

Abstract
Hepatitis C virus (HCV) infection is associated with increased thrombotic risk. Several mechanisms are involved including direct endothelial damage by the HCV virus, with activation of tissue factor, altered fibrinolysis and increased platelet aggregation and activation. In advanced stages, chronic HCV infection may evolve to liver cirrhosis, a condition in which alterations in the portal microcirculation may also ultimately lead to thrombin activation, platelet aggregation, and clot formation. Therefore in advanced HCV liver disease there is an increased prevalence of thrombotic phenomena in portal vein radicles. Increased thrombin formation may activate hepatic stellate cells and promote liver fibrosis. In addition, ischemic changes derived from vascular occlusion by microthrombi favor the so called parenchymal extinction, a process that promotes collapse of hepatocytes and the formation of gross fibrous tracts. These reasons may explain why advanced HCV infection may evolve more rapidly to end-stage liver disease than other forms of cirrhosis.
Keywords: Coagulation, Liver cirrhosis, Hepatitis C virus, Fibrogenesis, Parenchymal extinction, Portal thrombosis, Protein C
Core tip: Liver cirrhosis may be considered a prothrombotic condition despite it being associated with a low platelet count and deranged synthesis of clotting factors. When hepatitis C virus (HCV) is the etiological factor of liver cirrhosis, intrahepatic coagulation may be enhanced by several direct actions of HCV on the clotting system, platelet aggregation, and altered anticoagulation. The excessive thrombin generation may be related to increased fibrogenesis both by a direct effect of thrombin on hepatic stellate cells and fibrosis related to ischemic parenchymal extinction.
INTRODUCTION
Liver damage in hepatitis C virus (HCV) infection includes a wide range of clinical entities. Chronic infection may lead to chronic hepatitis, with more or less marked steatosis and/or steatohepatitis, cirrhosis, and hepatocarcinoma. In advanced stages of HCV infection, recent research has pointed out the importance of thrombotic events and microcirculatory changes within the liver. As in other forms of cirrhosis, but probably with a higher prevalence, in HCV-infected patients thrombin formation with accompanying microthrombosis of portal vein radicles takes place, a phenomenon that may accelerate the natural history of the disease. The factors involved in this process, and the clinical relevance of this prothrombotic situation are reviewed in the present work.
HEMOSTATIC SYSTEM
Coagulant and anticoagulant pathways: A schematic overview
Hemostasis, in its wide sense, involves adhesion, aggregation, and activation of platelets (primary hemostasis), and the formation of a fibrin clot as the final product of an autocatalytic process initiated by the damaged endothelium. As shown in Figure 1, thrombin, a protein derived from the action of activated factor X on prothrombin, plays a central role. Thrombin promotes platelet activation and aggregation, and activates factor XI, factor VIII and factor V, creating a positive feed-back loop that underscores the need of a counter-regulatory system. This is composed by antithrombin, tissue factor pathway inhibitor, thrombomodulin, liver synthesized vitamin-K dependent protein C and its endothelial receptor, and protein S. Thrombomodulin is a transmembrane protein located on endothelial cells, in intimate connection with endothelial protein C receptor (Figure 2). Thrombomodulin acts as a thrombin receptor. Once thrombin and protein C bind to thrombomodulin and protein C receptor, protein C becomes quickly activated by thrombin, a process highly dependent on thrombomodulin. Once activated, protein C binds to protein S. This complex inhibits activated factor V and factor VIII, leading to decreased thrombin generation (Figure 2). Thrombin generation is also quenched by the presence of antithrombin, a protease with many actions, also synthesized in the liver, and tissue factor pathway inhibitor (Figure 1), whose levels are not decreased, but even increased in cirrhosis[1]. Antithrombin strongly inhibits thrombin, but also inhibits the activated forms of factors XII, XI, X, IX and VII; plasmin and kallikrein, trypsin and C1[2-4]. Tissue factor pathway inhibitor forms a complex with factor Xa, inactivating it; and also blocking the tissue factor-factor VIIa complex. Like thrombomodulin, tissue factor inhibitor is an endothelial product[5] and, as discussed later, both may become altered in hepatitis C-dependent endothelial damage.
Figure 1.
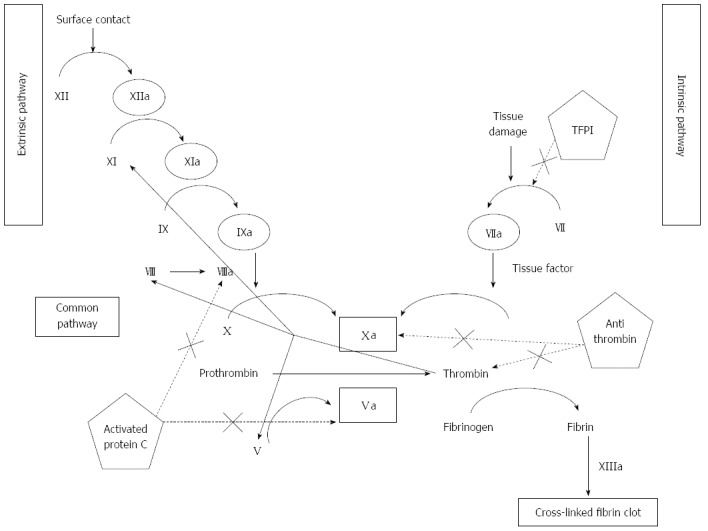
Coagulation cascade: Extrinsic, intrinsic, and common pathways. Coagulation factors are depicted in roman numerals with the suffix “a” denoting activated forms of the factors. Inhibitory pathways are shown in dotted lines. TFPI: Tissue factor pathway inhibitor.
Figure 2.
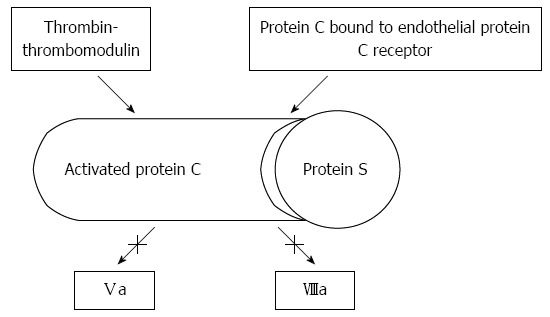
Anticoagulant effect of thrombomodulin, protein C, and protein S.
Platelets constitute another key component of hemostasis. Endothelial disruption exposes subendothelial collagen to the bloodstream, and this is followed by an avid binding of collagen to collagen-specific Ia/IIa platelet surface receptors. Another endothelial-derived multimeric protein, von Willebrand factor, firmly interacts with platelet glycoproteins Ib/IX/V and collagen fibrils. The function of this multimeric protein is controlled by a metalloprotease (ADAMTS 13), synthesized by several cells, including hepatic stellate cells and endothelial cells, among others[6]. Binding of collagen to glycoprotein VI receptor activate the platelets - a process also facilitated by thrombin which activates platelets by binding to other types of receptors, namely specific G protein receptors. Activated platelets release several products contained in their granules, including potent proinflammatory mediators, and finally aggregate and form a hemostatic plug. Aggregated platelets efficiently anchor fibrin, forming a plug that stops bleeding. Platelet membrane phospholipids bind to the gamma carboxy residues of activated factors IX and X, serving as a “platform” on which coagulation takes place. Calcium mediates this binding.
