

Abstract
AIM: To discover novel biomarkers for early diagnosis, prognosis or treatment of human colorectal cancer.
METHODS: iTRAQ 2D LC-MS/MS analysis was used to identify differentially expressed proteins (DEPs) in the human colonic epithelial carcinogenic process using laser capture microdissection-purified colonic epithelial cells from normal colon, adenoma, carcinoma in situ and invasive carcinoma tissues.
RESULTS: A total of 326 DEPs were identified, and four DEPs (DMBT1, S100A9, Galectin-10, and S100A8) with progressive alteration in the carcinogenic process were further validated by immunohistochemistry. The DEPs were involved in multiple biological processes including cell cycle, cell adhesion, translation, mRNA processing, and protein synthesis. Some of the DEPs involved in cellular process such as “translation” and “mRNA splicing” were progressively up-regulated, while some DEPs involved in other processes such as “metabolism” and “cell response to stress” was progressively down-regulated. Other proteins with up- or down-regulation at certain stages of carcinogenesis may play various roles at different stages of the colorectal carcinogenic process.
CONCLUSION: These findings give insights into our understanding of the mechanisms of colorectal carcinogenesis and provide clues for further investigation of carcinogenesis and identification of biomarkers.
Keywords: Colorectal Cancer, Proteome, Biomarker, Carcinogenesis
Core tip: In this study, we used iTRAQ 2D LC-MS/MS analysis to identify differentially expressed proteins (DEPs) in the human colonic epithelial carcinogenic process using laser capture microdissection-purified colonic epithelial cells from normal colon, adenoma, carcinoma in situ and invasive carcinoma tissues. A total of 326 DEPs were identified. Four DEPs (DMBT1, S100A9, Galectin-10, and S100A8) with progressive alteration in the carcinogenic process were further validated using immunohistochemistry. The DEPs were involved in multiple biological processes including cell cycle, cell adhesion, translation, mRNA processing, and protein synthesis. These findings give insights into our understanding of the mechanisms of colorectal carcinogenesis and provide clues for further investigation of carcinogenesis and identification of biomarkers.
INTRODUCTION
Colorectal cancer (CRC) is the third leading cause of cancer death, affecting over a million people worldwide per year in recent years[1,2]. The World Health Organization estimates a 77% increase in the number of newly diagnosed cases of CRC and an 80% increase in deaths from CRC by 2030[3]. Despite an improvement in relative survival of CRC at 5 years due to early diagnosis at initial stages and breakthroughs in treatment of stages II and III disease, CRC is still one of the most lethal malignancies and the 5-year survival rate for patients with metastasis is < 5%[4]. Most patients are diagnosed at an advanced stage and have a poor prognosis. At present, CRC diagnosis and therapy are still dependent upon descriptive classification and staging systems based on morphology/histology[5]. Remarkable achievements in the understanding of cellular and molecular mechanisms of colorectal carcinogenesis have been made in recent years. However, therapy for advanced colorectal cancer remains limited, and current screening methods including sigmoidoscopy and colonoscopy lack the required sensitivity and specificity[6,7]. Therefore, a comprehensive understanding of the mechanisms behind colorectal carcinogenesis will contribute to the improvement in early detection and prognosis and provide novel therapeutic targets.
Carcinogenesis is a multistep and complicated process characterized by genetic alterations, including chromosomal abnormalities, gene mutations, and epigenetic changes that disrupt normal cell growth and division[8]. A genetic model describing the transition from healthy colonic epithelia through dysplastic adenoma to malignant cancer has been proposed[9]. According to the model[10], the colonic carcinogenic process originates from normal colonic mucosa (NC) and then transforms sequentially from adenoma (AD), carcinoma in situ (CIS, equivalent to high-grade intraepithelial neoplasia), and ultimately to invasive colorectal cancer (ICC)[11]. A number of sequential genetic abnormalities, including gene mutations in APC, K-ras, and p53 and epigenetic changes, were identified at different stages of colorectal carcinogenesis[11,12].
In recent years, much progress has been made in understanding genetic changes in the colonic carcinogenesis process, and many studies have been conducted to analyze differentially expressed proteins (DEPs) between certain stages of colorectal carcinogenesis[13-15]. However, there has been no systematic comparison between typical stages across the carcinogenic process, and much less is known about dynamic alterations at the proteome level during the process. Tissue heterogeneity is the main problem for analysis of biological samples in the study of disease. Recent technological progress using laser capture microdissection (LCM) has made it possible to overcome this problem and to enrich the desired populations of cells from heterogeneous tissues[16-18].
Isobaric tags for relative and absolute quantification (iTRAQ) combined with two-dimensional liquid chromatography-tandem mass spectrometry (2D LC-MS/MS) is a highly sensitive and practical technology[19-21]. Compared to conventional proteomic technology such as 2D electrophoresis, the iTRAQ method has the following advantages. First, it can label proteins from up to eight samples in a single experiment. Second, it can resolve large proteins (> 200 kD), small proteins (< 10 kD), and proteins with extremes in isoelectric point[22]. Therefore, iTRAQ technology offered us a feasible method to simultaneously compare the proteomes of successive stages of colorectal carcinogenesis.
To clarify the dynamic patterns of DEPs during colorectal carcinogenesis and provide valuable information for further identification of biomarkers for prevention, treatment or early diagnosis of CRC, iTRAQ tagging followed by 2D LC-MS/MS was performed to identify DEPs among LCM-purified colonic epithelial carcinogenic tissues and explore their dynamic expression patterns. A total of 326 DEPs were identified among different stages to have distinct expression patterns during the carcinogenic process, and four top-ranked DEPs (DMBT1, S100A9, Galectin-10, and S100A8) were further validated by immunohistochemistry. To our knowledge, this is the first comprehensive study that systematically compares the dynamic alterations of proteins during the process of colorectal carcinogenesis by comparative proteomics.
MATERIALS AND METHODS
Sample collection
Twenty-seven cases of fresh colonic tissues (5 cases of NC, 8 cases of AD, 5 cases of CIS, 9 cases of ICC) collected between January 2011 and December 2012 were obtained from the Department of Surgery, Xiangya Hospital, Central South University, China and used for iTRAQ-labelling. The patients received neither chemotherapy nor radiotherapy before curative surgery and signed an informed consent form for the study, which was approved by the local ethical committee. All tissue specimens were obtained from surgical resection, and the normal colonic tissue samples were acquired from the resection edge furthest away from the lesion (≥ 10 cm). The tissue samples in the ICC group were from CRC patients with lymph node metastasis. All of the tissues were flash frozen in liquid nitrogen and stored at -80 °C until further use.
An independent set of formalin-fixed and paraffin-embedded archival tissue specimens, including 50 cases of NC, 50 cases of AD, 30 cases of CIS, and 63 cases of ICC, were obtained from colonoscopic or surgical resection at Xiangya Hospital, Central South University, China, and used for immunohistochemical staining. The parameters of patients and tissue specimens are shown in Supplementary Table 1.
