretreated CoQ10 (10 μM) for 24 hours prior to addition of Rotenone, upper panel, N=16) or H2O2 (lower panel, N=12. Apoptosis was determined by tunnel assay. Results represent mean ± SD *p ≤ 0.05.
CoQ10 inhibits oxidative stress-induced apoptosis in CD4+ and CD8+ T cells and CD19+ B cells
In order to determine whether protective effect of CoQ10 is different among various subsets of lymphocytes, we examined the protective effect of CoQ10 on oxidative stress-induced apoptosis in CD4+ and CD8+ T cell subsets and CD19+ B cells. Rotenone induced apoptosis in both CD4+ and CD8+ T cells and CD19+ B cells. CoQ10 inhibited rotenone induced apoptosis (Figure 3); and the magnitude of protective effect of CoQ10 was similar in on CD4+ and CD8+ T cells and CD19+ B cells.
Figure 3.
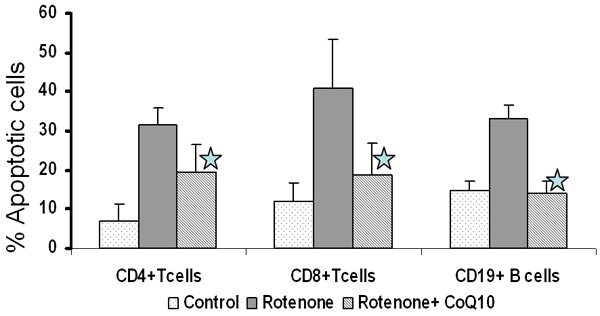
Effect of CoQ10 on rotenone-induced apoptosis in lymphocyte subsets. MNCs were pretreated CoQ10 (10 μM) for 24 hours prior to addition of rotenone. Apoptosis in CD4+ T and CD8+ T, CD19+ B lymphocyte subsets was determined by dual color flow cytometry. T cell subset results are the mean ± SD of 6 separate experiments each done on a separate subject. B cell results are the mean of 3 separate experiments *p ≤ 0.05 as compared to rotenone alone.
CoQ10 reverses apoptosis in lymphocytes by rectifying mitochondrial dysfunction induced by oxidative stressors
A key early contributing factor in oxidative stress-induced apoptosis is mitochondrial dysfunction. Mitochondrial dysfunction is characterized by increased production of superoxide anions, a reduction in inner transmembrane potential, increased permeability of the outer membrane by opening mitochondrial transport pore (MTP), a step critical for release of cytochrome c and sequential activation caspase-9 and caspase-3 that results in apoptotic cell death [9].
Both rotenone and H2O2 induce apoptosis by mitochondrial pathway Therefore in the subsequent experiments we used one reagent (rotenone) to determine whether CoQ10 reversed apoptosis by rectifying mitochondrial dysfunction. For this purpose, we measured production of reactive oxygen species (ROS), membrane potential and activation of caspase-9 and caspase-3 in lymphocyte subsets induced by rotenone.
Production of ROS
Control and CoQ10 pretreated Lymphocytes were exposed to rotenone for 15 minutes and intracellular ROS was measured with DHR123 by using FACSCalibur. Figure 4 shows that the exposure of lymphocytes to rotenone resulted in a significantly increased (P < 0.05) production of superoxide in CD4+ and CD8+ T cells and CD19+ B cells compared to unexposed cell, and pretreatment of lymphocytes with CoQ10 resulted in reduction of superoxide production to basal level.
Figure 4.
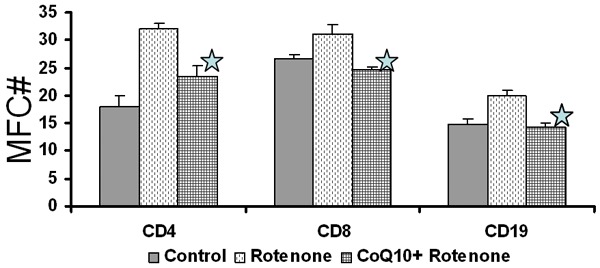
CoQ10 inhibits rotenone-induced ROS production. Control and CoQ10 pre-exposed MNCs were incubated with Rotenone and ROS production in lymphocyte subsets was measured by dual color flow cytometry Data are shown as mean ± SD of three separate experiments each done on a separate subject *p ≤ 0.05.