In order to avoid excessive thrombus formation, fibrinolysis is activated. This is also a tightly regulated process, by which an inactive protein - plasminogen - transforms into an active one - plasmin, which is able to destroy formed fibrin. A series of substances, such as tissue plasminogen activator, factor XIIa, and urokinase plasminogen activator transform plasminogen into plasmin, whereas several others block this effect. Indeed, plasminogen activator inhibitor, thrombin activatable fibrinolysis inhibitor (TAFI)[7], plasmin inhibitor, and histidine rich glycoprotein are all potent antifibrinolytic agents.
Thrombosis in HCV infection
There is considerable evidence that the hepatitis C virus is able to activate hemostasis through several mechanisms, one of the main processes being HCV-induced endothelial damage and/or activation. Several clinical observations that are outlined below support this evidence (Figure 3).
Figure 3.
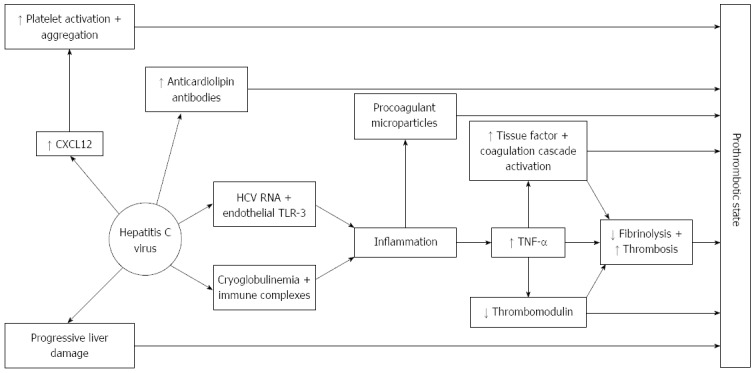
Hepatitis C virus has several effects on anticoagulant and procoagulant cascades, in addition to the effects on coagulation derived from liver cirrhosis development. (1) Hepatitis C virus (HCV) infection is associated with anticardiolipin antibodies which are related to thrombotic events; (2) Viral HCV RNA binds to toll-like receptors (TLR)-3 found in endothelial cells which leads to inflammation. Also, HCV infection is associated with cryoglobulinemia and thus immune complexes directed against viral RNA are formed. Inflammation generated by these two mechanisms lead to TNF-α secretion. TNF-α is an inducer of tissue factor expression, therefore exerting a prothrombotic effect by activating the coagulation cascade and also downregulates thrombomodulin expression; and (3) In HCV infection, CXCL12 is up-regulated in the endothelium of blood vessels formed in active inflammatory foci. CXCL12 is a potent promoter of platelet aggregation and adhesion.
In 1996, Prieto et al[8] reported an increased prevalence of anticardiolipin antibodies among HCV patients. This increased prevalence was related to prior thrombotic episodes, portal hypertension and thrombocytopenia. In a study on 201 patients, 124 of them with HCV infection, Biron et al[9] found a 33% prevalence of antiphospholipid antibodies that were significantly associated with increased liver fibrosis assessed by METAVIR fibrosis score. A relationship between HCV infection and prothrombotic state was also evidenced by Enger et al[10] (2014), who found that HCV infection was associated with various kinds of thromboembolic diseases in a cohort of 22733 HCV infected individuals in the United States. The incidence rate ratio among non-cirrhotic HCV infected patients was 1.44 (95%CI: 1.31-1.58) for any thrombotic event, and reached 6.06 (95%CI: 2.04-18.01) for portal vein thrombosis. Wang et al[11] compared 3686 HCV-infected patients with 14744 subjects without HCV or HBV infection and followed them during more than 5 years. They found that HCV infection was associated with a hazard ratio of deep vein thrombosis of 1.96 (95%CI: 1.03-3.73). Chen et al[12] in 2013 found a higher prevalence of pulmonary hypertension among HCV patients, portal vein thrombosis (a feature commonly present in their series of HCV patients) being an independent factor. However, other authors have failed to find a relationship between antiphospholpid antibodies and thrombotic episodes in HCV patients[13,14], although antiphospholipid antibodies were more frequently observed among HCV patients. Already in 1995, Violi et al[15] performed a study on 18 patients with liver cirrhosis and 36 controls and found that venous thrombosis was associated with HCV infection, positive antiphospholipid antibodies, and increased rate of thrombin generation.
Main mechanisms involved
The mechanisms that may contribute to thrombosis in HCV infection include: systemic inflammation due to viral infection, direct infection of endothelial cells, viral-induced down regulation of physiological anticoagulant mechanisms, and alterations of fibrinolysis and thrombin generation dependent upon tissue factor generation by the infected endothelium[16]. The generation of antiphospholipid antibodies associated with HCV infection may also favor the generation of a procoagulant milieu[8], and immune complexes, usually in relation to cryoglobulinemia, may also trigger thrombotic phenomena[17].
Direct effect of HCV on anticoagulant and procoagulant pathways
Activation of tissue factor is of primary importance in the initiation of the coagulation cascade; and, as previously mentioned, damaged endothelium is the main source of tissue factor. Endothelial damage takes place in HCV infected patients, mainly by two main mechanisms. Firstly, today it is well known that HCV viral RNA binds to toll-like receptor (TLR)-3 in endothelial cells leading to inflammation[18] and generating enhanced expression of both tumor necrosis factor (TNF)-α and TNF receptor 2. In advanced stages of the disease, endothelial cells also express the chemokine CXCL 12 that recruits immune cells[19]. Additionally, endothelial damage associated with cryoglobulinemia is due to a type 3 hypersensitivity reaction with formation of immune complexes of antibodies directed against viral RNA. These immune complexes activate endothelial cells[20]. Inflammation generated by either of these mechanisms is accompanied by TNF-α secretion. TNF-α is an inducer of tissue factor expression, therefore exerting a prothrombotic effect[21] and also downregulates thrombomodulin expression[22]. Cytokines decrease fibrinolytic properties of the endothelial cells[23]. In addition, tissue factor is present in increased amounts in microparticles in patients with chronic pure HCV infection[24], contributing in this way to enhanced coagulation.
HCV infection and platelets
Hemostatic platelet function is also enhanced. In patients affected by HCV infection, CXCL12 is up-regulated in the endothelium of blood vessels formed in active inflammatory foci. It is elevated in the plasma of patients with marked fibrosis and avidly binds to CXCR4 overexpressed by liver infiltrating lymphocytes[19]. CXCL12 is a potent promoter of platelet aggregation and adhesion[25], and, indeed, increased platelet activation and aggregation have been described among HCV patients[26,27].
Perhaps this increased platelet aggregation may explain some striking features described in HCV patients regarding platelet count. Thrombocytopenia is more marked in HCV-infected patients than among those affected by other forms of chronic liver disease in a similar stage of severity[28]. The reasons are not well understood[29,30], and there are reports in which thrombocytopenia improves after treatment with interferon alpha[31]. This effect is seen despite the fact that thrombocytopenia is a well known side effect of interferon alpha, which strongly suggests a direct causal relationship between thrombocytopenia and HCV infection. Theoretically, thrombocytopenia could be due to decreased production, but results regarding thrombopoietin levels are disparate. Español et al[32], in 2000, found normal values in 23 HCV patients with chronic hepatitis compared with 43 controls, but there are also reports pointing to decreased thrombopoietin levels in relation with progression of liver disease[33,34], and restoration of megakariopoiesis after successful liver transplantation[35]. These data suggest that liver function is essential for the maintenance of normal thrombopoietin levels, although it has been also reported that thrombopoietin may be degraded in excess by the enlarged spleen[36]. Thrombocytopenia could also be related to hypersplenism, but although splenic sequestration surely plays a role in advanced stages of liver disease, thrombocytopenia is already evident before spleen enlargement ensues. Antiplatelet antibodies have been described in HCV infection[37], but their pathogenetic role in thrombocytopenia is debatable[38]. However, in some studies the severity of the disease is accompanied by a progressive decrease of platelet production and an increase in platelet antibodies. Platelet count was inversely related to liver fibrosis and directly to viral load[39].