Tissue processing and LCM
To exclude the interference of stromal elements and adjoining cells, LCM[18] was used to purify the target cells. All NC, AD, CIS and ICC tissues were stained with haematoxylin and eosin and independently evaluated by two experienced pathologists. LCM was performed to purify the cells of interest from each type of tissue according to our previous procedure[17]. Briefly, frozen sections (8 μm thick) of all tissues were prepared using a Leica CM 1900 cryostat at -25 °C. The sections were placed on membrane-coated glass slides (Leica), fixed in 75% alcohol for 30 s, and stained with 0.5% violet-free methyl green (Sigma). The stained sections were air-dried and then subjected to LCM. Each cell population was determined to be 95% homogeneous by microscopic visualization of the captured cells (Supplementary Figure 1).
Protein extraction
The microdissected cells were dissolved in lysis buffer (7 mol/L urea, 2 mol/L thiourea, 65 mmol/L dithiothreitol, 0.1 mmol/L phenylmethylsulfonyl fluoride) at 4 °C for 1 h and then centrifuged at 12000 rpm for 30 min at 4 °C. The supernatant was collected, and the protein concentration was determined by the 2D Quantification Kit (Amersham Biosciences). To diminish the effects of biological variation on the proteomic results, equal amounts of proteins from each individual sample in various types of tissue (NC, AD, CIS and ICC) were pooled to generate a sample for the corresponding type of tissue. Four pooled protein samples (corresponding to the four types of tissues) were obtained for iTRAQ labelling.
Protein digestion and labelling with iTRAQ reagents
Trypsin digestion and iTRAQ labelling were performed according to the manufacturer’s protocol (Applied Biosystems). In brief, 100 μg protein of each pooled sample was reduced, alkylated, and then digested overnight at 37 °C with trypsin (mass spectrometry grade; Promega). The samples were then labelled with iTRAQ reagents as follows: iTRAQ reagent 113, AD; iTRAQ reagent 114, NC; iTRAQ reagent 115, CIS; and iTRAQ reagent 116, ICC. The labelled digests were then mixed and dried.
Off-line 2D LC-MS/MS
The mixed peptides were fractionated according to the procedure described in our previous study[23]. A total of 10 SCX fractions were collected. Each fraction was dried down by the rotary vacuum concentrator, dissolved in buffer C (5% acetonitrile, 0.1% formic acid), and analysed on Triple TOF 5600 systems (Applied Biosystems) in information dependent mode. Briefly, peptides were separated on reverse-phase columns (ZORBAX 300SB-C18 column, 5 μm, 300 Å, 0.1 mm × 15 mm; Micromass) using an Eksigent 1D PLUS system (Applied Biosystems). Peptides were separated by a linear gradient mobile phase A (5% acetonitrile, 0.1% formic acid) and mobile phase B (95% acetonitrile, 0.1% formic acid) from 5 to 40 of mobile phase B in 120 min at a flow rate of 300 nL/min. Survey scans were acquired from 400-1500 with up to 15 precursors selected for MS/MS and dynamic exclusion for 20 s. Each SCX fraction was analysed in duplicate.
Data analysis
Analyst QS 1.1 (Applied Biosystems) was used for data acquisition, and ProteinPilot 4.2 (Applied Biosystems) was used for protein identification and quantification. The precursor tolerance and the iTRAQ fragment tolerance were both set at 0.2 Da. The data analysis parameters were set as follows: Sample type, Itraq 4 plex (peptide labelled); Cys alkylation, MMTS; Digestion, Trypsin; Instrument, Triple TOF 5600; Species, Homo sapiens; ID Focus, Biological modifications; Database, Uniprot human database (release Apr 2013); Search Effort, Thorough; Max missed cleavages, 2; FDR Analysis, Yes; User Modified Parameter Files, No; Bias Correction, Auto; Background Correction, Yes. Identified proteins were grouped by the software to minimize redundancy. All peptides used for the calculation of protein ratios were unique to the given protein or proteins within the group, and peptides that were common to other isoforms or proteins of the same family were ignored.
The protein confidence threshold cutoff was 1.3 (unused ProtScore) with at least one peptide with 95% confidence. The average iTRAQ ratios from the duplicate experiments were calculated for each protein[23,24]. The confidence level of the altered expression of proteins was calculated by ProteinPilot as a P-value, which allows the results to be evaluated based on the confidence level of expression change. In addition, false discovery rate (FDR) for the protein identification was calculated by searching against a concatenated reversed database[25].
Determination of cutoff threshold for significant fold changes in iTRAQ experiments
In a comprehensive iTRAQ experiment, the variations are composed of technical, experimental and biological variations[26]. According to previous reports[26-28], the cutoff threshold for meaningful fold changes over experimental errors can be determined by using the experimental replicate method. Particularly, the main variation in this study was experimental variation, whereas the biological variation was minimized by sample pooling effect. The variations in our method using experimental replicates are considered to be the actual variations in the iTRAQ experiment.
The number of shared quantified proteins in the 2 iTRAQ experiments was 379 based on the following selection criteria: containing more than 2 unique peptides (> 95%), P-value < 0.05, and EF < 2. These 379 proteins were used initially to determine the experimental variations and to confirm the threshold for meaningful fold changes.
Experimental variations for the 113/114, 115/114, and 116/114 reporter ions were calculated using the ratios of the 379 common quantified proteins between the first and second iTRAQ experiments; the experimental variations for the reporter ions were r2 = 0.8499, r2 = 0.8382, and r2 = 0.9283, respectively (Supplementary Figure 2A). In addition, the cutoff threshold for meaningful fold changes (cutoff for DEPs) in the expression ratios of 113/114, 115/114, and 116/114 were determined using the experimental replicate method described in previous studies[27,28]. Accordingly, 90% of the identified proteins in the 2 iTRAQ experiments fell within 50% of the respective experimental variation (Supplementary Figure 2B). Therefore, only fold changes > 1.5 or < 0.67 were considered significant.
Differential proteome expression during colorectal carcinogenesis
To identify the DEPs in the colorectal carcinogenic process, protein expression profiles between two stages of this process (AD, CIS or ICC vs NC; CIS vs AD; ICC vs CIS) were compared. The proteins that met the following criteria were confidently considered as DEPs: (1) proteins were repeatedly identified by the two experiments; (2) proteins were identified based on ≥ 2 peptides; (3) an averaged ratio-fold change > 1.5 or < 0.67 between two stages; (4) proteins in CIS or ICC should be differentially expressed compared with NC; and (5) proteins should be differentially expressed in at least one of the three comparisons between adjacent stages (AD vs NC, CIS vs AD and ICC vs CIS).
Cluster analysis of differential protein expression profiles
The averaged iTRAQ values obtained in two experimental replicates for each protein of the four types of tissues (NC, AD, CIS, and ICC) were log2-transformed. Total DEPs were normalized and clustered with J-express 2012 (http://jexpress.bioinfo.no) into nine categories by using the K-means algorithm with the Pearson correlation distance.
Bioinformatics analysis
The DEPs were first annotated by GO from Biological Process using David software (http://david.abcc.ncifcrf.gov/). The GO terms were considered to be significantly enriched when the corrected P-value was less than 0.05. Pathway analyses of each K-means cluster of proteins were performed using REACTOME software (http://www.reactome.org/). The REACTOME performs an enrichment test to determine whether any Reactome pathways are enriched in the submitted data. A binomial test was used to calculate the probability. The P-values are corrected for multiple testing (Benjamini-Hochberg procedure) that arises from evaluating the submitted list of identifiers against every pathway. The pathway with the corrected P-value less than 0.05 was considered to be significantly enriched.
