

Abstract
Mutations of the gene GNAS have been shown to activate the adenylate cyclase gene and lead to constitutive cAMP signaling. Several preliminary reports have suggested a role for GNAS gene mutations during colorectal carcinogenesis, particularly mucinous carcinomas. The aim of this study was to clarify the incidence of GNAS mutations in adenomas (tubular, tubulovillous, and villous), carcinomas with residual adenoma, and carcinomas, and to relate these findings to mutations of the KRAS gene and to the mucinous status of the tumors. We used standard PCR techniques and direct gene sequencing to evaluate tumors for gene mutations. No GNAS mutations were identified in 25 tubular adenomas, but were present in 6.4% of tubulovillous adenomas and 11.2% of villous adenomas. A GNAS mutation was found in 9.7% of the benign portion of carcinoma with residual adenoma, but in none of 86 carcinomas. A similar trend was seen for KRAS mutation across the five groups of tumors. GNAS mutations may function as an important driver mutation during certain phases of colorectal carcinogenesis, but may then be lost once the biological advantage gained by the mutated gene is no longer necessary to sustain or advance tumor development.
Keywords: GNAS gene mutations, colon adenomas, colon carcinoma, colon carcinogenesis, mucinous carcinomas
Introduction
Systematic analyses of variants in cancer have revealed numerous potential candidate cancer genes. A non-synonymous base change of the gene (GNAS) that encodes for the stimulatory G-protein alpha subunit is one candidate gene. [1]. The protein acts as a ubiquitously expressed signal transducer that transmits hormonal and growth factor signals to effector proteins; particularly, activation of the membrane-associated enzyme adenylate cyclase [2]. Mutations occurring at codon 201 of GNAS activate the adenylate cyclase gene and lead to constitutive cAMP signaling [3].
In vitro techniques employing cell lines have suggested that mutant GNAS may play a direct role in mucin production [4,5]. GNAS mutations are found frequently in several tumor types in which mucin production is prominent, such as appendiceal mucinous neoplasms [6,7] and intraductal papillary neoplasms of the pancreas [8] and of the bile ducts [9]. Further, GNAS mutations were reported to be frequent in villous adenomas [10].
The aim of this study was to clarify the incidence of GNAS mutations in a large group of tubular, tubulovillous, and villous adenomas, carcinomas with residual adenoma, and carcinomas of the colorectum, and to relate these findings to mutations of the KRAS gene and to the mucinous status of the tumors.
Materials and methods
The current study utilized samples collected for a prior study. The computerized records of the hospital Pathology Department were used to identify adenomas and carcinomas. All adenomas were from patients known to be free of both colorectal cancer and multiple adenomas. The tubular adenomas represented ones we recently studied. The other tumors were previously evaluated for KRAS mutation [11]. All tubulovillous and villous adenomas from that study are included for this study, while the adenocarcinomas with residual adenomas represented 84% of those previously studied. The carcinomas were selected to include all 31 mucinous carcinomas previously studied, plus a random sampling of the previously studied non-mucinous carcinomas.
Our previous study was approved by the hospital Institutional Review Board, and the current study was independently approved with a waiver of HIPAA privacy authorization for anonymized tissue analysis. All samples were archived material from our Department of Pathology. Clinical material primarily reflected a suburban community of middle economic level, with substantial representations from various minority groups (Asian, African-American) of both middle and low economic status. One clinical pathologist reviewed all histological slides and indicated the areas for molecular study, including an area of normal tissue paired with each tumor. Histological slides stained with hematoxylin and eosin (H&E) and stored DNA samples were available for all cases. Criteria for differentiation of adenomas followed the World Health Organization criteria with respect to villous component: tubular adenomas, <20%; tubulovillous adenomas, 20-80%; and villous adenomas, >80%. Mucinous carcinoma was diagnosed based upon the World Health Organization definition: in mucinous adenocarcinomas the presence of pools of extracellular mucin covers more than 50% of the tumor [12]. Right side colonic segments were defined as cecum, ascending, hepatic flexure, transverse; and the left side was considered the splenic flexure, descending, sigmoid and rectum. All authors had access to the study data and had reviewed and approved the final manuscript.