Mitochondrial membrane potential
The effect of CoQ10 on mitochondrial membrane potential was measured by uptake of TMRE dye using FACSCalibur. Figure 5 shows that rotenone (A) induced a significant decrease (P < 0.05) in mitochondrial membrane potential (depolarization) in CD4+ and CD8+ T cells and CD19+ B cells and CoQ10 prevented mitochondrial membrane depolarization in lymphocyte subsets induced by rotenone.
Figure 5.
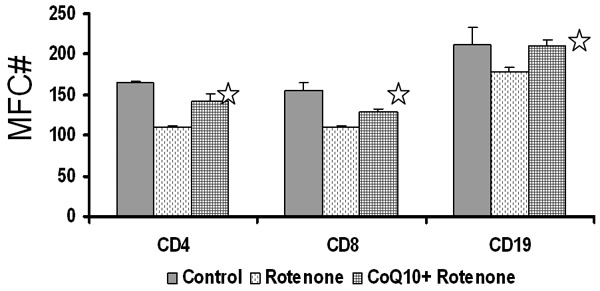
Effect of CoQ10 on oxidative stress-induced mitochondrial membrane potentials. MNCs pretreated with CoQ10 were exposed rotenone and mitochondrial membrane potentials were determined in CD19+ B cells and CD4+ T and CD8+ T cells by TMRE dye and FACS. CoQ10 inhibited mitochondrial membrane depolarization induced by rotenone *p ≤ 0.05.
Caspase 3 and caspase 9 activation
Depolarization of mitochondrial membrane is associated with the release of cytochrome c, which forms apoptosome by binding to APAF-1, that in turn activates initiator caspase-9, subsequently executioner caspase-3 to induced apoptosis, we measured rotenone-induced activation of both caspase-9 and caspase-3 in CoQ10 pretreated and untreated cells using flow cytometry. Figure 6 show that rotenone increased the activation of caspase-9 and caspase-3, in CD4+ and CD8+ T cells and CD19+ B cells and CoQ10 pretreatment inhibited activation of both caspase-9 and caspase-3.
Figure 6.
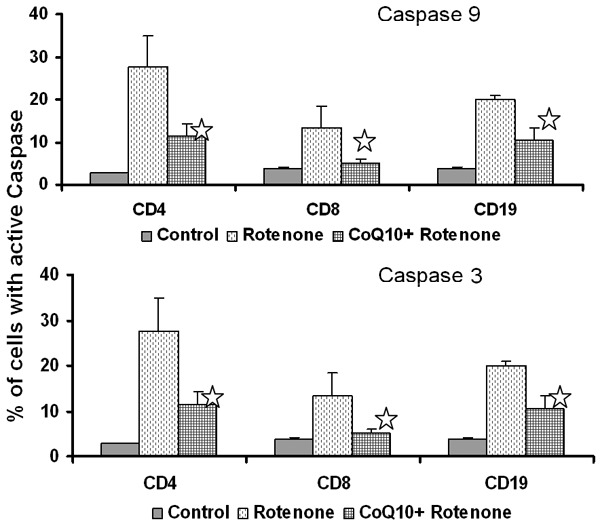
Inhibitory effect of CoQ10 on oxidative stress-induced caspase activation. Control and CoQ10 preexposed MNCs were incubated with rotenone and caspase 9 and caspase 3 activities in lymphocyte subsets were determined by flow cytometry. Data are shown as mean ± SD of three separate experiments p ≤ 0.05.
Discussion
Oxidative stress is caused by the persistent production of ROS. ROS are byproducts of oxidative phosphorylation and mainly produced in mitochondrial electron transport chain complexes I and III [10,11]. Increased ROS production has been shown to affect various mitochondrial parameters including membrane potential, and permeability transition pore activation [3-5]. These changes lead to mitochondrial outer membrane permeabilization, resulting in the release of apoptogenic proteins to induce apoptosis by caspase dependent and caspase independent pathways [9]. Oxidative stress is associated with altered functions of immune cells [12-14], autoimmune diseases and immune senescence and triggers inflammation [15-18].