Immunohistochemistry and evaluation of staining
Immunohistochemistry was performed according to the procedure described in our previous study[26]. Briefly, the sections were incubated with anti-DMBT1 (1:200; Santa Cruz), anti-S100A9 (1:200; Santa Cruz), anti-Galectin-10 (1:200; Abcam), or anti-S100A8 (1:200; Santa Cruz) antibody overnight at 4 °C, and they were then incubated with a biotinylated secondary antibody followed by avidin-biotin peroxidase complex (DAKO) according to the manufacturer’s instructions. Finally, tissue sections were incubated with 3’,3’-diaminobenzidine until a brown colour developed and were then counterstained with Harris’ modified haematoxylin. The evaluation of immunostaining was performed as previously described[29]. A score (ranging from 0-6) was obtained for each case. A combined staining score of ≤ 2 was considered to be negative staining (no expression); a score between 3 and 4 was considered to be moderate staining (expression); and a score between 5 and 6 was considered to be strong staining (high expression).
Statistical analysis
SPSS software (IBM, v19) was used for statistical analyses. Numerical variables with normal distribution were compared using unpaired t-tests or paired t-tests. Non-normal distribution data were compared using Wilcoxon rank sum tests. A P-value less than 0.05 was considered statistically significant.
RESULTS
Identification of quantified proteins during colorectal carcinogenesis
A total of 3211 non-redundant proteins were identified at a minimum confidence level of 95% (unused ProtScore > 1.3) in two iTRAQ experiments. Among these, 2374 proteins were repeatedly identified in the two experiments. In total, 3123 proteins were quantified, and 2319 of them were commonly quantified in the two iTRAQ experiments (Supplementary Figure 3). The detailed protein identification and quantification data are shown in Supplementary Table 2. Using the concatenated target-decoy database search strategy as detailed by Elias and Gygi[25], a 0% rate of false positives was estimated, which further strengthened the reliability of our data.
To define significant changes in protein expression, fold-changes > 1.5 or < 0.66 were established as cutoff values, which were determined using the experimental replicate method as described in the Methods section. A total of 326 DEPs were found according to the selection criteria, of which 199 were found in AD/NC, 307 in CIS/NC, and 228 were in ICC/NC. There were 141 (43%) common DEPs among all three tumor stages compared to NC. All the DEPs in AD/NC group were found in CIS/NC or ICC/NC groups, while there were 50 DEPs specific to CIS/NC, and 9 DEPs specific to ICC/NC (Figure 1). Out of 326 DEPs, 199 were found in AD/NC (86 upregulated and 113 downregulated), 239 were found in CIS/AD (102 upregulated and 137 downregulated), and 244 in ICC/CIS (141 upregulated and 103 downregulated) (Table 1).
Figure 1.
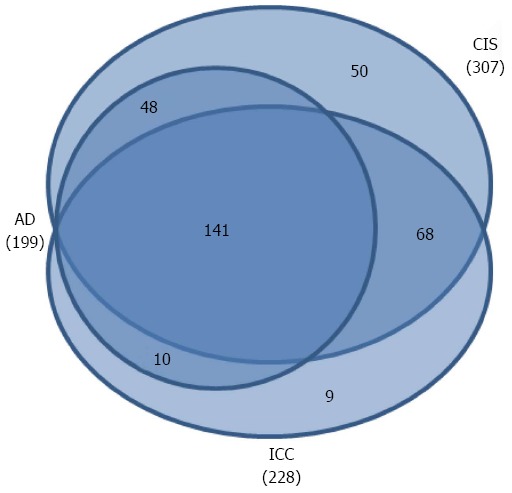
Venn diagrams of comparisons of the differentially expressed proteins from the different stages compared to the NC group.
Table 1.
Number of protein expression changes in comparison between adjacent stages of colon carcinogenesis process
| Comparison |
Protein expression |
|
| Downregulated | Upregulated | |
| AD:NC (113:114) | 113 | 86 |
| CIS:AD (115:113) | 137 | 102 |
| ICC:CIS (116:115) | 103 | 141 |
To obtain a biological view of the DEPs, GO enrichment analysis was employed to discover the significant biological processes. In the comparison of proteome expression in AD (113 reporter) vs NC (114 reporter), out of 199 DEPs, 86 up-regulated proteins were involved in gene expression processes such as “DNA replication”, “DNA metabolic process” and “translation”, whereas 113 down-regulated proteins were involved in energy and metabolism processes such as “blood coagulation”, “response to external stimulus” and “complement activation” (Figure 2A).
Figure 2.

Functional distribution of differentially expressed proteins between adjacent stages of the colon carcinogenesis process. Functional classification of differentially expressed proteins from AD vs NC, CIS vs AD and ICC vs CIS were assigned to “biological process” subcategories. Only significant subcategories for “biological process” are presented. Each subcategory is presented as the percentage of up- and down-regulated proteins.
Upon comparison of the proteomes of CIS (115 reporter) vs AD (113 reporter), we identified 239 DEPs, 102 and 137 of which were up-regulated and down-regulated, respectively. Notably, the down-regulated proteins were involved in energy metabolism pathways such as “respiratory electron transport chain”, “fatty acid beta-oxidation” and “generation of precursor metabolites and energy”, whereas the up-regulated proteins were involved in biological process such as “cell cycle”, “cell-cell adhesion” and “cell-matrix adhesion” (Figure 2B).
Between the expression patterns of ICC (116 reporter) vs CIS (115 reporter), 244 DEPs included 141 up-regulated and 103 down-regulated proteins. The bioinformatics analysis indicated that the up-regulated proteins mainly participate in “complement activation”, “fatty acid beta-oxidation” and “coenzyme metabolic processes”, whereas the down-regulated proteins participate in biological processes associated with “mRNA splicing”, “translation” and “cell-matrix adhesion” (Figure 2C).
Cluster analysis of DEPs and functional analysis
Among the DEPs, some were only differentially expressed in one stage, while others were differentially expressed in multiple stages. To explore the dynamics of DEPs and gain more insights into their biological significance in colorectal carcinogenic processes, K-means clustering and REACTOME pathway analysis were performed. The expression pattern K-means clustering analysis of DEPs showed stage-specific and co-regulated expression profiles.