We defined carcinomas with residual adenoma as tumors in which both a benign component and an invasive malignant component were present within the surgically removed specimen. We focused our study of carcinomas with residual adenoma to those with villous or tubulovillous benign areas. We excluded tumors with only tubular benign areas, as no tubular adenomas had demonstrated a GNAS mutation. The percentage of the tumor that was benign compared to malignant varied from 20% to 80%, and tumors were included as long as isolated areas of both components were evaluable. All carcinomas were studied prior to the administration of any radiation or chemotherapy. The pathological assessment did not provide information as to the clinical stage of the cancers, and reported stages are pathological stage only. Reference to all histological and molecular analyses was by coded numbers.
DNA extraction and purification
All tissue specimens were formalin-fixed and paraffin-embedded. Histological slides stained with H&E were examined and the area of relevant tissue was identified and marked, as was an area of normal tissue. Paraffin blocks were available for all cases. For cases of carcinoma with residual adenoma, different slides (and therefore different paraffin blocks) were used for studying the benign and malignant portions. Consecutive unstained slides from the blocks were prepared and the corresponding areas were isolated under a dissecting microscope by manual dissection. Neoplastic cells were carefully curetted, with the contamination from non-neoplastic cells (stoma, vascular) estimated at 30-50% by histological examination. All curetted samples contained an estimated 2000 to 10,000 epithelial cells. The paraffin wax was removed by xylene and ethanol washes. Cellular material was lysed in a proteinase K buffer solution. DNA was isolated and purified using the QIAamp DNA Mini Kit (Qiagen Inc., Valencia, CA). DNA concentration was determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE).
Sequence analysis of the GNAS and KRAS genes
The codon 201 region in exon 8 of the GNAS oncogene was amplified using the primer set 5’-ACTGTTTCGGTTGGCTTTGGTGA-3’ (forward) and 5’-AGGGACTGGGGTGAATGTCAAGA-3’ (reverse). The codon 12/13 region in exon 2 of the KRAS gene was amplified using the primer set 5’-AAGGCCTGCTGAAAATGACTG-3’ (forward) and 5’-GGTCCTGCACCAGTAATATGCA-3’ (reverse).
PCR was performed in 50 µl volumes with AmpliTaq Gold polymerase and ABI reagents (Applied Biosystems, Foster City, CA) using 100 ng of template DNA, 50 pmols of primer, 2.0 mM MgCl2, and 200 µM each dNTP on a GeneAmp PCR System 9700 (Applied Biosystems, Foster City, CA). PCR consisted of an initial 8 minute denaturation at 94°C, followed by 40 total cycles of a 30 second denaturation at 94°C, a 30 second annealing, and a one minute elongation at 72°C, with a final 30 minute extension at 72°C. The annealing temperature was stepped down at 62°C, 60°C, and 58°C for 5, 15, and 20 cycles, respectively. The post-PCR products were purified using the QIAquick PCR Purification Kit (Qiagen Inc., Valencia, CA) prior to sequencing. The sequencing reactions were performed in 20 µl volumes using 0.25X BigDye Terminator Cycle Sequencing Reagents (Applied Biosystems, Foster City, CA), 5.0 pmol of the reverse GNAS primer, and 1.0 µl of the purified PCR reaction. Reactions were run on a GeneAmp PCR System 9700 (Applied Biosystems, Foster City, CA) for 25 cycles using a two-minute extension time. The sequencing reaction fragments were cleaned using isopropanol precipitation. Sequencing products were separated by capillary electrophoresis with an ABI 3130 Genetic Analyzer and the data were processed with Sequencing Analysis v5.2 (Applied Biosystems, Foster City, CA) software. This method can detect as low as 10% mutant cells within a background of wild type cells.
Statistical methods
The Chi square method was used to compare clinical characteristics across the five tumor groups. Analysis of variance was used to compare the age distribution across groups. Fisher’s Exact test (2-sided) was used to compare gene mutations across the groups.
Results
All analyses were performed on stored DNA from previously studied colorectal tumors. These included: 1. tubular adenomas, 2. tubulovillous adenomas, 3. villous adenomas, 4. carcinomas with residual adenoma, and 5. carcinomas (without residual adenoma). Carcinomas with residual adenoma were resected specimens containing histologically confirmed areas of an adenoma as well as invasive cancer. The patient’s gender, age and tumor location are shown in Table 1. The mean age for each group of tumors is different (p = 0.01), with the mean age of patients with cancer the oldest. Both genders are equally represented across the five tumor groups (p = 0.29). Right-sided tumors are more frequent for the carcinoma with residual adenoma group than the other four tumor groups (p = 0.02). Although each group of tumors was specifically selected, the age ranges are broadly consistent with established findings. Our samples for villous adenomas and cancer contained more females than males and more right than left-sided tumors, reflecting perhaps specific inclusion of mucinous cancers.