In any case, increased coagulation and decreased fibrinolysis, together with enhanced platelet aggregation may underlie the aforementioned well described relationship between HCV infection and venous thrombosis. When liver damage evolves during the natural history of chronic HCV hepatitis, further mechanisms add to those described.
Prothrombotic alterations associated with liver cirrhosis
Complications of liver cirrhosis probably account for most of the hospital admissions of patients with advanced HCV liver disease. As other forms of liver cirrhosis, cirrhosis in patients affected by chronic HCV infection is characterized by progressive fibrous tissue deposition in the liver that separates groups of hepatocytes forming nodules with distorted vascular architecture and variable degrees of necrosis and liver cell regeneration[40].
Two syndromes converge in this disease: liver failure and portal hypertension, leading to a constellation of clinical and laboratory alterations, some of them theoretically associated with increased bleeding risk. Among these, the most outstanding features related to portal hypertension include the development of oesophageal varices, hypertensive gastritis, hemorrhoids, and hypersplenism; secondary thrombocytopenia is usually found in these patients. Liver failure impedes correct synthesis of clotting factors, such as prothrombin or, in later stages, fibrinogen. Therefore, both syndromes may cause bleeding diathesis. However, in stark contrast, thromboembolic events are not unusual in cirrhosis[41,42], with an overall incidence of 0.8% of non portal vein thrombosis in a study on 2074 cirrhotic patients[43]. In that study, although 5 out of the 17 affected patients showed antiphospholipid antibodies, none of them showed mutation of factor V and/or prothrombin, so no other classic prothrombotic abnormality was identified. On the other hand, the incidence of portal vein thrombosis is by far higher among cirrhotics than among the general population, reaching prevalence values of 0.6%-5%, increasing up to 40% among patients with advanced disease, or 10%-25% according to other reports[44]. Local factors, such as venous stasis and portal hypertension-related endothelial dysfunction are clearly involved in this type of venous thrombosis, but it is worth of note that considering all the forms of venous thromboembolic disease together, cirrhotic patients showed an increased risk, that was more marked among patients with cirrhosis categorized as Child class C, despite a more deranged prothrombin activity[45].
Therefore, liver cirrhosis, despite a usually observed low platelet count and deranged synthesis of some clotting factors, may be considered a prothrombotic condition.
Anticoagulant and procoagulant pathways in cirrhosis
Independent of etiology, recent research has pointed out that in liver cirrhosis several alterations predispose to an increased thrombotic risk, especially at the portal vein.
Synthesis of antithrombotic proteins, such as antithrombin, protein C and protein S is more intensely deranged than that of procoagulant proteins. As mentioned earlier, protein C plays a major role in controlling coagulation. In its active form it degrades several coagulation factors, especially factor V, leading to a decrease in thrombin production. Decreased synthesis of protein C by an impaired liver function may favor ongoing thrombin formation[46]. Binding of thrombomodulin to protein C increases the speed of protein C activation. Interestingly, in a previous study we found raised levels of thrombomodulin among cirrhotics, in the face of decreased protein C, protein S and antithrombin[47]. However, thrombomodulin-bound thrombin also shows a prothrombotic effect, since it inhibits fibrinolysis by cleaving TAFI into its active form[48]. Therefore, raised thrombomodulin, via its action on TAFI, can be viewed as another factor potentially involved in the procoagulant milieu of liver cirrhosis.
Thrombin activation may be aggravated in some situations in which anticoagulant pathways are further impaired. Factor V Leiden is a common (2%-15% prevalence among Caucasians) autosomal dominant trait[49]. It carries a single mutation at position 506 that makes it resistant to the degradative action of activated protein C. As a consequence, the action of factor Va on thrombin synthesis increases, leading to a procoagulant state. Indeed, factor V Leiden is associated with an increased risk of portal vein thrombosis both in patients with and without cirrhosis[50]-although there are studies that do not support this finding[51]. In addition, in patients with HCV infection who also bear factor V Leiden polymorphism there is an increased rate of liver fibrous tissue deposition[52], whose underlying mechanisms will be discussed later. Poujol-Robert et al[53], in 2004, reported an increased odds ratio for cirrhosis among patients with HCV infection and factor V Leiden mutation, and Papatheodoridis et al[54] (2003) found that the presence of activated protein C resistance was associated with more intense fibrosis in patients with chronic viral hepatitis. Moreover, factor V Leiden also carries an increased risk of fibrosis in other tissues, as shown by Xu et al[55] (2001) in pulmonary fibrosis that developed in bleomycin-treated mice carrying the factor V Leiden mutation: both homozygous and heterozygous animals showed a nearly 40% increase in hydroxyproline excretion compared to wild-type mice.
Other factors may contribute to this pro-coagulant effect. Persistent or chronic inflammation is a thrombophilic condition, characterized by raised fibrinogen and factor VIII, which are main contributors to this procoagulant milieu. Cirrhotics show raised levels of factor VIII[56]. Also, cirrhotics have raised von Willebrand factor, which may favor a greater platelet adhesion[57]. Lipoprotein receptor-related protein is responsible for catabolism of factor VIII. Its expression is decreased in cirrhotics[58]. In a similar fashion, ADAMTS-13, a metalloprotease involved in the catabolism of von Willebrand factor, is reduced in patients with liver cirrhosis[59]. Increased fibrinolysis related to decreased PAI-1 levels in relation to t-PA were also reported in cirrhotics[60], and a parallel deficiency in other mediators, such as TAFI, probably contributes[61]. It is currently accepted that hyperfibrinolysis may affect 30%-50% of cirrhotics with advanced disease[62].
Endothelial alterations of the portal vein radicles are well described in liver cirrhosis[63]. Endotoxaemia possibly plays a relevant role in endothelial alterations[64], independent on the eventual direct effects of HCV infection. As mentioned above, altered endothelium promotes coagulation by activation of tissue factor. In cirrhotics there is also an increase in the expression of several adhesion molecules, including platelet-endothelial cell adhesion molecule-1 (PECAM-1), L-selectin and P-selectin[65], and, as just mentioned, increased levels of von Willebrand factor[57].
Activated endothelial cells, as well as monocytes and platelets, also lead to the formation of microparticles that also carry tissue factor. In addition, platelet derived microparticles are able to transfer the GIIb-IIIa platelet receptor to leukocytes, a feature which leads to the activation of the nuclear transcription factor kappa B, inducing gene transcription of proinflammatory mediators[66]. In addition platelet microparticles are able to carry factor V[67]. Some studies point to an increased production of microparticles derived from leukocytes, lymphocytes, erythrocytes or even hepatocytes in liver cirrhosis[68]; despite some assertions[69], other researchers have failed to find raised platelet-derived microparticles in cirrhotic patients[70].
In summary, cirrhotics show more depressed levels of anticoagulants than those of procoagulants; although the role of microparticles in liver cirrhosis is unclear, portal hypertension-related endothelial damage and endotoxin-mediated cytokine activation, together with altered fibrinolysis in some cases, all contribute to the prothrombotic state of these patients, with increased thrombin formation.