The 326 DEPs were classified into 9 clusters by the K-means clustering algorithm (Figure 3, Supplementary Table 3). According to the overall tendency, the nine clusters were arbitrarily categorized into three groups. Group 1 consists of clusters 1 and 5, in which the abundance of DEPs increased at all three stages of AD, CIS and IC, and exhibited the highest expression in CIS or IC. Group 2 consists of cluster 2 and 7, in which the abundance of DEPs decreased at all three stages of AD, CIS and IC, and exhibited the lowest expression in CIS or IC. Group 3 consists of clusters 3, 4, 6, 8 and 9, in which the abundance of proteins fluctuated during the colorectal carcinogenic process and was significantly up-regulated or down-regulated only in certain stages. Ideally, the proteins within each cluster are co-regulated proteins, and may have similar biological functions during colorectal carcinogenesis. Pathway analysis with REACTOME revealed that proteins in clusters 1 and 5 were mainly involved in “translation”, “EPH-Ephrin signaling” and “Sema4D induced cell migration and growth-cone collapse”, etc., whereas the proteins in clusters 2 and 7 were associated with “O2/CO2 exchange in erythrocytes”, “glutathione synthesis and recycling,” and “TCA cycle” (Figure 4, Supplementary Tables 4 and 5). Interestingly, the abundance of proteins in clusters 8 and 9, which were mainly involved in pathways related with integrin and complement, were reduced first, and then increased. These proteins may exert different or even opposite functions at different stages of colorectal carcinogenesis through the associated pathways. The DEPs in each cluster were involved in multiple pathways, which indicated that multiple cellular pathways participated in the carcinogenic process, implying the complexity of the process.
Figure 3.
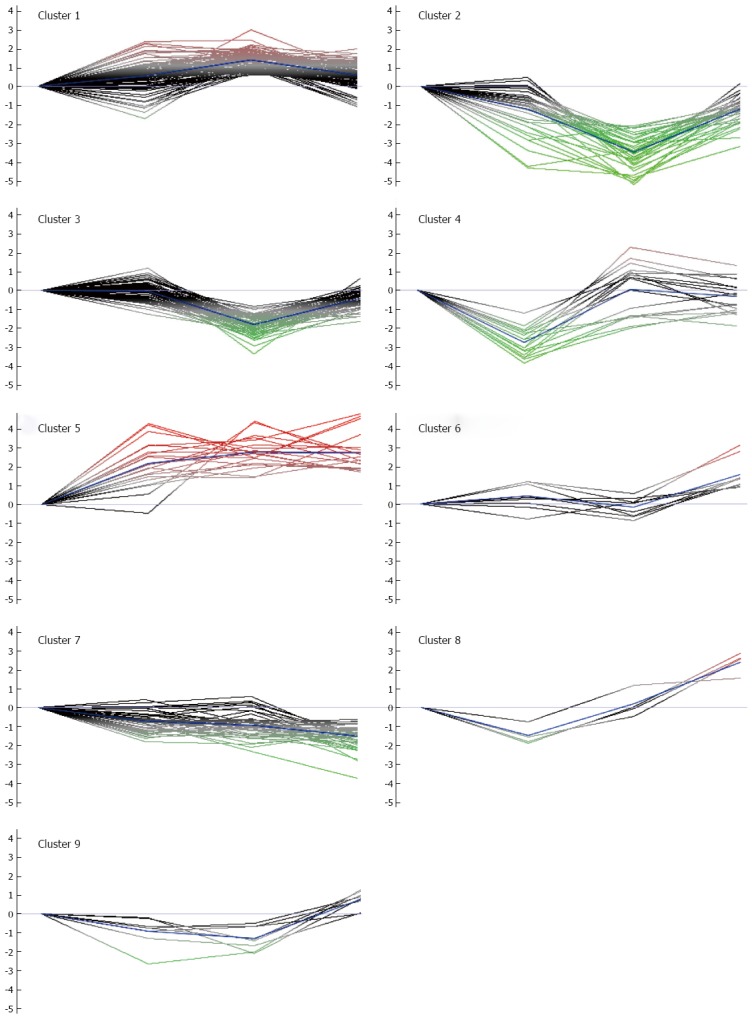
K-mean clusters of differentially expressed proteins. These proteins could be clustered into nine clusters. According to the average tendency, the nine clusters can be arbitrarily categorized into three groups. Group 1 includes clusters 1 and 5, in which the abundance of proteins progressively increased during the colon carcinogenic process. Group 2 consists of clusters 2 and 7, in which the abundance of proteins progressively decreased during the process. Group 3 consists of clusters 3, 4, 6, 8 and 9, in which the abundance of proteins was significantly up-regulated or down-regulated in certain stages of the process.
Figure 4.
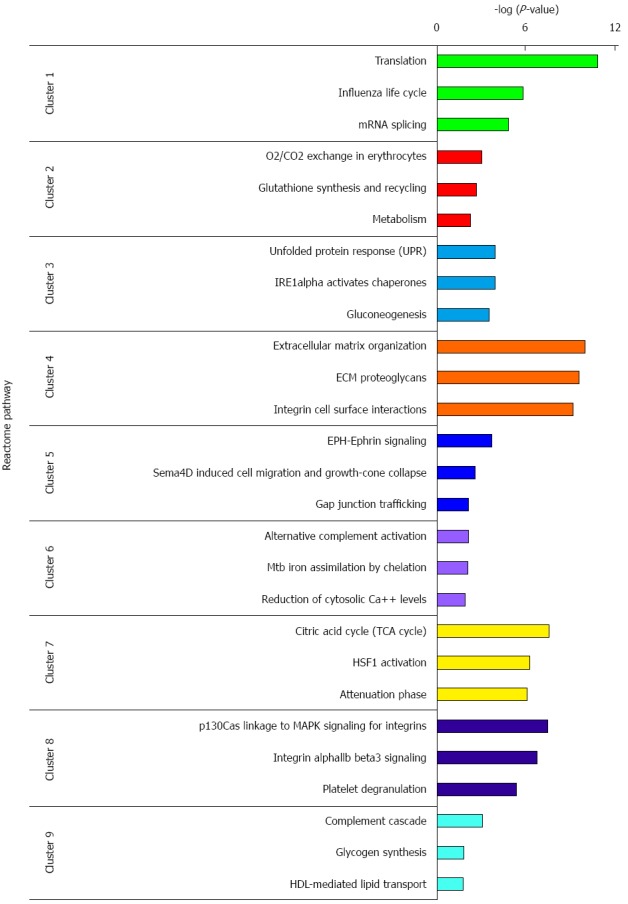
REACTOME pathway analysis of differential expressed proteins in each cluster. Only top 3 significant pathways are presented. Each pathway is presented as negative logarithm of P-value.
Immunohistochemistry
Four of the top ranked proteins (DMBT1, S100A9, Galectin-10, and S100A8), which had expression levels that were progressively up-regulated during colorectal carcinogenic process, were chosen for immunohistochemical verification (Table 2). An independent set of archival tissue specimens including NC, AD, CIS and ICC were used for detection of the expression levels of the four proteins by immunohistochemistry. As shown in Figure 5 and Supplementary Table 3, expression levels of the four proteins significantly increased from early stage, AD, until late stage, ICC, but their expression patterns were not identical. For example, although Galectin-10 expression increased in all pathological stages (P < 0.05 for AD vs NC, CIS vs NC, and ICC vs NC), the expression level was lower at CIS than at AD or (P < 0.05 for CIS vs AD and ICC vs CIS). As another example, S100A8 expression was significantly higher at ICC than at AD or CIS (P < 0.05 for ICC vs AD and ICC vs CIS), although it also increased in all three pathological stages (P < 0.05 for AD vs NC, CIS vs NC and ICC vs AD).
Table 2.