Table 1.
Clinical characteristics of colorectal tumors assayed for GNAS mutation
| Tubular adenoma | Tubulovillous adenoma | Villous adenoma | Carcinoma with adenoma | Carcinoma | P value | |
|---|---|---|---|---|---|---|
|
|
||||||
| No. (%) | No. (%)* | No. (%)* | No. (%) | No. (%) | ||
| No tumors | 25 | 31 | 98 | 62 | 86 | |
| Gender | ||||||
| Male | 16 (64) | 15 (52) | 41 (43) | 25 (40) | 38 (44) | 0.29 |
| Female | 9 (36) | 14 (48) | 55 (57) | 37 (60) | 48 (56) | |
| Age | 62.1 | 64.0 | 67.6 | 67.7 | 70.6 | 0.01 |
| Age range | 44-78 | 48-82 | 40-92 | 36-88 | 24-95 | |
| Location | ||||||
| Right | 14 (56) | 12 (39) | 54 (55) | 45 (73) | 54 (63) | 0.02 |
| Left | 11 (44) | 19 (61) | 44 (45) | 17 (27) | 32 (37) | |
Two people had two adenomas each.
A total of 25 tubular adenomas were studied. No GNAS mutations were detected in any of the tubular adenomas, although several were found to have a KRAS mutation (Table 2). Two of the 31 (6%) tubulovillous adenomas demonstrated a GNAS mutation (Figure 1), with 22 of the 31 (71%) tubulovillous adenomas harboring a KRAS mutation. Among villous adenomas, 11 of 98 (11%) demonstrated a GNAS mutation. A total of 60 (61%) of the villous adenomas contained a KRAS mutation. Ten villous adenomas with a GNAS mutation contained abundant intracellular mucin, involving over 50% of the tumor. Mucin status was not available for one villous adenoma. Of the 11 villous adenomas with a GNAS mutation, 7 (64%) also demonstrated a KRAS mutation, while 4 demonstrated wild type KRAS. Five of the villous adenomas with a GNAS mutation were found in the right colon and 6 were from the left colon.
Table 2.
GNAS and KRAS findings in colorectal tumors
| Tubular adenoma | Tubulovillous adenoma | Villous adenoma | Carcinoma with residual adenoma | Carcinom | P value | ||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Benign | Malignant | ||||||
|
|
|||||||
| No. (%) | No. (%) | No. (%) | No. (%) | No. (%) | No. (%) | ||
| No. | 25 | 31 | 98 | 62 | 86 | ||
| GNAS | |||||||
| Mutated | 0 (0) | 2 (6.4) | 11+ (11.2) | 6+ (9.7) | 3 (4.8) | 0 (0) | 0.53 |
| Wild | 25 (100) | 29 (93.6) | 87 (88.8) | 56 (90.3) | 59 (95.2) | 86 (100) | |
| GNAS Mutation type | |||||||
| R201H | 2 | 7 | 5 | 2 | |||
| R201C | 5 | 1 | 1 | ||||
| R201S | 1 | ||||||
| KRAS | |||||||
| Mutated | 4 (16) | 22 (71) | 60 (61) | 33 (53) | 39 (63) | 27 (31) | <0.0001 |
| Wild | 21 (84) | 9 (29) | 38 (39) | 29 (47) | 23 (37) | 59 (69) | |
One tumor had two different mutations.
Figure 1.