Platelets are also altered in liver cirrhosis, both in number and function. Decreased platelet number may be due to splenic pooling due to portal hypertension, lower thrombopoietin levels, and increased consumption due to endotoxaemia, increased antiplatelet antibody production, and increased coagulation activation[69], but no significant bleeding takes place if platelet count is over 60000/fl[71]. Qualitative platelet alterations include defective adhesion[72], partially compensated by increased von Willebrand factor; decreased aggregation in response to normal stimuli, such as ADP, ristocetin, thrombin, collagen or epinephrine[73] and altered function[26] which progresses as liver function worsens. The aforementioned increased production of prothrombotic platelet-derived microparticles may compensate for these defects, so that hemostasis does not become significantly altered. On the contrary, portal thrombotic events may occur if a thrombopoietin receptor agonist (Eltrombopag) is administered[74], despite a median maximum platelet count of 148000/fl.
All these factors add to the previously mentioned direct effects of HCV infection, explaining why in HCV liver cirrhosis the thrombotic complications are more frequently observed than in cirrhosis of other etiologies.
CONSEQUENCES OF INCREASED THROMBIN FORMATION
Thrombin and fibrous tissue deposition: Direct effects on fibrogenesis
Thrombin not only plays a central role in the coagulation system. The importance of increased thrombin activation in patients with liver cirrhosis resides in the fact that thrombin may be directly involved in fibrogenesis (Figure 4). Thrombin exerts this action after binding to a group of receptors called protease activator receptors (PAR). There are four of such type of receptors (1-4), which become activated by several different proteases including thrombin (which activates PAR 1, 3, 4), and tryptase, which activates PAR-2, providing a rational basis to sustain the finding of liver fibrosis in the context of systemic mastocytosis[75]. PAR-1 receptors are present in the liver, and increase their expression in advanced liver disease[76] and along the transformation of stellate cells into myofibroblasts[77]. Indeed, hepatic stellate cells are one of the main cells in which these receptors become up-regulated in chronic liver disease. Binding of PAR with their ligands on stellate cells leads to proliferation and activation of these cells, that secrete monocyte chemoattractant protein 1 (MCP-1), increase deposition of extracellular matrix and the expression of receptors for platelet derived growth factor (PDGF) and transforming growth factor (TGF)-β[78]. In addition MCP-1 attracts monocytes, that produce tissue factor which contributes to more thrombin generation[79].
Figure 4.
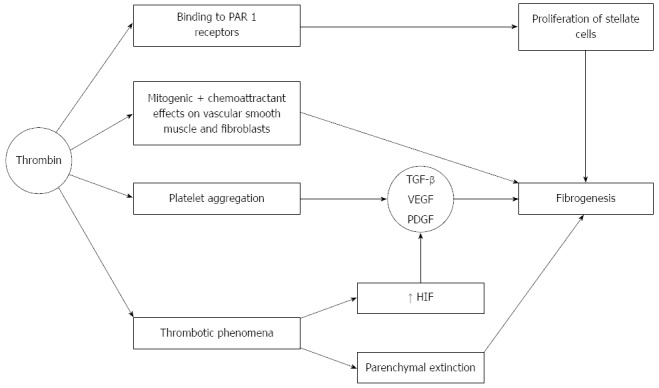
Effects on thrombin on fibrogenesis. Thrombin binds to PAR-1 receptors on hepatic stellate cells which leads to proliferaton and activation of these cells Thrombin also promotes platelet aggregation. Platelet alpha granules are rich in several growth factors, including TGF-β, which in turn promotes fibrogenesis. Vascular endothelial growth factor (VEGF) and PDGF also play contributory roles. Thrombin also exerts mitogenic and chemoattractant effects on vascular smooth muscle cells and fibroblasts. Finally, thrombotic phenomena also occur within the liver, leading to ischemic parenchymal injury, and substitution of parenchyma by fibrous tissue (the so called parenchymal extinction). When a clot provokes ischemia, VEGF, PDGF and TGF-β are activated probably via an increase in hypoxia-inducible-factor (HIF), which is raised in cirrhosis in relation to portal microthrombotic phenomena.
Another well known effect of thrombin is the ability to promote platelet aggregation. Platelet alpha granules are rich in several growth factors, including TGF-β, which in turn promotes fibrogenesis. Vascular endothelial growth factor (VEGF) and PDGF also play contributory roles. PDGF is a very strong mitogen for stellate cells and it also promotes fibrogenic activity by these cells[80]. PDGF upregulates the expression of matrix metalloproteinase (MMP)-2, MMP-9 and tissue inhibitor of metalloproteinase (TIMP)-1 and downregulates that of collagenase[40].
Thrombin also exerts mitogenic and chemoattractant effects on vascular smooth muscle cells and fibroblasts[81]. Factor Xa also activates PAR-1 and PAR-2, and may lead to fibrous tissue deposition. It was shown that incubation of fibroblasts with factor Xa led to a 12.6-fold increase in TGF-β expression, an increase which was by far more intense than that elicited when the cells were stimulated by thrombin[82].
Therefore, thrombin, both directly and indirectly, together with other activated coagulation factors, may play a role in the progression of liver cirrhosis, via the described effect on fibrogenesis.
Thrombin and clot formation: Parenchymal extinction and fibrogenesis
In addition to thrombosis of major veins, microthrombotic phenomena also occur within the liver, leading to ischemic parenchymal injury, and substitution of parenchyma by fibrous tissue (the so called parenchymal extinction[83]). This phenomenon consists in the ischemic collapse of hepatocytes between portal vein radicles and hepatic central venule, and becomes strongly exacerbated if congestive phenomena coexist, such as heart failure[84]. Coalescence of neighbouring tracts could lead to the formation of gross fibrous tracts and the evolution to cirrhosis[85]. When a clot provokes ischemia, VEGF, PDGF and TGF-β are activated[86] probably via an increase in hypoxia-inducible-factor (HIF), which is raised in cirrhosis due to increased portal resistance[87].
However, there are studies that do not support a pathogenetic role of portal vein thrombosis on progression of liver disease[88], but, as discussed above, factor V Leiden seems to accelerate progression of fibrosis in HCV infected patients. In other studies, other prothrombotic conditions such as hyperhomocysteinaemia[89] or mutations in factor XIII (both as an isolated finding or in combination with PAI-1 4G/5G mutation) also constitute a risk factor for an increased rate of liver fibrosis development in patients affected with HCV or chronic hepatitis B[90]. In other studies, the mutation associated with increased fibrosis progression rate was the prothrombin G20210 A mutation[91].
These data support the importance of microthrombotic phenomena in the progression of liver disease, especially in HCV-infected patients, in whom the endothelial changes promoted by HCV may be considered triggering factors, aggravated in later stages of the disease by the endothelial changes secondary to portal hypertension.
Therapeutic future prospects
Based on the aforementioned data, low molecular weight heparin has been advocated as a therapeutic option in patients with cirrhosis, with promising results: enoxaparin for 48 wk not only significantly prevented the development of portal vein thrombosis in patients with cirrhosis with Child-Pugh scores between 7 and 10, but it also associated with a decreased probability of decompensation of liver disease[92]. In that study, 18 out of 34 randomized to enoxaparin and 18 out of 36 in the placebo group were HCV-infected patients. Experimental research in rats has shown that treatment with heparin reduced severity of biochemical and histological changes induced in rats with oral carbon tetrachloride[93]. The possible role of anticoagulation in the treatment of advanced liver disease, especially in HCV-induced cirrhosis, is a promising idea that warrants confirmation[94].