The major function of mitochondrial respiratory chain (MRC), which is located in the inner mitochondrial membrane, is to synthesize ATP via oxidative phosphorylation [19]. MRC is consists of 5 enzyme complex I-V. CoQ10 is the predominant form of ubiquinone and serves as an electron carrier in MRC [20]. CoQ10, in addition to oxidative phosphorylation, may be required for other functions including potent lipid soluble antioxidant [21], regulation of permeability transition pore opening and maintenance of body temperature via its role as a co-factor for the mitochondrial uncoupling proteins, as an anti-oxidant, and other cellular functions including, DNA replication and repair [22].
CoQ10, ubiquinone, is ubiquitously present in all eukaryotic cells. The biosynthesis of CoQ10 requires at least 15 genes. Primary CoQ10 deficiency as a result of mutations of some of these genes is associated with a heterogenous group of disorders ranging from cerebellar ataxia to severe infantile form of steroid resistant nephrotic syndrome [23,24]. These primary CoQ10 deficiencies are rare; however, secondary immunodeficiencies of CoQ10 are more frequent. CoQ10 deficiencies are associated with increased oxidative stress and apoptosis of various cell types via mitochondrial pathway [25-27]. A number of stimuli trigger mitochondrial pathway of apoptosis, which is associated with depolarization of mitochondrial potentials, opening of MTP, and release of two important apoptogenic proteins, the cytochrome c and apoptosis-inducing factor (AIF), mediating cell death via caspase-dependent and caspase-independent pathways respectively [9,28]. Lopez et al [26] showed increased oxidative stress and apoptosis in CoQ10-deficient human fibroblasts, and exogenous CoQ10 protected from both oxidative stress and apoptosis. Duran-Prado [29] demonstrated that CoQ10 protects human endothelial cells by beta-amyloid-induced oxidative stress and apoptosis. CoQ10 inhibited calcium release from the mitochondria due to its effect on the opening of MTP. Li et al [30] using mitochondrial I complex inhibitor, rotenone, reported an increase in ROS production and increased caspase-independent apoptosis via translocation of AIF to the nucleus in murine hippocampal HT22 cells. CoQ10 inhibited both increased production of rotenone-induced ROS production, nuclear translocation of AIF, and apoptosis. In the present study, we have demonstrated that rotenone induced increased production of ROS, depolarization of mitochondrial membrane, activation of caspase-9 and caspase-3, and increased apoptosis of lymphocytes. CoQ10 inhibited increased ROS production, prevented depolarization of mitochondrial membrane, and inhibited activation of caspase-9, caspase3, and and apoptosis. The difference in rotenone-induced caspase-independent apoptosis observed by Li et al [29] and caspase-dependent apoptosis observed by us may be due to differences in target cell types (hippocampal versus lymphocytes).
Alleva et al [6] showed that CoQ10 reduced H2O2 but not anti-Fas- and TRAIL-induced apoptosis in Jurkat T cells by blocking the generation of ROS and mitochondrial membrane depolarization. This may be explained by the fact that death-receptor pathway is independent of mitochondrial pathway and does not depend on oxidative stress. These investigators did not examine the effect of CoQ10 on mitochondrial pathway of apoptosis. In this study, we have demonstrated that CoQ10 attenuated oxidative stress-induced apoptosis in B lymphocytes and T lymphocyte subsets from healthy subjects. Furthermore, we showed that the inhibitory effect of CoQ10 on lymphocyte subsets apoptosis is associated with its ability to inhibit increased production of ROS, by blocking mitochondrial membrane depolarization, and inhibiting caspase-9 and caspase-3 activation induced by rotenone Similar findings have beed reported by Naderi et al (who showed that water soluble CoQ10 inhibits mitochondrial apoptotic events in Fibroblasts and HEK293 cells exposed to H2O2 [31]. In summary, CoQ10, a natural chemical compound, attenuates oxidative stress-induced apoptosis in T cell subsets by rectifying mitochondrial dysfunction. This suggest that CoQ10 may have therapeutic potential in disorders of lymphocytes associated with excess oxidative stress and apoptosis.
Acknowledgements
This work in part was supported by an unrestricted gift from Richard and Aida Demirjian.
Disclosure of conflict of interest
None.