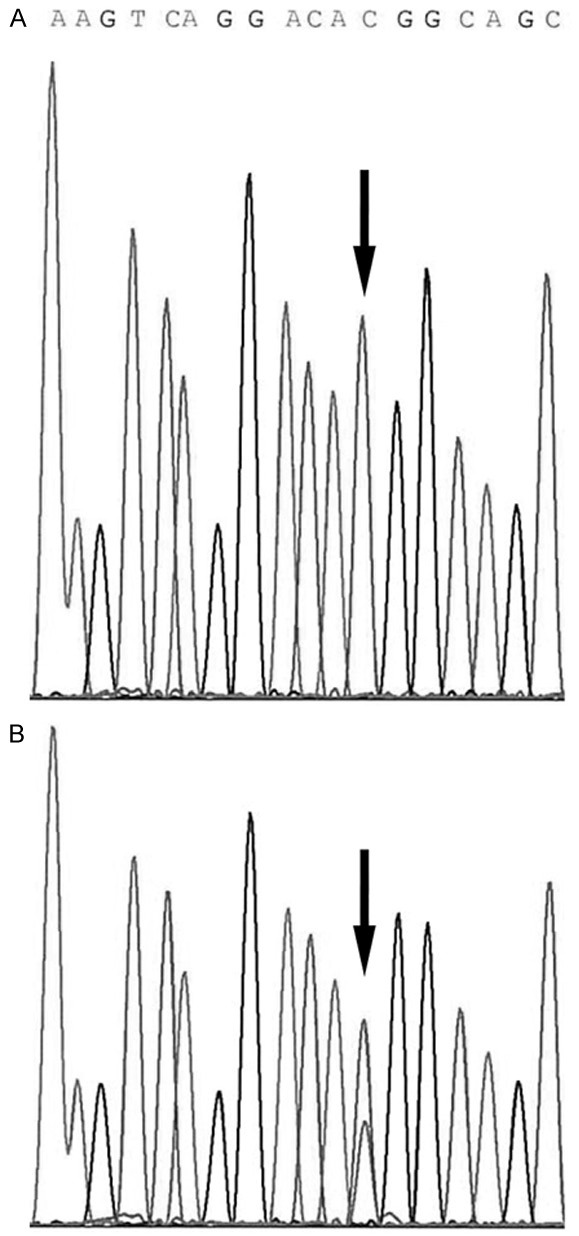
DNA anti-sense sequence analysis of GNAS from codon 204 through 199. A. DNA from normal tissue. The black arrow indicates the wild-type pattern showing a single peak at the 602 nucleotide position of codon 201. B. DNA from a villous adenoma. The arrow indicates a heterozygous mutant peak under the wild-type peak.
The pathological stage of the 62 carcinomas with residual adenoma were: stage 0 = 4, stage 1 = 22, stage 2 = 9, stage 3 = 19, stage 4 = 4 and unknown stage = 4. A total of 21 of these 62 tumors (34%) contained mucin within the cancerous part of the tumor, with 9 tumors having over 50% mucin and 12 tumors with 30-50% mucin. Among these 62 carcinomas with residual adenoma, 6 (10%) demonstrated a GNAS mutation in the benign part of the tumor, with a GNAS mutation in just two of the six associated malignant portions (Table 3). Of these six tumors, three demonstrated a KRAS mutation in the benign portions. Of the four tumors with GNAS mutation in the benign part but GNAS wild type in the cancerous part, there was extensive cellular material in two of the cancer sections studied, while two cancer samples had more limited cellular material associated with extensive mucin; however, both of these two samples contained sufficient cellular DNA to demonstrate a KRAS mutation.
Table 3.
Carcinomas with residual adenoma showing a GNAS mutation in one or both portions, with KRAS mutation status and mucin status
| Tumor # | Adenomatous part | Carcinomatous part | |||
|---|---|---|---|---|---|
|
|
|||||
| GNAS mut | KRAS | GNAS mut | Mucin | KRAS | |
| 1 | R201C | Mutated | R201C | <50%* | Mutated |
| 2 | R201H | Wild | WILD | <50% | Wild |
| 3 | R201H | Wild | WILD | <50% | Mutated |
| 4 | R201H/R201S | Wild | R201H | <50% | Mutated |
| 5 | R201H | Mutated | WILD | <50% | Mutated |
| 6 | R201H | Mutated | WILD | <50% | Mutated |
| 7 | WILD | Mutated | R201H | >50% | Mutated |
Carcinomas with >50% mucin meet WHO criteria for mucinous adenocarcinoma.
Four of the six carcinomas with residual adenoma showing a GNAS mutation were from the right colon and 2 were from the left colon. The benign portions of these six tumors demonstrated between 30%-50% intracellular mucin, and a similar degree of mucin was present in the cancerous part of five of the six tumors. The benign portion of three of the six carcinomas with residual adenoma was villous and was tubulovillous in the other three tumors. One carcinoma with residual adenoma demonstrated no GNAS mutation in the benign portion but was mutated in the malignant portion. This tumor contained >50% mucin in the cancerous portion (Figure 2). Overall, a KRAS mutation was found in 63% of the benign portions of the carcinomas with residual adenoma and in 53% of the malignant portions (Table 2).