Top 10 differential proteins (up-regulated and down-regulated) between different stages
| No. | GN | Protein name | Cluster | AD:NC | CIS:NC | ICC:NC |
| 1 | DMBT1 | Deleted in malignant brain tumors 1 protein | 5 | 3.12 | 3.43 | 4.82 |
| 2 | S100A9 | Protein S100-A9 | 5 | 2.56 | 2.52 | 4.67 |
| 3 | CLC | Galectin-10 | 5 | 4.21 | 2.50 | 4.54 |
| 4 | S100A8 | Protein S100-A8 | 5 | 1.51 | 1.46 | 3.72 |
| 5 | MPO | Myeloperoxidase | 6 | 1.22 | 0.09 | 3.12 |
| 6 | OLFM4 | Olfactomedin-4 | 5 | 3.89 | 3.01 | 3.02 |
| 7 | LDHA | L-lactate dehydrogenase A chain | 5 | 1.93 | 3.69 | 2.92 |
| 8 | SERPINB5 | Serpin B5 | 5 | 4.32 | 2.69 | 2.78 |
| 9 | EPX | Eosinophil peroxidase | 5 | 3.16 | 2.82 | 2.73 |
| 10 | COL12A1 | Collagen alpha-1(XII) chain | 5 | 0.57 | 4.35 | 2.68 |
| 11 | TNC | Tenascin | 5 | -0.48 | 4.41 | 2.33 |
| 12 | HNRNPA1 | Heterogeneous nuclear ribonucleoprotein A1 | 5 | 2.69 | 3.55 | 2.16 |
| 13 | MUC2 | Mucin-2 | 2 | -0.80 | -5.21 | -0.39 |
| 14 | CA2 | Carbonic anhydrase 2 | 2 | -2.86 | -5.08 | -0.62 |
| 15 | DCN | Decorin | 4 | -3.67 | -2.03 | -1.06 |
| 16 | COL14A1 | Collagen alpha-1(XIV) chain | 2 | -4.25 | -3.42 | -1.06 |
| 17 | LUM | Lumican | 4 | -3.90 | -1.40 | -1.21 |
| 18 | CA1 | Carbonic anhydrase 1 | 2 | -2.63 | -4.96 | -1.74 |
| 19 | ITLN1 | Intelectin-1 | 2 | -2.18 | -5.00 | -1.91 |
| 20 | ASPN | Asporin | 4 | -3.59 | -1.36 | -1.91 |
| 21 | OGN | Mimecan | 2 | -4.35 | -4.71 | -2.29 |
| 22 | GSTP1 | Glutathione S-transferase P | 7 | -1.36 | -1.06 | -2.73 |
| 23 | CKB | Creatine kinase B-type | 2 | -1.26 | -2.98 | -2.76 |
| 24 | PFN1 | Profilin-1 | 7 | -0.47 | 0.30 | -2.84 |
| 25 | CHGA | Chromogranin-A | 2 | -3.44 | -4.84 | -3.20 |
| 26 | TPI1 | Triosephosphate isomerase | 7 | -0.88 | -2.32 | -3.75 |
Figure 5.
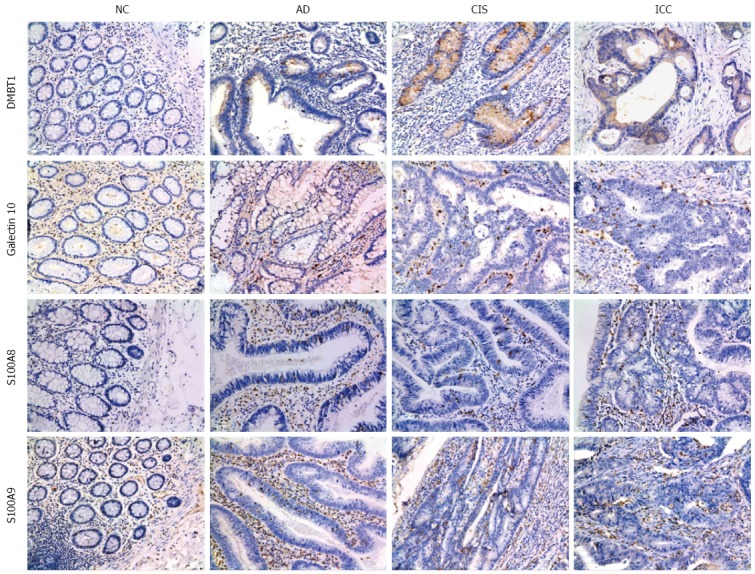
Representative results of immunohistochemistry show the expression of DMBT1, S100A9, Galectin-10 and S100A8 in the NC, AD, CIS and ICC (Original magnification, × 200). DMBT1 immunostaining in NC, AD, CIS and ICC. Negative staining was observed in NC, moderate in AD tissues, and strong cytoplasmic staining in CIS and ICC tissues. S100A9 immunostaining in NC, AD, CIS and ICC. Negative staining was found in NC, weak intralesional staining in AD, moderate intralesional staining in CIS and strong intralesional staining in ICC. Galectin-10 immunostaining in NC, AD, CIS and ICC. Negative staining was found in NC, weak staining in AD and CIS, and moderate staining in ICC. S100A8 immunostaining in NC, AD, CIS and ICC. Negative staining was observed in NC, moderate intralesional staining in AD and CIS, and strong intralesional staining in ICC.
DISCUSSION
Colorectal carcinogenesis has been a typical model for multistage carcinogenesis. The colorectal carcinogenic process includes several typical pathological stages: AD, CIS and ICC. Previous studies mostly focused on genes and acquired a great deal of information supporting the multistage model of colorectal carcinogenesis. With the advent of proteomics, researchers have made efforts to study alterations of the proteomes between certain stages of carcinogenesis[30,31]. Compared to previous reports on colorectal carcinogenesis, our current study mainly investigated the characteristics and dynamics of DEPs throughout multiple typical stages of the colorectal carcinogenic processes. Protein quantification by iTRAQ is a very useful technique to monitor relative changes in proteins in a variety of settings, such as multiple stages of cancer development. Some limitations of iTRAQ technique include underestimation of ratios, limited dynamic range (fold changes of < 2 orders of magnitude), and relatively expensive reagents[32,33]. Since iTRAQ underestimates ratios, we expect that the actual ratio change of up-regulation or down-regulation would be more than that we reported.
There were 199 DEPs founded between AD vs NC. GO enrichment analysis indicated that proteins associated with DNA replication, translation, protein complex biogenesis and assembly were significantly up-regulated in AD, whereas proteins associated with blood coagulation, response to external stimulus, complement activation and cell adhesion, etc., were down-regulated in AD. Previous genetic analysis by Suppression Subtractive Hybridization (SSH) also found that genes involved in DNA replication, complement activation and cell adhesion were significantly differentially expressed in AD[34]. This is in accordance with our findings at the protein level.
Previous studies compared the proteomes between colorectal adenomas and carcinomas, and found that the DEPs participated in RNA processing, translation, and cell adhesion[35,36]. In the present study, we also showed that during the transition from AD to CIS, the DEPs were associated with cell-cell adhesion, cell-matrix adhesion, translation and RNA metabolic processing. In addition, our results showed that the up-regulated proteins were associated with cell cycle and cellular component movement, while the down-regulated proteins were associated with respiratory electron transport chain and fatty acid beta-oxidation. Previous studies using genome-wide mRNA expression profile analysis showed that the pathway of fatty acid metabolism is down-regulated in CRC compared to adenomas[37]. In addition to CRC, proteins involved in fatty acid β-oxidation were also found to be down-regulated in pancreatic cancer cells[38,39].