Footnotes
Conflict-of-interest statement: The authors declare that there are no conflicts of interest regarding this manuscript. They also declare that they have not received any funding for this study.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: February 25, 2016
First decision: March 21, 2016
Article in press: April 15, 2016
P- Reviewer: Jin B, Utama A S- Editor: Ma YJ L- Editor: A E- Editor: Ma S
References
- 1.Potze W, Arshad F, Adelmeijer J, Blokzijl H, van den Berg AP, Meijers JC, Porte RJ, Lisman T. Decreased tissue factor pathway inhibitor (TFPI)-dependent anticoagulant capacity in patients with cirrhosis who have decreased protein S but normal TFPI plasma levels. Br J Haematol. 2013;162:819–826. doi: 10.1111/bjh.12462. [DOI] [PubMed] [Google Scholar]
- 2.Hepner M, Karlaftis V. Antithrombin. Methods Mol Biol. 2013;992:355–364. doi: 10.1007/978-1-62703-339-8_28. [DOI] [PubMed] [Google Scholar]
- 3.Levy JH, Sniecinski RM, Welsby IJ, Levi M. Antithrombin: anti-inflammatory properties and clinical applications. Thromb Haemost. 2016;115:712–728. doi: 10.1160/TH15-08-0687. [DOI] [PubMed] [Google Scholar]
- 4.Sheffield WP, Bhakta V. The M358R variant of α1-proteinase inhibitor inhibits coagulation factor VIIa. Biochem Biophys Res Commun. 2016;470:710–713. doi: 10.1016/j.bbrc.2016.01.069. [DOI] [PubMed] [Google Scholar]
- 5.Mast AE. Tissue Factor Pathway Inhibitor: Multiple Anticoagulant Activities for a Single Protein. Arterioscler Thromb Vasc Biol. 2016;36:9–14. doi: 10.1161/ATVBAHA.115.305996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng XL. Structure-function and regulation of ADAMTS-13 protease. J Thromb Haemost. 2013;11 Suppl 1:11–23. doi: 10.1111/jth.12221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouma BN, Mosnier LO. Thrombin activatable fibrinolysis inhibitor (TAFI) at the interface between coagulation and fibrinolysis. Pathophysiol Haemost Thromb. 2003;33:375–381. doi: 10.1159/000083832. [DOI] [PubMed] [Google Scholar]
- 8.Prieto J, Yuste JR, Beloqui O, Civeira MP, Riezu JI, Aguirre B, Sangro B. Anticardiolipin antibodies in chronic hepatitis C: implication of hepatitis C virus as the cause of the antiphospholipid syndrome. Hepatology. 1996;23:199–204. doi: 10.1002/hep.510230201. [DOI] [PubMed] [Google Scholar]
- 9.Biron C, Andréani H, Blanc P, Ramos J, Ducos J, Guigue N, Michel H, Larrey D, Schved JF. Prevalence of antiphospholipid antibodies in patients with chronic liver disease related to alcohol or hepatitis C virus: correlation with liver injury. J Lab Clin Med. 1998;131:243–250. doi: 10.1016/s0022-2143(98)90096-8. [DOI] [PubMed] [Google Scholar]
- 10.Enger C, Forssen UM, Bennett D, Theodore D, Shantakumar S, McAfee A. Thromboembolic events among patients with hepatitis C virus infection and cirrhosis: a matched-cohort study. Adv Ther. 2014;31:891–903. doi: 10.1007/s12325-014-0138-4. [DOI] [PubMed] [Google Scholar]
- 11.Wang CC, Chang CT, Lin CL, Lin IC, Kao CH. Hepatitis C Virus Infection Associated With an Increased Risk of Deep Vein Thrombosis: A Population-Based Cohort Study. Medicine (Baltimore) 2015;94:e1585. doi: 10.1097/MD.0000000000001585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen HS, Xing SR, Xu WG, Yang F, Qi XL, Wang LM, Yang CQ. Portopulmonary hypertension in cirrhotic patients: Prevalence, clinical features and risk factors. Exp Ther Med. 2013;5:819–824. doi: 10.3892/etm.2013.918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harada M, Fujisawa Y, Sakisaka S, Kawaguchi T, Taniguchi E, Sakamoto M, Sumie S, Sasatomi K, Koga H, Torimura T, et al. High prevalence of anticardiolipin antibodies in hepatitis C virus infection: lack of effects on thrombocytopenia and thrombotic complications. J Gastroenterol. 2000;35:272–277. doi: 10.1007/s005350050345. [DOI] [PubMed] [Google Scholar]
- 14.Ordi-Ros J, Villarreal J, Monegal F, Sauleda S, Esteban I, Vilardell M. Anticardiolipin antibodies in patients with chronic hepatitis C virus infection: characterization in relation to antiphospholipid syndrome. Clin Diagn Lab Immunol. 2000;7:241–244. doi: 10.1128/cdli.7.2.241-244.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Violi F, Ferro D, Basili S, Artini M, Valesini G, Levrero M, Cordova C. Increased rate of thrombin generation in hepatitis C virus cirrhotic patients. Relationship to venous thrombosis. J Investig Med. 1995;43:550–554. [PubMed] [Google Scholar]
- 16.Galli L, Gerdes VE, Guasti L, Squizzato A. Thrombosis Associated with Viral Hepatitis. J Clin Transl Hepatol. 2014;2:234–239. doi: 10.14218/JCTH.2014.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toschi V, Fiorini GF, Motta A, Renoldi P, Paracchini ML, Gibelli A. Clinical significance of endothelial damage markers in essential mixed cryoglobulinemia. Acta Haematol. 1991;86:90–94. doi: 10.1159/000204810. [DOI] [PubMed] [Google Scholar]
- 18.Pircher J, Czermak T, Merkle M, Mannell H, Krötz F, Ribeiro A, Vielhauer V, Nadjiri J, Gaitzsch E, Niemeyer M, et al. Hepatitis C virus induced endothelial inflammatory response depends on the functional expression of TNFα receptor subtype 2. PLoS One. 2014;9:e113351. doi: 10.1371/journal.pone.0113351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wald O, Pappo O, Safadi R, Dagan-Berger M, Beider K, Wald H, Franitza S, Weiss I, Avniel S, Boaz P, et al. Involvement of the CXCL12/CXCR4 pathway in the advanced liver disease that is associated with hepatitis C virus or hepatitis B virus. Eur J Immunol. 2004;34:1164–1174. doi: 10.1002/eji.200324441. [DOI] [PubMed] [Google Scholar]
- 20.Falasca K, Mancino P, Ucciferri C, Dalessandro M, Zingariello P, Lattanzio FM, Petrarca C, Martinotti S, Pizzigallo E, Conti P, et al. Inflammatory cytokines and S-100b protein in patients with hepatitis C infection and cryoglobulinemias. Clin Invest Med. 2007;30:E167–E176. doi: 10.25011/cim.v30i5.2892. [DOI] [PubMed] [Google Scholar]
- 21.Grignani G, Maiolo A. Cytokines and hemostasis. Haematologica. 2000;85:967–972. [PubMed] [Google Scholar]
- 22.Ishii H, Horie S, Kizaki K, Kazama M. Retinoic acid counteracts both the downregulation of thrombomodulin and the induction of tissue factor in cultured human endothelial cells exposed to tumor necrosis factor. Blood. 1992;80:2556–2562. [PubMed] [Google Scholar]