Figure 2.
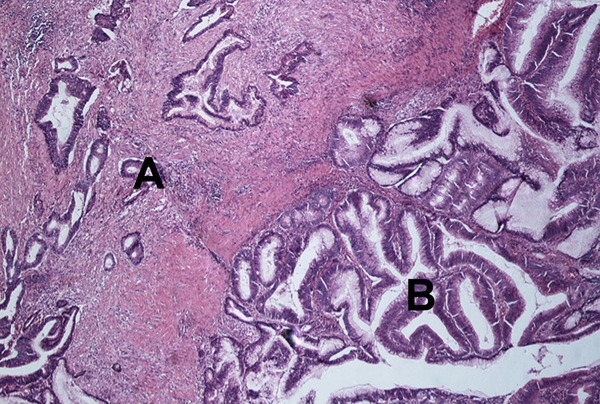
Hematoxylin and eosin stain of a carcinoma with residual adenoma. Area A indicates invasive carcinoma. Area B is part of the residual tubulovillous adenoma. 400X light microscope.
The 86 colorectal carcinomas studied included 15 with greater than 50% mucin, 16 with between 30-50% mucin, and 55 lacking significant mucin. The stages of the cancers were: stage 1 = 21, stage 2 = 21, stage 3 = 40, stage 4 = 3, and unknown stage = 1. All colorectal carcinomas, regardless of mucin status or stage, demonstrated wild type GNAS. A total of 27 (31%) of the carcinomas demonstrated a KRAS mutation (Table 2).
Discussion
Colorectal mucinous adenocarcinoma is a subtype of colorectal carcinoma that contains prominent mucin production. The frequency of mucinous carcinoma varies, but is approximately 10% of colorectal cancers; the clinical significance of mucinous carcinoma is controversial [13,14]. The suggestion that mucinous colorectal carcinomas develop through a genetic pathway that is different from that of non-mucinous carcinomas is supported by the identification of gene mutations specific to tumors producing mucus [15], as well as more generalized genetic differences [16]. Since GNAS mutations are found frequently in other mucinous tumors, it is reasonable to consider the possibility that a mutation in the gene GNAS is present in mucinous colorectal cancer.
Several published studies suggest a role generally for GNAS gene mutations in the process of colorectal carcinogenesis. One study using transgenic Apc(Min/+) mice reported that GNAS mutations promoted the formation of intestinal adenomas through augmentation of the Wnt and ERK1/2 MAPK pathway in the intestinal epithelium [17]. A study of human tumors re-ported a GNAS mutation in 20 of 24 villous adenomas [10].
Another study of 12 carcinomas with residual adenoma found GNAS mutation in both the benign and malignant portions in 3 tumors, and in just the benign portion for 6 additional tumors [18]. However, other studies have found GNAS mutations to be uncommon in human colorectal cancer, occurring in just 2% in one study [10], and in just 0.47% of a larger study of advanced colorectal cancer [19]. A recent paper reported a GNAS mutation in 6 of 311 (1.9%) colorectal cancers. Five of these 6 cancers had a mucinous phenotype. The authors then selected an additional 19 mucinous colon carcinomas, and found a GNAS mutation in 4 (21%) [20].
We did not detect GNAS mutations in villous adenomas with the frequency (83%) reported by Yamada [10]. However, our sample size is four times larger, and there are differences in the source of material and ethnicity between our two populations. A recent paper reported a GNAS mutation in 0 of 17 tubular adenomas, 3 of 20 (15%) tubulovillous adenomas, and 6 of 13 (46%) villous adenomas from United States patients [21], results that parallel our findings. The authors further report a GNAS mutation in just 10 of 428 (2.3%) colorectal carcinomas, and 7 of 8 of the carcinomas with a GNAS mutation available for histological review revealed a prominent villous morphology. Furthermore, five of these 8 tumors arose in a contiguous villous adenoma [21], similar to our findings.