References
- 1.Cui H, Kong Y, Zhang H, editors. Oxidative Stress, Mitochondrial Dysfunction, and Aging Journal of Signal Transduction. 2012. Article ID 646354, 13 pages http://dx.doi.org/10.1155/2012/646354. [DOI] [PMC free article] [PubMed]
- 2.Kirkinezosa IG, Moraes CT. Reactive oxygen species and mitochondrial diseases. Semin Cell Dev Biol. 2001;12:449–457. doi: 10.1006/scdb.2001.0282. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong JS, Whiteman M, Rose P, Jones DP. The Coenzyme Q10 Analog Decylubiquinone Inhibits the Redox-activated Mitochondrial Permeability Transition: Role of Mitochondrial respiratory complex III. J Biol Chem. 2003;278:49079–49084. doi: 10.1074/jbc.M307841200. [DOI] [PubMed] [Google Scholar]
- 4.Fontaine E, Ichas F, Bernardi P. Regulation of the Permeability Transition Pore in Skeletal Muscle Mitochondria:Modulation by electron flow through respiratory chain complex I. J Biol Chem. 1998;273:25734–25740. doi: 10.1074/jbc.273.20.12662. [DOI] [PubMed] [Google Scholar]
- 5.Papucci L, Schiavone N, Witort E, Donnini M, Lapucci A, Tempestini A, Formigli L, Zecchi-Orlandini S, Orlandini G, Carella G, Brancato R, Capaccioli S. Coenzyme Q10 prevents apoptosis by inhibiting mitochondrial depolarization independently of its free radical scavenging property. J Biol Chem. 2003;278:28220–28228. doi: 10.1074/jbc.M302297200. [DOI] [PubMed] [Google Scholar]
- 6.Allevaa R, Tomasetti M, Anderac L, Gellertd N, Borghia B, Weberd C, Murphye MP, Neuzild J. Coenyme Q blocks biochemical but not receptor mediated apoptosis by increasing mitochondrial antioxidant protection. FEBS Letters. 2001;503:46–50. doi: 10.1016/s0014-5793(01)02694-1. [DOI] [PubMed] [Google Scholar]
- 7.Hoppe U, Bergemanna J, Diembecka W, Ennen J, Gohlaa S, Harris I, Jacob J, Kielholz J, Mei W, Pollet D, Schachtschabel D, Sauermanna G, Schreiner V, Stäb F, Steckel F. Coenzyme Q10, a cutaneous antioxidant and energizer. Biofactors. 1999;9:371–378. doi: 10.1002/biof.5520090238. [DOI] [PubMed] [Google Scholar]
- 8.Tomasetti M, Littarru GP, Stocker R, Alleva R. Coenzyme Q10 enrichment decreases oxidative DNA damage in human lymphocytes. Free Radic Biol Med. 1999;27:1027–1032. doi: 10.1016/s0891-5849(99)00132-x. [DOI] [PubMed] [Google Scholar]
- 9.Green DR. At the gates of death. Cancer Cell. 2006;9:328–330. doi: 10.1016/j.ccr.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 10.Halliwell B. Reactive oxygen species in living systems: Source, biochemistry, and role in human disease. Am J Med. 1991;91:14S–22S. doi: 10.1016/0002-9343(91)90279-7. [DOI] [PubMed] [Google Scholar]
- 11.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bashir S, Harris G, Denman MA, Blake DR, Winyard PG. Oxidative DNA damage and cellular sensitivity to oxidative stress in human autoimmune diseases. Ann Rheum Dis. 1993;52:659–666. doi: 10.1136/ard.52.9.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glaser R, Kiecolt-Glaser JK. Stress-induced immune dysfunction: implications for health. Nat Rev Immunol. 2005;5:243–251. doi: 10.1038/nri1571. [DOI] [PubMed] [Google Scholar]
- 14.Larbi A, Kempf J, Pawelec G. Oxidative stress modulation and T cell activation. Exp Gerontol. 2007;42:852–858. doi: 10.1016/j.exger.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 15.Finkel T, Holbrook NJ. -Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 16.Halda A, Lotharius J. Oxidative stress and inflammation in Parkinson’s disease: is there a causal link? Experimental neurology. 2005;193:279–290. doi: 10.1016/j.expneurol.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 17.Lih-Brody L, Powell SR, Collier KP, Reddy GM, Cerchia R, Kahn E, Wessman GS, Katz S, Floyd RA, McKinley MJ, Fisher SE, Mullin GE. Increased oxidative stress and decreased antioxidant defenses in mucosa of inflammatory bowel disease. Dig Dis Sci. 1996;41:2078–2086. doi: 10.1007/BF02093613. [DOI] [PubMed] [Google Scholar]