- 23.Schleef RR, Bevilacqua MP, Sawdey M, Gimbrone MA, Loskutoff DJ. Cytokine activation of vascular endothelium. Effects on tissue-type plasminogen activator and type 1 plasminogen activator inhibitor. J Biol Chem. 1988;263:5797–5803. [PubMed] [Google Scholar]
- 24.Hodowanec AC, Lee RD, Brady KE, Gao W, Kincaid S, Plants J, Bahk M, Mackman N, Landay AL, Huhn GD. A matched cross-sectional study of the association between circulating tissue factor activity, immune activation and advanced liver fibrosis in hepatitis C infection. BMC Infect Dis. 2015;15:190. doi: 10.1186/s12879-015-0920-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gear AR, Camerini D. Platelet chemokines and chemokine receptors: linking hemostasis, inflammation, and host defense. Microcirculation. 2003;10:335–350. doi: 10.1038/sj.mn.7800198. [DOI] [PubMed] [Google Scholar]
- 26.Witters P, Freson K, Verslype C, Peerlinck K, Hoylaerts M, Nevens F, Van Geet C, Cassiman D. Review article: blood platelet number and function in chronic liver disease and cirrhosis. Aliment Pharmacol Ther. 2008;27:1017–1029. doi: 10.1111/j.1365-2036.2008.03674.x. [DOI] [PubMed] [Google Scholar]
- 27.Panasiuk A, Prokopowicz D, Zak J, Matowicka-Karna J, Osada J, Wysocka J. Activation of blood platelets in chronic hepatitis and liver cirrhosis P-selectin expression on blood platelets and secretory activity of beta-thromboglobulin and platelet factor-4. Hepatogastroenterology. 2001;48:818–822. [PubMed] [Google Scholar]
- 28.Dieterich DT, Spivak JL. Hematologic disorders associated with hepatitis C virus infection and their management. Clin Infect Dis. 2003;37:533–541. doi: 10.1086/376971. [DOI] [PubMed] [Google Scholar]
- 29.Tana MM, Zhao X, Bradshaw A, Moon MS, Page S, Turner T, Rivera E, Kleiner DE, Heller T. Factors associated with the platelet count in patients with chronic hepatitis C. Thromb Res. 2015;135:823–828. doi: 10.1016/j.thromres.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weksler BB. Review article: the pathophysiology of thrombocytopenia in hepatitis C virus infection and chronic liver disease. Aliment Pharmacol Ther. 2007;26 Suppl 1:13–19. doi: 10.1111/j.1365-2036.2007.03512.x. [DOI] [PubMed] [Google Scholar]
- 31.Rajan S, Liebman HA. Treatment of hepatitis C related thrombocytopenia with interferon alpha. Am J Hematol. 2001;68:202–209. doi: 10.1002/ajh.1180. [DOI] [PubMed] [Google Scholar]
- 32.Español I, Gallego A, Enríquez J, Rabella N, Lerma E, Hernández A, Pujol-Moix N. Thrombocytopenia associated with liver cirrhosis and hepatitis C viral infection: role of thrombopoietin. Hepatogastroenterology. 2000;47:1404–1406. [PubMed] [Google Scholar]
- 33.Adinolfi LE, Giordano MG, Andreana A, Tripodi MF, Utili R, Cesaro G, Ragone E, Durante Mangoni E, Ruggiero G. Hepatic fibrosis plays a central role in the pathogenesis of thrombocytopenia in patients with chronic viral hepatitis. Br J Haematol. 2001;113:590–595. doi: 10.1046/j.1365-2141.2001.02824.x. [DOI] [PubMed] [Google Scholar]
- 34.Giannini E, Borro P, Botta F, Fumagalli A, Malfatti F, Podestà E, Romagnoli P, Testa E, Chiarbonello B, Polegato S, et al. Serum thrombopoietin levels are linked to liver function in untreated patients with hepatitis C virus-related chronic hepatitis. J Hepatol. 2002;37:572–577. doi: 10.1016/s0168-8278(02)00274-x. [DOI] [PubMed] [Google Scholar]
- 35.Goulis J, Chau TN, Jordan S, Mehta AB, Watkinson A, Rolles K, Burroughs AK. Thrombopoietin concentrations are low in patients with cirrhosis and thrombocytopenia and are restored after orthotopic liver transplantation. Gut. 1999;44:754–758. doi: 10.1136/gut.44.5.754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rios R, Sangro B, Herrero I, Quiroga J, Prieto J. The role of thrombopoietin in the thrombocytopenia of patients with liver cirrhosis. Am J Gastroenterol. 2005;100:1311–1316. doi: 10.1111/j.1572-0241.2005.41543.x. [DOI] [PubMed] [Google Scholar]
- 37.Nagamine T, Ohtuka T, Takehara K, Arai T, Takagi H, Mori M. Thrombocytopenia associated with hepatitis C viral infection. J Hepatol. 1996;24:135–140. doi: 10.1016/s0168-8278(96)80021-3. [DOI] [PubMed] [Google Scholar]
- 38.Panzer S, Seel E, Brunner M, Körmöczi GF, Schmid M, Ferenci P, Peck-Radosavljevic M. Platelet autoantibodies are common in hepatitis C infection, irrespective of the presence of thrombocytopenia. Eur J Haematol. 2006;77:513–517. doi: 10.1111/j.0902-4441.2006.t01-1-ejh2888.x. [DOI] [PubMed] [Google Scholar]
- 39.Olariu M, Olariu C, Olteanu D. Thrombocytopenia in chronic hepatitis C. J Gastrointestin Liver Dis. 2010;19:381–385. [PubMed] [Google Scholar]
- 40.Zhou WC, Zhang QB, Qiao L. Pathogenesis of liver cirrhosis. World J Gastroenterol. 2014;20:7312–7324. doi: 10.3748/wjg.v20.i23.7312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tripodi A, Mannucci PM. The coagulopathy of chronic liver disease. N Engl J Med. 2011;365:147–156. doi: 10.1056/NEJMra1011170. [DOI] [PubMed] [Google Scholar]
- 42.Gulley D, Teal E, Suvannasankha A, Chalasani N, Liangpunsakul S. Deep vein thrombosis and pulmonary embolism in cirrhosis patients. Dig Dis Sci. 2008;53:3012–3017. doi: 10.1007/s10620-008-0265-3. [DOI] [PubMed] [Google Scholar]
- 43.García-Fuster MJ, Abdilla N, Fabiá MJ, Fernández C, Oliver V. [Venous thromboembolism and liver cirrhosis] Rev Esp Enferm Dig. 2008;100:259–262. doi: 10.4321/s1130-01082008000500002. [DOI] [PubMed] [Google Scholar]
- 44.Tsochatzis EA, Senzolo M, Germani G, Gatt A, Burroughs AK. Systematic review: portal vein thrombosis in cirrhosis. Aliment Pharmacol Ther. 2010;31:366–374. doi: 10.1111/j.1365-2036.2009.04182.x. [DOI] [PubMed] [Google Scholar]
- 45.Aggarwal A, Puri K, Liangpunsakul S. Deep vein thrombosis and pulmonary embolism in cirrhotic patients: systematic review. World J Gastroenterol. 2014;20:5737–5745. doi: 10.3748/wjg.v20.i19.5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tripodi A, Salerno F, Chantarangkul V, Clerici M, Cazzaniga M, Primignani M, Mannuccio Mannucci P. Evidence of normal thrombin generation in cirrhosis despite abnormal conventional coagulation tests. Hepatology. 2005;41:553–558. doi: 10.1002/hep.20569. [DOI] [PubMed] [Google Scholar]