All 31 of the carcinomas with >30% mucin we studied were GNAS wild type. Thus, our findings do not specifically correlate with the mucinous status of the colorectal tumors. Rather, our data indicate a specific phase of colorectal carcinogenesis during which GNAS may be mutated within the evolving tumor. This phase correlates with the villous features of colorectal tumors. GNAS mutations appear within a few tubulovillous adenomas and are also found within villous adenomas and the villous or tubulovillous parts of carcinomas with residual adenoma. The frequency of mutation then lessens within the carcinomatous portion of the carcinomas with residual adenoma, as the benign adenoma is progressively replaced by carcinoma, and the mutation is not detected in the carcinomas lacking residual adenomatous tissue.
A similar increase and then decrease in the frequency of a particular gene mutation during colorectal carcinogenesis is also noted for our KRAS data, and as we reported in more detail previously [11]. Our data and the literature data summarized above clearly demonstrate that GNAS mutation is not detected in tubular adenomas. However, GNAS mutation is found in tubulovillous and villous adenomas, but then less frequently in carcinomas. When a GNAS mutation is detected in carcinomas, it is most likely found in those cancers with residual adenoma, or villous or mucinous histology.
It is possible that a GNAS mutation is just one of several possible pathways contributing to excess mucin production in colorectal tumors with villous features.
It is not clear what accounts for the fall-off in the frequency of GNAS mutation with advancing tumor type. However, a similar finding has been reported for mutations in the APC gene in gastric neoplasms [22]. In this study, the authors found a higher APC mutation rate for gastric adenomas (76%) than for gastric adenocarcinomas (4%).
One possible explanation for our findings is that GNAS mutations (and KRAS mutations) may function as important driver mutations during a certain phase of colorectal carcinogenesis, but then may be lost once the biological advantage gained by the mutated gene is no longer necessary to sustain or advance tumor development, as previously suggested [10].
This theory postulates that as an adenoma, the GNAS mutation confers a selective growth advantage, and it is a “driver” mutation. However, if this mutation were of no growth advantage in the later stages of carcinogenesis, then the development of additional random changes could result in the loss of the allele with the GNAS mutation.
Indeed, it has been suggested that the selective advantage of any given driver mutation is low, 0.4% [23], and that there is a resulting high “extinction rate” for driver mutations, as high as 99% by one estimate [24].
Another possible explanation for the observed data is that adenomas with a GNAS mutation fail to become cancerous more frequently than those without a GNAS mutation. It is known from cross sectional data that not all adenomas are destined to advance to malignancy [25]. However, the decrease in frequency of GNAS mutation from adenoma to carcinoma is quite large, and would appear to exceed the difference between the incidence of adenoma and carcinoma detection. Furthermore, it is unlikely that very many of carcinomas with residual adenomatous tissue containing a GNAS mutation would remain both asymptomatic and also fail to progress pathologically.
On the cellular level, it is possible that adenomas contain two populations of tumor cells, one with a GNAS mutation and one that is GNAS wild type. For example, studies have shown that not all areas of a colorectal tumor necessarily contain identical KRAS findings [26]. Growth of the tumor beyond a certain point might involve loss of the GNAS mutated cells and continued growth of the cells with wild-type GNAS. Indeed, a study of melanomas reported that malignant cells with BRAF gene mutations do not outgrow cells with wild-type BRAF, and the authors suggested that the cells with BRAF mutation undergo senescence [27]. We have no data on variations in the GNAS findings across individual colon tumors, but it is an infrequent finding for KRAS, and it is unlikely that two competing clones, one with a GNAS mutation and one with wild type, would explain the large difference in GNAS findings between adenomas and carcinomas.
In conclusion, our genetic epidemiological data on GNAS mutation during colorectal carcinogenesis mirrors our previous findings for KRAS gene mutation, with the gene mutation present within specific histological stages of tumorigeneis; but with further progression into a carcinoma, the mutation is lost. Thus, a given mutation, once acquired, need not persist indefinitely within the evolving tumor, even a key driver mutation.
Acknowledgements
The authors thank Dr. Errol Berman for review of histological slides and The Harvey Nussbaum Foundation of Saint Barnabas Medical Center and the June Bleiwise Foundation for financial support.