Between CIS and ICC, the proteins associated with complement activation were up-regulated in ICC. The multiple roles that complement proteins play in carcinogenesis, including functions that facilitate cancer metastasis such as promotion of angiogenesis, invasion and migration, have been reviewed[40,41]. Furthermore, recent studies demonstrate that activation of the complement C5 component C5a and its receptor C5aR can promote cancer cell invasion[42,43].
DMBT1 (deleted in malignant brain tumours 1) gene is located in 10q25.3-q26.1, a region with frequent LOH in many types of human cancers. Therefore, DMBT1 was proposed to be a tumour suppressor[44]. DMBT1 deletion occurs in brain tumours[45], and down-regulation of DMBT1 was reported in mammary tumours, oral squamous cell carcinoma, skin cancers and other tumour types[46,47]. However, there are also a number of reports showing that DMBT1 was up-regulated in cancers. For example, Dolznig et al[48] found that DMBT1 expression was increased in colonic samples compared to normal controls. In our present study, we also found that DMBT1 expression was up-regulated from early to late stages of colonic carcinogenesis. In addition to CRC, DMBT1 expression was also up-regulated during oesophageal carcinogenesis[49]. Since DMBT1 expression was up-regulated in some cancer types while down-regulated in others, the mechanism by which DMBT1 contributes to carcinogenesis could be tissue-specific, and needs more investigation.
S100A9 and S100A8 proteins are members of a family of Ca2+ binding proteins. Both proteins are often co-expressed and form a heterodimer to exert their biological functions. Overexpression of S100A8 and S100A9 has been associated with carcinogenesis[50-52]. For example, Stulík et al[53] reported that S100A9/A8 were up-regulated in human colon carcinoma. S100A9/A8 could activate MAPK and NF-κB pathways in colon tumour progression[50]. Our results showed that S100A9/A8 expression was gradually up-regulated during carcinogenesis. The tendency of both protein expression levels during colonic carcinogenesis was similar, which was consistent with the performance of their biological function as a heterodimer. Our study suggested that both proteins are associated with colorectal carcinoma progression, which supported the therapeutic strategies of blocking S100A9/A8 activity for either inflammatory diseases or cancer[52].
Galectins are a family of proteins characterized by their binding specificity for β-galactoside sugars. The best understood galectin in cancer is Galectin-3. Galectin-3 has been shown to play important roles in tumorigenesis processes, including transformation, metastasis and invasion[54,55]. As for Galectin-10, Ågesen et al[56] demonstrated that Galectin-10 was the most differentially expressed gene, with 10-fold higher expression in early- vs late-onset CRC, and was important in the development of early-onset CRC.
The present work investigated for the first time the dynamic expression patterns of DEPs in multistage carcinogenesis of CRC using quantitative proteomic methods. We systematically compared the characteristics and dynamics of the expressed proteins across the various stages of colorectal carcinogenesis. The findings reported here provide a basis for discovery of candidate biomarkers for early diagnosis of CRC, and give clues for further investigation of the mechanisms of colorectal carcinogenesis and for discovery of new therapeutic targets.
COMMENTS
Background
Colorectal cancer is the third leading cause of cancer death in the world. Colorectal carcinogenesis is a multistep and complicated process, from normal colonic mucosa, adenoma, carcinoma in situ, and ultimately to invasive colorectal carcinoma.
Research frontiers
In recent years, much progress has been made in understanding genetic changes in the colonic carcinogenesis process, and many studies have been conducted to analyze differentially expressed proteins between certain stages of colorectal carcinogenesis
Innovations and breakthroughs
In this study, proteomic analysis was used to identify differentially expressed proteins in the human colonic epithelial carcinogenic process, including normal colon, adenoma, carcinoma in situ and invasive carcinomas tissues. A total of 326 proteins were identified to be differentially expressed. The differential expression of four proteins (DMBT1, S100A9, Galectin-10, and S100A8) was validated using immunohistochemistry.
Applications
These findings give insights into our understanding of the mechanisms of colorectal carcinogenesis. In addition, these studies provide a list of differentially expressed protein as potential biomarkers.
Peer-review
This paper provides practical and novel proteomic information that is currently not known with respect to the expression of various proteins during colon carcinogenesis. It is well written, and data presented are interesting.
Footnotes
Supported by the National “973” Project of China, No. 2011CB910704; and National Natural Science Foundation of China, No. 81372904, No. 81570537 and No. 81272971.
Institutional review board statement: The study was reviewed and approved by the XiangYa Hospital Institutional Review Board.
Conflict-of-interest statement: No conflict of interest.
Data sharing statement: Technical appendix, and dataset available from the corresponding author at email yonghengchen@gmail.com.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: December 7, 2015
First decision: December 30, 2015
Article in press: March 18, 2016
P- Reviewer: Shiao SL, Uppara M, Zoller M S- Editor: Qi Y L- Editor: Wang TQ E- Editor: Ma S
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 3.Jemal A, Center MM, DeSantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010;19:1893–1907. doi: 10.1158/1055-9965.EPI-10-0437. [DOI] [PubMed] [Google Scholar]
- 4.Tol J, Koopman M, Cats A, Rodenburg CJ, Creemers GJ, Schrama JG, Erdkamp FL, Vos AH, van Groeningen CJ, Sinnige HA, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med. 2009;360:563–572. doi: 10.1056/NEJMoa0808268. [DOI] [PubMed] [Google Scholar]
- 5.Cardoso J, Boer J, Morreau H, Fodde R. Expression and genomic profiling of colorectal cancer. Biochim Biophys Acta. 2007;1775:103–137. doi: 10.1016/j.bbcan.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 6.de Wijkerslooth TR, Bossuyt PM, Dekker E. Strategies in screening for colon carcinoma. Neth J Med. 2011;69:112–119. [PubMed] [Google Scholar]