- 47.Raya-Sánchez JM, González-Reimers E, Rodríguez-Martín JM, Santolaria-Fernández F, Molina-Pérez M, Rodríguez-Moreno F, Martínez-Riera A. Coagulation inhibitors in alcoholic liver cirrhosis. Alcohol. 1998;15:19–23. doi: 10.1016/s0741-8329(97)00082-7. [DOI] [PubMed] [Google Scholar]
- 48.Martin FA, Murphy RP, Cummins PM. Thrombomodulin and the vascular endothelium: insights into functional, regulatory, and therapeutic aspects. Am J Physiol Heart Circ Physiol. 2013;304:H1585–H1597. doi: 10.1152/ajpheart.00096.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dahlbäck B. Advances in understanding pathogenic mechanisms of thrombophilic disorders. Blood. 2008;112:19–27. doi: 10.1182/blood-2008-01-077909. [DOI] [PubMed] [Google Scholar]
- 50.Qi X, Ren W, De Stefano V, Fan D. Associations of coagulation factor V Leiden and prothrombin G20210A mutations with Budd-Chiari syndrome and portal vein thrombosis: a systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2014;12:1801–12.e7. doi: 10.1016/j.cgh.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 51.Saugel B, Lee M, Feichtinger S, Hapfelmeier A, Schmid RM, Siveke JT. Thrombophilic factor analysis in cirrhotic patients with portal vein thrombosis. J Thromb Thrombolysis. 2015;40:54–60. doi: 10.1007/s11239-014-1124-z. [DOI] [PubMed] [Google Scholar]
- 52.Wright M, Goldin R, Hellier S, Knapp S, Frodsham A, Hennig B, Hill A, Apple R, Cheng S, Thomas H, et al. Factor V Leiden polymorphism and the rate of fibrosis development in chronic hepatitis C virus infection. Gut. 2003;52:1206–1210. doi: 10.1136/gut.52.8.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Poujol-Robert A, Boëlle PY, Poupon R, Robert A. Factor V Leiden as a risk factor for cirrhosis in chronic hepatitis C. Hepatology. 2004;39:1174–1175. doi: 10.1002/hep.20166. [DOI] [PubMed] [Google Scholar]
- 54.Papatheodoridis GV, Papakonstantinou E, Andrioti E, Cholongitas E, Petraki K, Kontopoulou I, Hadziyannis SJ. Thrombotic risk factors and extent of liver fibrosis in chronic viral hepatitis. Gut. 2003;52:404–409. doi: 10.1136/gut.52.3.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu Z, Westrick RJ, Shen YC, Eitzman DT. Pulmonary fibrosis is increased in mice carrying the factor V Leiden mutation following bleomycin injury. Thromb Haemost. 2001;85:441–444. [PubMed] [Google Scholar]
- 56.Tripodi A, Primignani M, Chantarangkul V, Dell’Era A, Clerici M, de Franchis R, Colombo M, Mannucci PM. An imbalance of pro- vs anti-coagulation factors in plasma from patients with cirrhosis. Gastroenterology. 2009;137:2105–2111. doi: 10.1053/j.gastro.2009.08.045. [DOI] [PubMed] [Google Scholar]
- 57.Tripodi A. Hemostasis abnormalities in cirrhosis. Curr Opin Hematol. 2015;22:406–412. doi: 10.1097/MOH.0000000000000164. [DOI] [PubMed] [Google Scholar]
- 58.Hollestelle MJ, Geertzen HG, Straatsburg IH, van Gulik TM, van Mourik JA. Factor VIII expression in liver disease. Thromb Haemost. 2004;91:267–275. doi: 10.1160/TH03-05-0310. [DOI] [PubMed] [Google Scholar]
- 59.Feys HB, Canciani MT, Peyvandi F, Deckmyn H, Vanhoorelbeke K, Mannucci PM. ADAMTS13 activity to antigen ratio in physiological and pathological conditions associated with an increased risk of thrombosis. Br J Haematol. 2007;138:534–540. doi: 10.1111/j.1365-2141.2007.06688.x. [DOI] [PubMed] [Google Scholar]
- 60.Ferguson JW, Helmy A, Ludlam C, Webb DJ, Hayes PC, Newby DC. Hyperfibrinolysis in alcoholic cirrhosis: relative plasminogen activator inhibitor type 1 deficiency. Thromb Res. 2008;121:675–680. doi: 10.1016/j.thromres.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 61.Colucci M, Binetti BM, Branca MG, Clerici C, Morelli A, Semeraro N, Gresele P. Deficiency of thrombin activatable fibrinolysis inhibitor in cirrhosis is associated with increased plasma fibrinolysis. Hepatology. 2003;38:230–237. doi: 10.1053/jhep.2003.50277. [DOI] [PubMed] [Google Scholar]
- 62.Leebeek FW, Rijken DC. The Fibrinolytic Status in Liver Diseases. Semin Thromb Hemost. 2015;41:474–480. doi: 10.1055/s-0035-1550437. [DOI] [PubMed] [Google Scholar]
- 63.Adams DH, Burra P, Hubscher SG, Elias E, Newman W. Endothelial activation and circulating vascular adhesion molecules in alcoholic liver disease. Hepatology. 1994;19:588–594. doi: 10.1002/hep.1840190308. [DOI] [PubMed] [Google Scholar]
- 64.Echeverría C, Montorfano I, Tapia P, Riedel C, Cabello-Verrugio C, Simon F. Endotoxin-induced endothelial fibrosis is dependent on expression of transforming growth factors β1 and β2. Infect Immun. 2014;82:3678–3686. doi: 10.1128/IAI.02158-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reinhart K, Bayer O, Brunkhorst F, Meisner M. Markers of endothelial damage in organ dysfunction and sepsis. Crit Care Med. 2002;30:S302–S312. doi: 10.1097/00003246-200205001-00021. [DOI] [PubMed] [Google Scholar]
- 66.Salanova B, Choi M, Rolle S, Wellner M, Luft FC, Kettritz R. Beta2-integrins and acquired glycoprotein IIb/IIIa (GPIIb/IIIa) receptors cooperate in NF-kappaB activation of human neutrophils. J Biol Chem. 2007;282:27960–27969. doi: 10.1074/jbc.M704039200. [DOI] [PubMed] [Google Scholar]
- 67.Alberio L, Safa O, Clemetson KJ, Esmon CT, Dale GL. Surface expression and functional characterization of alpha-granule factor V in human platelets: effects of ionophore A23187, thrombin, collagen, and convulxin. Blood. 2000;95:1694–1702. [PubMed] [Google Scholar]
- 68.Rautou PE, Bresson J, Sainte-Marie Y, Vion AC, Paradis V, Renard JM, Devue C, Heymes C, Letteron P, Elkrief L, et al. Abnormal plasma microparticles impair vasoconstrictor responses in patients with cirrhosis. Gastroenterology. 2012;143:166–76.e6. doi: 10.1053/j.gastro.2012.03.040. [DOI] [PubMed] [Google Scholar]
- 69.Tapper EB, Robson SC, Malik R. Coagulopathy in cirrhosis - the role of the platelet in hemostasis. J Hepatol. 2013;59:889–890. doi: 10.1016/j.jhep.2013.03.040. [DOI] [PubMed] [Google Scholar]
- 70.Rautou PE, Vion AC, Valla D, Boulanger CM. Circulating platelet derived microparticles are not increased in patients with cirrhosis. J Hepatol. 2013;59:912. doi: 10.1016/j.jhep.2013.05.048. [DOI] [PubMed] [Google Scholar]
- 71.Tripodi A, Primignani M, Chantarangkul V, Clerici M, Dell’Era A, Fabris F, Salerno F, Mannucci PM. Thrombin generation in patients with cirrhosis: the role of platelets. Hepatology. 2006;44:440–445. doi: 10.1002/hep.21266. [DOI] [PubMed] [Google Scholar]