References
- 1.Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JK, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PV, Ballinger DG, Sparks AB, Hartigan J, Smith DR, Suh E, Papadopoulos N, Buckhaults P, Markowitz SD, Parmigiani G, Kinzler KW, Velculescu VE, Vogelstein B. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–13. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 2.Weinstein LS, Liu J, Sakamoto A, Xie T, Chen M. Minireview: GNAS: normal and abnormal functions. Endocrinology. 2004;145:5459–64. doi: 10.1210/en.2004-0865. [DOI] [PubMed] [Google Scholar]
- 3.Landis CA, Masters SB, Spada A, Pace AM, Bourne HR, Vallar L. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature. 1989;340:692–6. doi: 10.1038/340692a0. [DOI] [PubMed] [Google Scholar]
- 4.Nishikawa G, Sekine S, Ogawa R, Matsubara A, Mori T, Taniguchi H, Kushima R, Hiraoka N, Tsuta K, Tsuda H, Kanai Y. Frequent GNAS mutations in low-grade appendiceal mucinous neoplasms. Br J Cancer. 2013;108:951–8. doi: 10.1038/bjc.2013.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laburthe M, Augeron C, Rouyer-Fessard C, Roumagnac I, Maoret JJ, Grasset E, Laboisse C. Functional VIP receptors in the human mucus-secreting colonic epithelial cell line CL. 16E. Am J Physiol. 1989;256:G443–50. doi: 10.1152/ajpgi.1989.256.3.G443. [DOI] [PubMed] [Google Scholar]
- 6.Zauber P, Marotta S, Sabbath-Solitare M. GNAS mutations are associated with mucin production in low-grade appendiceal mucinous neoplasms, villous adenomas, and carcinomas. Human Pathol. 2015;46:339. doi: 10.1016/j.humpath.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 7.Singhi AD, Davidson JM, Choudry HA, Pingpank JF, Ahrendt SA, Holtzman MP, Zureikat AH, Zeh HJ, Ramalingam L, Mantha G, Nikiforova M, Barlett DL, Pai RK. GNAS is frequently mutated in both low-grade and high-grade disseminated appendiceal mucinous neoplasms but does not affect survival. Human Pathol. 2014;45:1737–43. doi: 10.1016/j.humpath.2014.04.018. [DOI] [PubMed] [Google Scholar]
- 8.Amato E, dal Molin M, Mafficini A, Yu J, Malleo G, Rusev B, Fassan M, Antonello D, Sadakari Y, Castelli P, Zamboni G, Maitra A, Salvia R, Hruban RH, Bassi C, Capelli P, Lawlor RT, Goggins M, Scarpa A. Targeted next-generation sequencing of cancer genes dissects the molecular profiles of intraductal papillary neoplasms of the pancreas. J Pathol. 2014;233:217–27. doi: 10.1002/path.4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsai JH, Yuan RH, Chen YL, Liau JY, Jeng YM. GNAS is frequently mutated in a specific subgroup of intraductal papillary neoplasms of the bile duct. Am J Surg Pathol. 2013;37:1862–70. doi: 10.1097/PAS.0b013e3182986bb5. [DOI] [PubMed] [Google Scholar]
- 10.Yamada M, Sekine S, Ogawa R, Taniguchi H, Kushima R, Tsuda H, Kanai Y. Frequent activating GNAS mutations in villous adenoma of the colorectum. J Pathol. 2012;228:113–8. doi: 10.1002/path.4012. [DOI] [PubMed] [Google Scholar]
- 11.Zauber P, Marotta S, Sabbath-Solitare M. KRAS gene mutations are more common in colorectal villous adenomas and in situ carcinomas than in carcinomas. Int J Mol Epidemiol Genet. 2013;4:1–10. [PMC free article] [PubMed] [Google Scholar]
- 12.Hamilton SR, Bosman FT, Boffetta P, Ilyas M, Morreau H, Nakamura SI, Quirke P, Riboli E, Sobin LH. Carcinoma of the colon and rectum. In: Bosmann FT, Carneiro F, Hruban RH, Theise ND, editors. WHO classification of tumours of the digestive system. Lyon: IARC Press; 2010. pp. 134–46. [Google Scholar]
- 13.Mekenkamp LJ, Heesterbeek KJ, Koopman M, Tol J, Teerenstra S, Venderbosch S, Punt CJ, Nagtegaal ID. Mucinous adenocarcinomas: poor prognosis in metastatic colorectal cancer. Eur J Cancer. 2012;48:501–9. doi: 10.1016/j.ejca.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 14.Hogan J, Burke JP, Samaha G, Condon E, Waldron D, Faul P, Coffey JC. Overall survival is improved in mucinous adenocarcinoma of the colon. Int J Colorectal Dis. 2014;29:563–9. doi: 10.1007/s00384-013-1826-2. [DOI] [PubMed] [Google Scholar]