- 18.Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic Biol Med. 2010;49:1603–1616. doi: 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rahman S, Hanna MG. Mitochondrial disease disorders: diagnosis and new treatment in mitochondrial diseases. J Neurol Neurosurg Psychiatry. 2009;80:943–953. doi: 10.1136/jnnp.2008.158279. [DOI] [PubMed] [Google Scholar]
- 20.Ernster L, Dallner G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim Biophys Acta. 1995;1271:195–204. doi: 10.1016/0925-4439(95)00028-3. [DOI] [PubMed] [Google Scholar]
- 21.Bentinger M, Brismar K, Dallner G. The antioxidant role of Coenzyme Q. Mitochondrion. 2007;75:741–751. doi: 10.1016/j.mito.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 22.López-Martín JM, Salviati L, Trevisson E, Montini G, DiMauro S, Quinzii C, Navas P. Missense mutation of the CoQ2 gene causes defects of bioenergetics and defects of bioenergetics and de novo pyrimidine synthesis. Hum Mol Genet. 2007;16:1091–1097. doi: 10.1093/hmg/ddm058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Desbats MA, Lunardi G, Doimo M, Trivisson E, Salviati L. Genetic bases and clinical manifestations of coenzyme Q10 (CoQ10) deficiency. J Inherit Metab Dis. 2015;38:145–156. doi: 10.1007/s10545-014-9749-9. [DOI] [PubMed] [Google Scholar]
- 24.Doimo M, Desbats MA, Cerqua C, Cassina M, Trevisson E, Salviati L. Genetics of Coenzyme Q10 Deficiency. Mol Syndromol. 2014;5:156–162. doi: 10.1159/000362826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Di Giovanni S, Mirabella M, Spinazzola A, Crociani P, Silvestri G, Broccolini A, Tonali P, Di Mauro S, Serividie S. Coenzyme Q10 reverses pathological phenotype and reduces apoptosis in familial CoQ10 deficiency. Neurology. 2001;57:515–518. doi: 10.1212/wnl.57.3.515. [DOI] [PubMed] [Google Scholar]
- 26.Lopez LC, Luna-Sanchez M, Garcia-Corzo L, Quinzii CM, Hirano M. Pathomechanisms in coenzyme Q10-deficient human fibroblasts. Mol Syndromol. 2014;5:163–169. doi: 10.1159/000360494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quinzii CM, López LC, Gilkerson RW, Dorado B, Coku J, Naini AB, Hirano M. Reactive oxygen species, oxidative stress, and cell death correlate with level of CoQ10 deficiency. FASEB J. 2010;24:3733–3743. doi: 10.1096/fj.09-152728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martinou JC, Green DR. Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol. 2001;2:67–71. doi: 10.1038/35048069. [DOI] [PubMed] [Google Scholar]
- 29.Durán-Prado M, Frontiñán J, Santiago-Mora R, Peinado JR, Parrado-Fernández C, Gómez-Almagro MV, Alcaín FJ. Coenzyme Q10 protects human endothelial cells from β-amyloid uptake and oxidative stress-induced injury. PLoS One. 2014;9:e109223. doi: 10.1371/journal.pone.0109223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Chen G, Ma W, Li PA. Water-soluble coenzyme Q10 inhibits nuclear translocation of apoptosis inducing factor and cell death caused by mitochondrial complex I inhibititon. Int J Mol Sci. 2014;15:13388–13400. doi: 10.3390/ijms150813388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Naderi J, Somayajulu-Nitu M, Mukerji A, Sharda P, Sikorska M, Borowy-Borowski H, Antonsson B, Pandey S. Water soluble formulations of Coenzyme q10 inhibits bax-induced destabilization of mitochondria in mammalian cells. Apoptosis. 2006;11:1359–69. doi: 10.1007/s10495-006-8417-4. [DOI] [PubMed] [Google Scholar]