- 72.Ordinas A, Escolar G, Cirera I, Viñas M, Cobo F, Bosch J, Terés J, Rodés J. Existence of a platelet-adhesion defect in patients with cirrhosis independent of hematocrit: studies under flow conditions. Hepatology. 1996;24:1137–1142. doi: 10.1053/jhep.1996.v24.pm0008903388. [DOI] [PubMed] [Google Scholar]
- 73.Thomas DP, Ream VJ, Stuart RK. Platelet aggregation in patients with Laennec’s cirrhosis of the liver. N Engl J Med. 1967;276:1344–1348. doi: 10.1056/NEJM196706152762403. [DOI] [PubMed] [Google Scholar]
- 74.Afdhal NH, Giannini EG, Tayyab G, Mohsin A, Lee JW, Andriulli A, Jeffers L, McHutchison J, Chen PJ, Han KH, et al. Eltrombopag before procedures in patients with cirrhosis and thrombocytopenia. N Engl J Med. 2012;367:716–724. doi: 10.1056/NEJMoa1110709. [DOI] [PubMed] [Google Scholar]
- 75.Wendum D, Prevot S, Poujol-Robert A, Rosmorduc O, Cabane J, Fouillard L, Flejou JF. [Liver involvement revealing systemic mastocytosis: report of two cases] Gastroenterol Clin Biol. 2004;28:80–83. doi: 10.1016/s0399-8320(04)94851-8. [DOI] [PubMed] [Google Scholar]
- 76.Marra F, DeFranco R, Grappone C, Milani S, Pinzani M, Pellegrini G, Laffi G, Gentilini P. Expression of the thrombin receptor in human liver: up-regulation during acute and chronic injury. Hepatology. 1998;27:462–471. doi: 10.1002/hep.510270221. [DOI] [PubMed] [Google Scholar]
- 77.Gaça MD, Zhou X, Benyon RC. Regulation of hepatic stellate cell proliferation and collagen synthesis by proteinase-activated receptors. J Hepatol. 2002;36:362–369. doi: 10.1016/s0168-8278(01)00285-9. [DOI] [PubMed] [Google Scholar]
- 78.Marra F, Grandaliano G, Valente AJ, Abboud HE. Thrombin stimulates proliferation of liver fat-storing cells and expression of monocyte chemotactic protein-1: potential role in liver injury. Hepatology. 1995;22:780–787. [PubMed] [Google Scholar]
- 79.Edwards RL, Levine JB, Green R, Duffy M, Mathews E, Brande W, Rickles FR. Activation of blood coagulation in Crohn’s disease. Increased plasma fibrinopeptide A levels and enhanced generation of monocyte tissue factor activity. Gastroenterology. 1987;92:329–337. [PubMed] [Google Scholar]
- 80.Pinzani M, Gesualdo L, Sabbah GM, Abboud HE. Effects of platelet-derived growth factor and other polypeptide mitogens on DNA synthesis and growth of cultured rat liver fat-storing cells. J Clin Invest. 1989;84:1786–1793. doi: 10.1172/JCI114363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Calvaruso V, Maimone S, Gatt A, Tuddenham E, Thursz M, Pinzani M, Burroughs AK. Coagulation and fibrosis in chronic liver disease. Gut. 2008;57:1722–1727. doi: 10.1136/gut.2008.150748. [DOI] [PubMed] [Google Scholar]
- 82.Kitasato L, Yamaoka-Tojo M, Hashikata T, Ishii S, Kameda R, Shimohama T, Tojo T, Ako J. Factor Xa in mouse fibroblasts may induce fibrosis more than thrombin. Int Heart J. 2014;55:357–361. doi: 10.1536/ihj.13-351. [DOI] [PubMed] [Google Scholar]
- 83.Wanless IR, Wong F, Blendis LM, Greig P, Heathcote EJ, Levy G. Hepatic and portal vein thrombosis in cirrhosis: possible role in development of parenchymal extinction and portal hypertension. Hepatology. 1995;21:1238–1247. [PubMed] [Google Scholar]
- 84.Wanless IR, Liu JJ, Butany J. Role of thrombosis in the pathogenesis of congestive hepatic fibrosis (cardiac cirrhosis) Hepatology. 1995;21:1232–1237. [PubMed] [Google Scholar]
- 85.Wanless IR, Shiota K. The pathogenesis of nonalcoholic steatohepatitis and other fatty liver diseases: a four-step model including the role of lipid release and hepatic venular obstruction in the progression to cirrhosis. Semin Liver Dis. 2004;24:99–106. doi: 10.1055/s-2004-823104. [DOI] [PubMed] [Google Scholar]
- 86.Corpechot C, Barbu V, Wendum D, Kinnman N, Rey C, Poupon R, Housset C, Rosmorduc O. Hypoxia-induced VEGF and collagen I expressions are associated with angiogenesis and fibrogenesis in experimental cirrhosis. Hepatology. 2002;35:1010–1021. doi: 10.1053/jhep.2002.32524. [DOI] [PubMed] [Google Scholar]
- 87.Moeller M, Thonig A, Pohl S, Ripoll C, Zipprich A. Hepatic arterial vasodilation is independent of portal hypertension in early stages of cirrhosis. PLoS One. 2015;10:e0121229. doi: 10.1371/journal.pone.0121229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nery F, Chevret S, Condat B, de Raucourt E, Boudaoud L, Rautou PE, Plessier A, Roulot D, Chaffaut C, Bourcier V, et al. Causes and consequences of portal vein thrombosis in 1,243 patients with cirrhosis: results of a longitudinal study. Hepatology. 2015;61:660–667. doi: 10.1002/hep.27546. [DOI] [PubMed] [Google Scholar]
- 89.Adinolfi LE, Ingrosso D, Cesaro G, Cimmino A, D’Antò M, Capasso R, Zappia V, Ruggiero G. Hyperhomocysteinemia and the MTHFR C677T polymorphism promote steatosis and fibrosis in chronic hepatitis C patients. Hepatology. 2005;41:995–1003. doi: 10.1002/hep.20664. [DOI] [PubMed] [Google Scholar]
- 90.Dik K, de Bruijne J, Takkenberg RB, Roelofs JJ, Tempelmans MJ, Dijkgraaf MG, Gelderblom HC, Reesink HW, Meijers JC, Jansen PL, et al. Factor XIII Val34Leu mutation accelerates the development of fibrosis in patients with chronic hepatitis B and C. Hepatol Res. 2012;42:668–676. doi: 10.1111/j.1872-034X.2011.00963.x. [DOI] [PubMed] [Google Scholar]
- 91.Maharshak N, Halfon P, Deutsch V, Peretz H, Berliner S, Fishman S, Zelber-Sagi S, Rozovski U, Leshno M, Oren R. Increased fibrosis progression rates in hepatitis C patients carrying the prothrombin G20210A mutation. World J Gastroenterol. 2011;17:5007–5013. doi: 10.3748/wjg.v17.i45.5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Villa E, Cammà C, Marietta M, Luongo M, Critelli R, Colopi S, Tata C, Zecchini R, Gitto S, Petta S, et al. Enoxaparin prevents portal vein thrombosis and liver decompensation in patients with advanced cirrhosis. Gastroenterology. 2012;143:1253–60.e1-4. doi: 10.1053/j.gastro.2012.07.018. [DOI] [PubMed] [Google Scholar]
- 93.Shah B, Shah G. Antifibrotic effect of heparin on liver fibrosis model in rats. World J Gastrointest Pharmacol Ther. 2012;3:86–92. doi: 10.4292/wjgpt.v3.i6.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jairath V, Burroughs AK. Anticoagulation in patients with liver cirrhosis: complication or therapeutic opportunity? Gut. 2013;62:479–482. doi: 10.1136/gutjnl-2012-303088. [DOI] [PubMed] [Google Scholar]