- 7.Alvarez-Chaver P, Otero-Estévez O, Páez de la Cadena M, Rodríguez-Berrocal FJ, Martínez-Zorzano VS. Proteomics for discovery of candidate colorectal cancer biomarkers. World J Gastroenterol. 2014;20:3804–3824. doi: 10.3748/wjg.v20.i14.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roth AD, Tejpar S, Delorenzi M, Yan P, Fiocca R, Klingbiel D, Dietrich D, Biesmans B, Bodoky G, Barone C, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol. 2010;28:466–474. doi: 10.1200/JCO.2009.23.3452. [DOI] [PubMed] [Google Scholar]
- 9.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 10.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 11.Worthley DL, Whitehall VL, Spring KJ, Leggett BA. Colorectal carcinogenesis: road maps to cancer. World J Gastroenterol. 2007;13:3784–3791. doi: 10.3748/wjg.v13.i28.3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet. 2014;383:1490–1502. doi: 10.1016/S0140-6736(13)61649-9. [DOI] [PubMed] [Google Scholar]
- 13.Shi Y, Li Z, Zheng W, Liu X, Sun C, Laugsand JB, Liu Z, Cui G. Changes of immunocytic phenotypes and functions from human colorectal adenomatous stage to cancerous stage: Update. Immunobiology. 2015;220:1186–1196. doi: 10.1016/j.imbio.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 14.Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–627. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- 15.Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 16.Espina V, Wulfkuhle JD, Calvert VS, VanMeter A, Zhou W, Coukos G, Geho DH, Petricoin EF, Liotta LA. Laser-capture microdissection. Nat Protoc. 2006;1:586–603. doi: 10.1038/nprot.2006.85. [DOI] [PubMed] [Google Scholar]
- 17.Yao H, Zhang Z, Xiao Z, Chen Y, Li C, Zhang P, Li M, Liu Y, Guan Y, Yu Y, et al. Identification of metastasis associated proteins in human lung squamous carcinoma using two-dimensional difference gel electrophoresis and laser capture microdissection. Lung Cancer. 2009;65:41–48. doi: 10.1016/j.lungcan.2008.10.024. [DOI] [PubMed] [Google Scholar]
- 18.Li M, Li C, Li D, Xie Y, Shi J, Li G, Guan Y, Li M, Zhang P, Peng F, et al. Periostin, a stroma-associated protein, correlates with tumor invasiveness and progression in nasopharyngeal carcinoma. Clin Exp Metastasis. 2012;29:865–877. doi: 10.1007/s10585-012-9465-5. [DOI] [PubMed] [Google Scholar]
- 19.Lin HC, Zhang FL, Geng Q, Yu T, Cui YQ, Liu XH, Li J, Yan MX, Liu L, He XH, et al. Quantitative proteomic analysis identifies CPNE3 as a novel metastasis-promoting gene in NSCLC. J Proteome Res. 2013;12:3423–3433. doi: 10.1021/pr400273z. [DOI] [PubMed] [Google Scholar]
- 20.Zhang PF, Zeng GQ, Hu R, Li C, Yi H, Li MY, Li XH, Qu JQ, Wan XX, He QY, et al. Identification of flotillin-1 as a novel biomarker for lymph node metastasis and prognosis of lung adenocarcinoma by quantitative plasma membrane proteome analysis. J Proteomics. 2012;77:202–214. doi: 10.1016/j.jprot.2012.08.021. [DOI] [PubMed] [Google Scholar]
- 21.Wang WS, Liu XH, Liu LX, Lou WH, Jin DY, Yang PY, Wang XL. iTRAQ-based quantitative proteomics reveals myoferlin as a novel prognostic predictor in pancreatic adenocarcinoma. J Proteomics. 2013;91:453–465. doi: 10.1016/j.jprot.2013.06.032. [DOI] [PubMed] [Google Scholar]
- 22.Wu WW, Wang G, Baek SJ, Shen RF. Comparative study of three proteomic quantitative methods, DIGE, cICAT, and iTRAQ, using 2D gel- or LC-MALDI TOF/TOF. J Proteome Res. 2006;5:651–658. doi: 10.1021/pr050405o. [DOI] [PubMed] [Google Scholar]
- 23.Zeng GQ, Zhang PF, Deng X, Yu FL, Li C, Xu Y, Yi H, Li MY, Hu R, Zuo JH, et al. Identification of candidate biomarkers for early detection of human lung squamous cell cancer by quantitative proteomics. Mol Cell Proteomics. 2012;11:M111.013946. doi: 10.1074/mcp.M111.013946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang WS, Liu XH, Liu LX, Jin DY, Yang PY, Wang XL. Identification of proteins implicated in the development of pancreatic cancer-associated diabetes mellitus by iTRAQ-based quantitative proteomics. J Proteomics. 2013;84:52–60. doi: 10.1016/j.jprot.2013.03.031. [DOI] [PubMed] [Google Scholar]
- 25.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 26.Gan CS, Chong PK, Pham TK, Wright PC. Technical, experimental, and biological variations in isobaric tags for relative and absolute quantitation (iTRAQ) J Proteome Res. 2007;6:821–827. doi: 10.1021/pr060474i. [DOI] [PubMed] [Google Scholar]
- 27.Jin J, Park J, Kim K, Kang Y, Park SG, Kim JH, Park KS, Jun H, Kim Y. Detection of differential proteomes of human beta-cells during islet-like differentiation using iTRAQ labeling. J Proteome Res. 2009;8:1393–1403. doi: 10.1021/pr800765t. [DOI] [PubMed] [Google Scholar]
- 28.Glen A, Gan CS, Hamdy FC, Eaton CL, Cross SS, Catto JW, Wright PC, Rehman I. iTRAQ-facilitated proteomic analysis of human prostate cancer cells identifies proteins associated with progression. J Proteome Res. 2008;7:897–907. doi: 10.1021/pr070378x. [DOI] [PubMed] [Google Scholar]
- 29.Liu YF, Zhang PF, Li MY, Li QQ, Chen ZC. Identification of annexin A1 as a proinvasive and prognostic factor for lung adenocarcinoma. Clin Exp Metastasis. 2011;28:413–425. doi: 10.1007/s10585-011-9380-1. [DOI] [PubMed] [Google Scholar]
- 30.Mu Y, Chen Y, Zhang G, Zhan X, Li Y, Liu T, Li G, Li M, Xiao Z, Gong X, et al. Identification of stromal differentially expressed proteins in the colon carcinoma by quantitative proteomics. Electrophoresis. 2013;34:1679–1692. doi: 10.1002/elps.201200596. [DOI] [PubMed] [Google Scholar]
- 31.Besson D, Pavageau AH, Valo I, Bourreau A, Bélanger A, Eymerit-Morin C, Moulière A, Chassevent A, Boisdron-Celle M, Morel A, et al. A quantitative proteomic approach of the different stages of colorectal cancer establishes OLFM4 as a new nonmetastatic tumor marker. Mol Cell Proteomics. 2011;10:M111.009712. doi: 10.1074/mcp.M111.009712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang H, Alvarez S, Hicks LM. Comprehensive comparison of iTRAQ and label-free LC-based quantitative proteomics approaches using two Chlamydomonas reinhardtii strains of interest for biofuels engineering. J Proteome Res. 2012;11:487–501. doi: 10.1021/pr2008225. [DOI] [PubMed] [Google Scholar]