- 15.Pastrello C, Santarosa M, Fornasarig M, Sigon R, Perin T, Giannini G, Boiocchi M, Viel A. MUC gene abnormalities in sporadic and hereditary mucinous colon cancers with microsatellite instability. Dis Markers. 2005;21:121–6. doi: 10.1155/2005/370908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Melis M, Ly Q, Nair R, Siegel E, McLoughlin J, Lewis J, Jensen E, Alvarado M, Eschrich S, Bloom G, Yeatman T, Shibata D. Colorectal mucinous adenocarcinomas are characterized by markers of differentiation and components of the mucin-producing machinery. Dis Colon Rectum. 2007;50:713–4. [Google Scholar]
- 17.Wilson CH, McIntyre RE, Arends MJ, Adams DJ. The activating mutation R201C in GNAS promotes intestinal tumourigenesis in Apc(Min/+) mice through activation of Wnt and ERK1/2 MAPK pathways. Oncogene. 2010;29:4567–75. doi: 10.1038/onc.2010.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sekine S, Ogawa R, Oshiro T, Kanemitsu Y, Taniguchi H, Kushima R, Kanai Y. Frequent lack of GNAS mutations in colorectal adenocarcinoma associated with GNAS-mutated villous adenoma. Genes, Chromosomes Cancer. 2014;53:366–72. doi: 10.1002/gcc.22147. [DOI] [PubMed] [Google Scholar]
- 19.Idziaszcyk S, Wilson CH, Smith CG, Adams DJ, Cheadle JP. Analysis of the frequency of GNAS codon 201 mutations in advanced colorectal cancer. Cancer Genet Cytogenet. 2010;202:67–9. doi: 10.1016/j.cancergencyto.2010.04.023. [DOI] [PubMed] [Google Scholar]
- 20.Stachler MD, Rinehart E, Lindeman N, Odze R, Srivastava A. Novel molecular insights from routine genotyping of colorectal carcinomas. Human Pathol. 2015;46:507–13. doi: 10.1016/j.humpath.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 21.Fecteau RE, Luttervbaugh J, Markowitz SD, Willis J, Guda K. GNAS mutations identify a set of right-sided, RAS mutant, villous colon cancers. PLoS One. 2014;9:e87966. doi: 10.1371/journal.pone.0087966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JH, Abraham SC, Kim HS, Nam JH, Choi C, Lee MC, Park CS, Juhng SW, Rashid A, Hamilton SR, Wu TT. Inverse relationship between APC gene mutation in gastric adenomas and development of adenocarcinoma. Am J Pathol. 2002;161:611–8. doi: 10.1016/S0002-9440(10)64216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bozic I, Antal T, Ohtsuki H, Carter H, Kim D, Chen S, Karchin R, Kinzler KW, Vogelstein B, Nowak MA. Accumulation of driver and passenger mutations during tumor progression. Proc Natl Acad Sci U S A. 2010;107:18545–50. doi: 10.1073/pnas.1010978107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Regoes RR. Population genetics meets cancer genomics. Proc Natl Acad Sci U S A. 2010;107:18241–2. doi: 10.1073/pnas.1013177107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Regula J, Rupinski M, Kraszewska E, Polkowski M, Pachlewski J, Orlowska J, Nowacki MP. Colonoscopy in colorectal cancer screening for detection of advanced neoplasia. N Engl J Med. 2006;355:1863–72. doi: 10.1056/NEJMoa054967. [DOI] [PubMed] [Google Scholar]
- 26.Losi L, Baisse B, Bouzourene H, Benhattar J. Evolution of intratumoral genetic heterogeneity during colorectal cancer progression. Carcinogenesis. 2005;26:916–22. doi: 10.1093/carcin/bgi044. [DOI] [PubMed] [Google Scholar]
- 27.Lin J, Goto Y, Murata H, Sakaizawa K, Uchiyama A, Saida T, Takata M. Polyclonality of BRAF mutations in primary melanoma and the selection of mutant alleles during progression. Brit J Cancer. 2011;104:464–8. doi: 10.1038/sj.bjc.6606072. [DOI] [PMC free article] [PubMed] [Google Scholar]