- 33.Shirran SL, Botting CH. A comparison of the accuracy of iTRAQ quantification by nLC-ESI MSMS and nLC-MALDI MSMS methods. J Proteomics. 2010;73:1391–1403. doi: 10.1016/j.jprot.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DU G, Fang X, Dai W, Zhang R, Liu R, Dang X. Comparative gene expression profiling of normal and human colorectal adenomatous tissues. Oncol Lett. 2014;8:2081–2085. doi: 10.3892/ol.2014.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Albrethsen J, Knol JC, Piersma SR, Pham TV, de Wit M, Mongera S, Carvalho B, Verheul HM, Fijneman RJ, Meijer GA, et al. Subnuclear proteomics in colorectal cancer: identification of proteins enriched in the nuclear matrix fraction and regulation in adenoma to carcinoma progression. Mol Cell Proteomics. 2010;9:988–1005. doi: 10.1074/mcp.M900546-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knol JC, de Wit M, Albrethsen J, Piersma SR, Pham TV, Mongera S, Carvalho B, Fijneman RJ, Meijer GA, Jiménez CR. Proteomics of differential extraction fractions enriched for chromatin-binding proteins from colon adenoma and carcinoma tissues. Biochim Biophys Acta. 2014;1844:1034–1043. doi: 10.1016/j.bbapap.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 37.Carvalho B, Sillars-Hardebol AH, Postma C, Mongera S, Terhaar Sive Droste J, Obulkasim A, van de Wiel M, van Criekinge W, Ylstra B, Fijneman RJ, et al. Colorectal adenoma to carcinoma progression is accompanied by changes in gene expression associated with ageing, chromosomal instability, and fatty acid metabolism. Cell Oncol (Dordr) 2012;35:53–63. doi: 10.1007/s13402-011-0065-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou W, Liotta LA, Petricoin EF. Cancer metabolism: what we can learn from proteomic analysis by mass spectrometry. Cancer Genomics Proteomics. 2012;9:373–381. [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou W, Capello M, Fredolini C, Racanicchi L, Piemonti L, Liotta LA, Novelli F, Petricoin EF. Proteomic analysis reveals Warburg effect and anomalous metabolism of glutamine in pancreatic cancer cells. J Proteome Res. 2012;11:554–563. doi: 10.1021/pr2009274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mamidi S, Höne S, Kirschfink M. The complement system in cancer: Ambivalence between tumour destruction and promotion. Immunobiology. 2015:Epub ahead of print. doi: 10.1016/j.imbio.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 41.Rutkowski MJ, Sughrue ME, Kane AJ, Mills SA, Parsa AT. Cancer and the complement cascade. Mol Cancer Res. 2010;8:1453–1465. doi: 10.1158/1541-7786.MCR-10-0225. [DOI] [PubMed] [Google Scholar]
- 42.Nitta H, Wada Y, Kawano Y, Murakami Y, Irie A, Taniguchi K, Kikuchi K, Yamada G, Suzuki K, Honda J, et al. Enhancement of human cancer cell motility and invasiveness by anaphylatoxin C5a via aberrantly expressed C5a receptor (CD88) Clin Cancer Res. 2013;19:2004–2013. doi: 10.1158/1078-0432.CCR-12-1204. [DOI] [PubMed] [Google Scholar]
- 43.Maeda Y, Kawano Y, Wada Y, Yatsuda J, Motoshima T, Murakami Y, Kikuchi K, Imamura T, Eto M. C5aR is frequently expressed in metastatic renal cell carcinoma and plays a crucial role in cell invasion via the ERK and PI3 kinase pathways. Oncol Rep. 2015;33:1844–1850. doi: 10.3892/or.2015.3800. [DOI] [PubMed] [Google Scholar]
- 44.Deichmann M, Mollenhauer J, Helmke B, Thome M, Hartschuh W, Poustka A, Näher H. Analysis of losses of heterozygosity of the candidate tumour suppressor gene DMBT1 in melanoma resection specimens. Oncology. 2002;63:166–172. doi: 10.1159/000063802. [DOI] [PubMed] [Google Scholar]
- 45.Pang JC, Dong Z, Zhang R, Liu Y, Zhou LF, Chan BW, Poon WS, Ng HK. Mutation analysis of DMBT1 in glioblastoma, medulloblastoma and oligodendroglial tumors. Int J Cancer. 2003;105:76–81. doi: 10.1002/ijc.11019. [DOI] [PubMed] [Google Scholar]
- 46.Wu W, Kemp BL, Proctor ML, Gazdar AF, Minna JD, Hong WK, Mao L. Expression of DMBT1, a candidate tumor suppressor gene, is frequently lost in lung cancer. Cancer Res. 1999;59:1846–1851. [PubMed] [Google Scholar]
- 47.Mori M, Shiraishi T, Tanaka S, Yamagata M, Mafune K, Tanaka Y, Ueo H, Barnard GF, Sugimachi K. Lack of DMBT1 expression in oesophageal, gastric and colon cancers. Br J Cancer. 1999;79:211–213. doi: 10.1038/sj.bjc.6690035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dolznig H, Rupp C, Puri C, Haslinger C, Schweifer N, Wieser E, Kerjaschki D, Garin-Chesa P. Modeling colon adenocarcinomas in vitro a 3D co-culture system induces cancer-relevant pathways upon tumor cell and stromal fibroblast interaction. Am J Pathol. 2011;179:487–501. doi: 10.1016/j.ajpath.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alvarez H, Opalinska J, Zhou L, Sohal D, Fazzari MJ, Yu Y, Montagna C, Montgomery EA, Canto M, Dunbar KB, et al. Widespread hypomethylation occurs early and synergizes with gene amplification during esophageal carcinogenesis. PLoS Genet. 2011;7:e1001356. doi: 10.1371/journal.pgen.1001356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ichikawa M, Williams R, Wang L, Vogl T, Srikrishna G. S100A8/A9 activate key genes and pathways in colon tumor progression. Mol Cancer Res. 2011;9:133–148. doi: 10.1158/1541-7786.MCR-10-0394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Srikrishna G. S100A8 and S100A9: new insights into their roles in malignancy. J Innate Immun. 2012;4:31–40. doi: 10.1159/000330095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Markowitz J, Carson WE. Review of S100A9 biology and its role in cancer. Biochim Biophys Acta. 2013;1835:100–109. doi: 10.1016/j.bbcan.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stulík J, Osterreicher J, Koupilová K, Knízek A, Bures J, Jandík P, Langr F, Dedic K, Jungblut PR. The analysis of S100A9 and S100A8 expression in matched sets of macroscopically normal colon mucosa and colorectal carcinoma: the S100A9 and S100A8 positive cells underlie and invade tumor mass. Electrophoresis. 1999;20:1047–1054. doi: 10.1002/(SICI)1522-2683(19990101)20:4/5<1047::AID-ELPS1047>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 54.Reticker-Flynn NE, Malta DF, Winslow MM, Lamar JM, Xu MJ, Underhill GH, Hynes RO, Jacks TE, Bhatia SN. A combinatorial extracellular matrix platform identifies cell-extracellular matrix interactions that correlate with metastasis. Nat Commun. 2012;3:1122. doi: 10.1038/ncomms2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Radosavljevic G, Volarevic V, Jovanovic I, Milovanovic M, Pejnovic N, Arsenijevic N, Hsu DK, Lukic ML. The roles of Galectin-3 in autoimmunity and tumor progression. Immunol Res. 2012;52:100–110. doi: 10.1007/s12026-012-8286-6. [DOI] [PubMed] [Google Scholar]
- 56.Ågesen TH, Berg M, Clancy T, Thiis-Evensen E, Cekaite L, Lind GE, Nesland JM, Bakka A, Mala T, Hauss HJ, et al. CLC and IFNAR1 are differentially expressed and a global immunity score is distinct between early- and late-onset colorectal cancer. Genes Immun. 2011;12:653–662. doi: 10.1038/gene.2011.43. [DOI] [PubMed] [Google Scholar]